Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain
the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in
Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles
and JavaScript.
Classic reaction re-engineered through molecular face recognition
A catalyst has been developed that recognizes the topology of just one face of a planar reaction intermediate. Remarkably, this enables one mirror-image isomer of the reaction product to be made selectively.
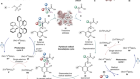
Complex 3D molecules are ubiquitous in daily life, with functions in everything from high-performance materials to smart medicines. Just as 3D shape often reflects function on the macroscopic scale, so, too, does it determine microscopic behaviour. When building 3D molecules for applications, chemists must therefore develop synthetic routes that ensure that each atom is correctly positioned in the final product. However, even if such precision is achieved, some molecules can be produced as mirror-image isomers (enantiomers), and their properties might vary widely, affecting their use in applications. In a paper in Nature, Wendlandt et al.1 report a reaction that not only enables the synthesis of single enantiomers of molecules, but does so using a reaction mechanism that was thought to be intrinsically lacking in enantioselectivity.

 Read the paper: Quaternary stereocentres via an enantioconvergent catalytic SN1 reaction
Read the paper: Quaternary stereocentres via an enantioconvergent catalytic SN1 reaction
 Precision pruning of molecules
Precision pruning of molecules
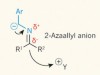 Natural polarity inverted
Natural polarity inverted