- NEWS AND VIEWS
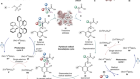
An exciting tool for asymmetric synthesis
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 564, 197-199 (2018)
doi: https://doi.org/10.1038/d41586-018-07625-1
References
Hölzl-Hobmeier, A. et al. Nature 564, 240-243 (2018).
Yang, C. & Inoue, Y. Chem. Soc. Rev. 43, 4123-4143 (2014).
Rodriguez, O. & Morrison, H. J. Chem. Soc. D Chem. Commun. 679 (1971).
Alonso, R. & Bach, T. Angew. Chem. Int. Ed. 53, 4368–4371 (2014).
Ley, C., Morlet-Savary, F., Jacques, P. & Fouassier, J. P. Chem. Phys. 255, 335–346 (2000).
Drucker, C. S., Toscano, V. G. & Weiss, R. G. J. Am. Chem. Soc. 95, 6482–6484 (1973).
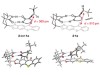 Read the paper: Catalytic deracemization of chiral allenes by sensitized excitation with visible light
Read the paper: Catalytic deracemization of chiral allenes by sensitized excitation with visible light
 Enzymes team up with light-activated catalysts
Enzymes team up with light-activated catalysts
 Light opens pathways for nickel catalysis
Light opens pathways for nickel catalysis