- NEWS AND VIEWS
Pressure regulates immune-cell function
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 573, 41-42 (2019)
doi: https://doi.org/10.1038/d41586-019-02339-4
References
Huang, C. & Niethammer, P. Immunity 48, 1006–1013 (2018).
O’Neill, L. A. J. & Hardie, D. G. Nature 493, 346–355 (2013).
Sadiku, P. & Walmsley, S. R. EMBO Rep. 20, e47388 (2019).
Taylor, C. T. & Colgan, S. P. Nature Rev. Immunol. 17, 774–785 (2017).
Solis, A. G. et al. Nature 573, 69–74 (2019).
Begandt, D., Thome, S., Sperandio, M. & Walzog, B. J. Leukoc. Biol. 102, 699–709 (2017).
Mead, J. & Whittenberger, J. L. J. Appl. Physiol. 5, 779–796 (1953).
Kaelin, W. G. Jr & Ratcliffe, P. J. Mol. Cell 30, 393–402 (2008).
Lin, N. & Simon, M. C. J. Clin. Invest. 126, 3661–3671 (2016).
Palazon, A., Goldrath, A. W., Nizet, V. & Johnson, R. S. Immunity 41, 518–528 (2014).
Liu, Y. V. et al. J. Biol. Chem. 282, 37064–37073 (2007).
Li, M. et al. FEBS Lett. 586, 3888–3893 (2012).
 Read the paper: Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity
Read the paper: Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity
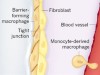 Macrophages form a protective cellular barrier in joints
Macrophages form a protective cellular barrier in joints
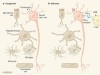 Infiltration of old brains by T cells causes dysfunction of neural stem cells
Infiltration of old brains by T cells causes dysfunction of neural stem cells