- NEWS AND VIEWS
Immune cells track hard-to-target brain tumours
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 565, 170-171 (2019)
doi: https://doi.org/10.1038/d41586-018-07728-9
References
Yarchoan, M. et al. N. Engl. J. Med. 377, 2500–2501 (2017).
Wolchok, J. D. et al. N. Engl. J. Med. 377, 1345–1356 (2017).
Yarchoan, M. et al. Nature Rev. Cancer 17, 209–222 (2017).
Keskin, D. B. et al. Nature 565, 234–239 (2019).
Hilf, N. et al. Nature 565, 240–245 (2019).
Ott, P. A. et al. Nature 547, 217–221 (2017).
Sahin, U. et al. Nature 547, 222–226 (2017).
Kinkead, H. L. et al. JCI Insight 3, e122857 (2018).
Friaetta, J. A. et al. Nature Med. 24, 563–571 (2018).
Popovic, A. et al. J. Clin. Invest. 128, 3209–3318 (2018).
Competing Interests
E.M.J. serves on the scientific advisory boards of four immunotherapy companies: CSTONE, Genocea, Adaptive Biotech and DragonFly. E.M.J. receives grant money from Aduro Biotech, Hertix and Bristol-Myers Squibb and has the potential to receive royalties on a vaccine licensed to Aduro Biotech. Also, E.M.J. is the president of the American Association for Cancer Research and serves as the chairperson of the National Cancer Advisory Board.
 Read the paper: Actively personalized vaccination trial for newly diagnosed glioblastoma
Read the paper: Actively personalized vaccination trial for newly diagnosed glioblastoma
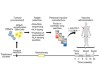 Read the paper: Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial
Read the paper: Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial
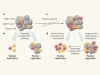 Precision T-cell therapy targets tumours
Precision T-cell therapy targets tumours
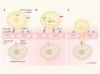 T cells engineered to home in on brain cancer
T cells engineered to home in on brain cancer