
Abstract
G protein-coupled receptors (GPCRs) mediate most of our physiological responses to hormones, neurotransmitters and environmental stimulants. They are considered as the most successful therapeutic targets for a broad spectrum of diseases. Multiple sclerosis (MS) is an inflammatory disease that is characterized by immune-mediated demyelination and degeneration of the central nervous system (CNS). It is the leading cause of non-traumatic disability in young adults. Great progress has been made over the past few decades in understanding the pathogenesis of MS. Numerous data from animal and clinical studies indicate that many GPCRs are critically involved in various aspects of MS pathogenesis, including antigen presentation, cytokine production, T-cell differentiation, T-cell proliferation, T-cell invasion, etc. In this review, we summarize the recent findings regarding the expression or functional changes of GPCRs in MS patients or animal models, and the influences of GPCRs on disease severity upon genetic or pharmacological manipulations. Hopefully some of these findings will lead to the development of novel therapies for MS in the near future.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS), also known as disseminated sclerosis or encephalomyelitis disseminata, is an inflammatory disease that is characterized by immune-mediated demyelination and neurodegeneration of the central nervous system (CNS). It leads to substantial disability through deficits of sensation and of motor, autonomic, and neurocognitive functions. The disease onset usually occurs in young adults between 20 to 40 years of age 1,2 with a prevalence that ranges between 2 and 150 per 100 000 3. The disease is usually not life-threatening, but its socioeconomic importance is second only to trauma in young adults 4,5.
CD4+ T-cell-mediated autoimmunity has long been accepted as one of the most important aspects of MS pathogenesis, especially for the early initiation of the disease 6,7. T-helper type 1 (Th1) cells, characterized by the production of interferon-γ (IFN-γ), have been considered the type of effector T-helper cells that mediate the pathogenesis of MS. Subsequent studies have revealed that the IL-17-expressing T-helper cells (Th17) are also involved and are at least as critical as Th1 cells in this disease. Mice with fewer Th17 cells are less susceptible to experimental autoimmune encephalomyelitis (EAE) 8,9, a mouse model of MS; and IL-17-expressing T cells have been found in lesions of brain tissues from patients with MS 10.
There are several types of MS, including Benign MS, Relapsing Remitting MS (RRMS), Secondary Progressive MS (SPMS), Primary Progressive MS (PPMS), and Malignant MS (also known as Marberg Variant MS). RRMS is the most frequent (85%-90%) form and affects women about twice as often as men. Patients tend to experience an attack or series of attacks (exacerbations) followed by complete or partial remission. Most RRMS patients later develop SPMS. At that stage, there are no real periods of remission, but only breaks in attack duration with no real recovery from symptoms. About 10%-15% of patients presenting with insidious disease onset and steady progression are termed PPMS 7. It is characterized by gradual clinical decline with no real or distinct periods of remission. Magnetic resonance imaging (MRI) 11 or histopathological evaluation 12,13 also revealed heterogeneity in morphological alterations of the brain in different patients. It is still not clear which factors may contribute to the different disease courses and the heterogeneity in clinical presentations. Complex genetic effects and environmental components that translate into different immune abnormalities and/or increased vulnerability of CNS tissue to inflammatory insults or reduced ability to repair damage are certainly involved. Relatives of people who have the disease have an increased risk; if a patient with MS has an identical twin, that twin's risk climbs to more than 25% 4,14. But when a team of US researchers compared the complete genomes of twin females with each other, they failed to find any genetic differences that might cause MS 15. This observation indicates that the environmental stress might also play an important role in eventually triggering the pathogenesis of MS.
G protein-coupled receptors (GPCRs), also known as 7-transmembrane receptors, are the largest family of cell surface receptors involved in transmitting extracellular environmental signals into the cells. These receptors are activated by a wide variety of stimulations, including light, odorant molecules, peptide and non-peptide neurotransmitters, hormones, growth factors, lipids, etc. 16. The GPCR family comprises approximately 2% of the human genome and remains a central focus in basic pharmacology studies and drug discovery efforts 17. After agonist binding, the activated receptors catalyze the exchange of GDP for GTP on the α-subunit of heterotrimeric G proteins (composed of α-, β-, and γ-subunits), which in turn engages conformational changes that lead to the dissociation of the Gα from the dimeric Gβγ subunits 18. Both the Gα- and Gβγ- subunits can convey the extracellular signals by activating or suppressing downstream effector molecules, such as adenylyl and guanylyl cyclases, phosphodiesterases, phospholipases, phosphoinositide 3-kinases (PI3K), ion channels and other signaling components 16.
GPCRs have emerged as the most important targets for human therapeutics due to their large numbers and critical roles in the physiology of vital systems, such as cardiovascular, nervous, immune, metabolic, and endocrine systems. The prominent roles of GPCRs in cancer are also well recognized 19,20. These receptors are the target of > 50% of the current therapeutic agents on the market, including more than a quarter of the 100 top-selling drugs with benefits in the range of several billion US dollars each year. Here, we review recent progress on the roles of GPCRs in the pathogenesis of MS and hope that some of these receptors might become new therapeutic targets for this disease in the near future.
GPCRs involved in MS or EAE
Adenosine receptors
Dysfunction of adenosinergic system has been implicated in the development of MS in humans and EAE in animals. Blood adenosine level decreases greatly in MS patients 21. Among the four known subtypes of adenosine receptors – referred to as A1, A2A, A2B, and A3, the role of A1 in MS pathology has been intensively studied in both clinical samples and animal models. A1 was selectively diminished on cells of monocyte/macrophage lineage in both brain and blood samples from MS patients. This reduction potentially led to increased macrophage activation and CNS inflammation 21,22. In animal model, the A1 knockout mice developed a severe progressive-relapsing form of EAE with extensive inflammation and demyelination in CNS compared with the corresponding controls 23. Conversely, treatment with the A1 receptor agonist ADAC reduced spinal cord injuries in EAE mice 23. Caffeine, a non-selective antagonist of adenosine receptors, has also been shown to alleviate EAE in mice and rat 23,24,25. It has been postulated that chronic treatment with caffeine may benefit EAE animals by upregulating A1 receptor and TGF-β, and suppressing IFN-γ 25. These results suggest that adenosine might act through the A1 receptor to suppress inflammation and that dysfunction of A1 contributes to the pathogenesis of MS.
On the other hand, a recent study unexpectedly discovered that mice with a genetic deficiency in CD73, an extracellular nucleotidase critical for the generation of extracellular adenosine, are highly resistant to MOG-induced brain and spinal cord injury 24. Such reduction in EAE severity was not due to the lack of responsiveness of T cells, since CD4+ T cells from CD73−/− mice secreted more proinflammatory cytokines than wild-type mice and were able to induce EAE when transferred into naive CD73+/+ recipients. This correlates well with other reports that adenosine is an anti-inflammatory mediator. It seems that adenosine concentration in the CNS, possibly surrounding the choroid plexus epithelium, is critical for pathogenic T-cell infiltration, as the CD73−/− mice had fewer infiltrating lymphocytes in their CNS compared with wild-type mice even though their T cells were highly activated. In the same study, the authors also found that pharmacological blockade of the A2A receptor with SCH58261 attenuates EAE pathology 24. These results are quite controversial because A2A receptor is recognized as a major mediator of anti-inflammatory responses. Activation of A2A has been reported to suppress key components of the inflammatory process, including leukocyte recruitment, phagocytosis, pro-inflammatory cytokine production, and immune cell proliferation 26,27. These findings have already led to the clinical testing of A2A agonists in the treatment of inflammatory diseases such as chronic obstructive pulmonary disease (COPD) and diabetic foot ulcer 27,28,29. The beneficial effect of A2A antagonist in EAE animals suggests that A2A receptors in the CNS might play an opposite role compared to the A2A receptors expressed on immune cells, though the functions of CNS A2A receptors are yet to be defined in autoimmune diseases. Using A2A knockout mice would certainly help to clarify the seemingly different roles of the periphery and CNS A2A receptors. It is quite surprising that no EAE studies have been conducted on the A2A knockout mice considering that these animals have been available for quite some time 30,31.
Unique among the four adenosine receptors, A2B is a low-affinity receptor for adenosine. Adenosine activates A1, A2A, and A3 receptors with EC50 values between 10 nM and 1 μM, whereas A2B receptor activation generally requires adenosine levels that exceed 10 μM 32. The physiological adenosine concentrations are lower than 1 μM, so activation of the A2B receptor is believed to require pathological conditions such as ischemia, trauma, inflammation or other types of stress 33. Though its functions in MS or EAE are not clear, A2B has been reported to play pro-inflammatory roles in both rodent and human asthma and COPD. Activation of A2B has been demonstrated to increase the production of IL-6 and IL-19 from mast cells, bronchial smooth-muscle cells, bronchial epithelial cells, and lung fibroblasts 34. In fact, CVT-6883, an A2B-selective antagonist, is under clinical investigation for the treatment of COPD 27. A2B is also believed to be involved in inflammatory bowel diseases. A2B receptors are upregulated in gut tissue during both human and murine colitis 35, and A2B blockade 36 or knockout 37 suppresses intestinal inflammation and attenuates the course of disease in murine colitis.
Though it is still unclear whether the A3 receptor is involved in MS, this receptor has been implicated to mediate the inhibition of TNF-α production by adenosine 38,39. Anti TNF-α drugs are remarkably effective in several autoimmune diseases, including rheumatoid arthritis, Crohn's disease, psoriasis and ankylosing spondylitis 40. Blocking TNF-α with antibodies or soluble TNF receptors also decreased EAE severity in animals 41. Unfortunately, such treatments have been found to be harmful rather than beneficial in human MS trials 42,43. A3 has also been found to be overexpressed in inflammatory tissues 44 and the PBMCs of arthritic animals 45. These findings warrant further investigations of this receptor in the pathogenesis of EAE or MS.
While the precise roles of adenosine and adenosine receptor subtypes in the development of MS and EAE remains to be clarified, the above findings clearly highlight the critical involvement of adenosine and adenosine receptors in inflammation and autoimmunity.
Adrenergic receptors
Accumulating evidence over the past few decades has documented that the brain communicates with the periphery immune system via two major pathways. The first pathway involves activation of the hypothalamic-pituitary-adrenal axis and the eventual secretion of corticosteroids from the adrenal cortex (reviewed in 46). The second pathway involves activation of the sympathetic nervous system (SNS) and the release of the various neural transmitters (reviewed in 47). As one of the major neural transmitters from the SNS, the catecholamine norepinephrine elicits its biological functions by activation of α1-, α2-, and β-adrenergic receptors 47.
Results from a series of studies show that the expression of adrenergic receptors, especially β-adrenergic receptors, changes significantly in both MS patients and EAE animals, when compared with the related controls. An early study with the Lewis rat acute EAE model indicated that in response to immune challenge, the splenic noradrenaline content fell significantly, accompanied by an increase in lymphocyte β-receptor density. These changes were considered as early indicators of immune reactivity 48. Similarly, in the MRL-lpr/lpr mouse, a genetic model of the human autoimmune disease systemic lupus erythematosus, noradrenergic innervation and noradrenaline content were reduced in the spleen prior to the onset of observed splenomegaly and remained reduced at all ages examined 49. In MS patients, increased β-adrenergic receptor density on PBMCs, including lymphocytes, has been well documented 50,51,52,53,54,55. This increase in β-adrenoreceptor density has been shown to be correlated with the expression of high affinity IL-2 receptors (IL-2R) on PBMCs and disease activity of RRMS. In vitro studies showed that β-agonist stimulation of PBMCs reduces the IL-2R expression and suppresses cell proliferation following mitogenic stimulation 51. This observation may indicate a recovery role for the enhanced β-adrenoceptor expression in MS. β2-adrenergic receptor is expressed on Th1 but not on Th2 cells 56. Given that cellular Th1 immune activity is considered to be one of the major contributors to disease activity in MS, increased expression of β2-adrenoreceptor may reflect activation of Th1 cells and a predominant cellular immune activity. Adrenergic receptors have also been reported to modulate cytokine production in dendritic cells (DCs) and affect their Th cell-priming ability 57. In particular, activation of β2-adrenergic receptors in DCs hampered IL-12, but stimulated IL-10 production resulting in reduced migration and Th1 priming 58,59. In contrast, more recent studies indicated that activation of β2-adrenergic receptors in DCs might lead to a dominant Th2/Th17-promoting phenotype in response to immunogenic protein or pathogen stimulation 60.
Another interesting phenomenon is the lacking of the astrocytic β2-adrenergic receptor in MS patients. β2-adrenoceptor has been identified on all GFAP-positive astrocytes in white matter and the optic nerve of healthy human and normal animals 61,62, and astrocytes are the main cellular target of norepinephrine terminals in the brain 63. However, in MS patients, this receptor could neither be visualized on astrocytes in normal-appearing white matter nor in reactive astrocytes in chronic active and inactive plaques, although it was normally present on neurons 61,64. Astrocytes are considered the primary APCs of the CNS in EAE models. Mice astrocytes can express MHC class II and B-7 co-stimulatory molecules, which are necessary for the efficient activation of naive T cells 65, and have potential for processing and presenting CNS auto-antigens to pro-inflammatory T cells 66. In normal conditions, the expression of MHC class II molecules are tightly suppressed by norepinephrine via β2-adrenergic receptor activation 67,68. Norepinephrine also inhibits the astrocytic expression of proinflammatory cytokines through the IκBα/NFκB pathway 69,70. The loss of astrocytic β2-adrenergic receptor might explain the presence of MHC class II on astrocytes and the increased pro-inflammatory cytokine levels in MS lesions. What causes the loss of astrocytic β2-adrenergic receptor in MS patients remains unclear, though a 'hit and run' viral infection model has been proposed 71.
Compounds regulating the adrenergic receptors have been used to treat EAE and MS. Nonselective β-adrenergic agonist isoproterenol and the β2-specific agonist terbutaline significantly suppressed both the first acute attack and the number of relapses in EAE Lewis rats 72. Other β2-adrenergic agonists, such as salbutamol and albuterol, have been proposed to be used as add-on therapy in patients with MS 71,73,74,75. In a recent trial with albuterol as an add-on treatment to glatiramer acetate therapy, improvement in the MS functional composite and a delay in the time to the first relapse were observed in the glatiramer acetate plus albuterol group 76. Other modulators of adrenergic receptors have also been reported to benefit EAE animals or MS patients. Prazosin, an α1-adrenergic receptor antagonist, suppressed the clinical and histological expression of EAE in the Lewis rat 77,78,79. Tizanidine, an α2-adrenergic receptor agonist, is a very useful medication in patients suffering from spasticity caused by MS 80,81.
Cannabinoid receptors
The medicinal use of Cannabis sativa (marijuana) can be traced back for centuries. But the existence of an 'endocannabinoid system' has only gained appreciation in the past few decades. This system consists of endocannabinoids (arachidonoylethanolamine (AEA), 2-arachidonoyl glycerol, 2-arachidonyl glyceryl ether (noladin ether), N-arachidonoyl-dopamine, virodhamine, etc.), their synthesizing/degradation enzymes and their receptors. There are two major types of cannabinoid receptors, termed CB1 and CB2. But other receptors, such as vanilloid receptor, GPR55 and GPR119 have also been reported to be activated by cannabinoids 82,83,84. The CB1 receptor is mainly expressed in the CNS, but also in the lungs, liver and kidneys. The CB2 receptor is mainly expressed in the immune system and in hematopoietic cells. Recently, CB2 has also been described in microglia and neuronal progenitor cells, but with few exceptions, it is not expressed by neurons within the CNS 82,85.
Both CB1 and CB2 are Gαi-coupled GPCRs. Activation of these receptors leads to inhibition of adenylate cyclase activity, reduced cAMP level, decreased activity of PKA, and eventual reduction in cytokine production and synaptic transmission 86. Activation of these receptors showed protective effects in various EAE models. Using the Theiler's murine encephalomyelitis virus (TMEV) model, the synthetic cannabinoids WIN 55212-2, ACEA, and JWH-015 significantly improved the neurological deficits in a long-lasting way 87,88. In a rat EAE model, decreased endocannabinoid level was reported in the brain and activation of cannabinoid receptors reduced the neurological impairment 89. The non-selective cannabinoid receptor agonist WIN-2 was found to ameliorate the clinical signs and diminish the cell infiltration into the spinal cord in a passive EAE rat model 90
The CB1 receptor was the initial focus of attention for studies using cannabinoids to treat EAE, because the activation of CB1 was believed to inhibit synaptic transmissions, which might contribute to spasticity, tremor and paralysis in EAE 91,92,93. These studies provide objective evidence to support the claims of MS patients that cannabinoids may have a benefit in symptom management 94. The beneficial effects were further supported by recent clinical trials with medical cannabis extracts 95. More direct evidence for the protective roles of CB1 receptor came from a study in a conditional knockout EAE model in which the neuronal CB1 was selectively deleted. In such animals, the cannabinoid-mediated EAE suppression was abolished 96.
In the same study, Maresz et al. 96 also reported that CB2 receptor expressed by the pathogenic T cells was critical for controlling inflammation associated with EAE. CB2-deficient T cells in the CNS of EAE animals exhibited reduced levels of apoptosis, a higher rate of proliferation, and increased production of inflammatory cytokines, resulting in more severe clinical symptoms. Palazuelos et al. 97 also found that CB2 knockout mice showed exacerbated clinical score of EAE; and the underlying mechanism might involve the extensive recruitment of immature bone marrow-derived CD34+ myeloid progenitor cells towards the spinal cords in CB2 knockout EAE mice. The immunosuppressive effect of CB2 activation was also supported by studies with selective CB1/CB2 agonists/antagonists. Ni et al. 98 demonstrated that the therapeutic effect of WIN55212-2, a non-selective CB1/CB2 agonist, could be blocked by CB2 antagonist SR144528, but not by CB1 antagonist SR141716A. JWH-015, a cannabinoid with a relatively high selectivity for CB2, was reported to suppress microglial activation 99. O-1966, a selective CB2 agonist 100, was found to significantly improve the motor function in the chronic EAE model, the remitting-relapsing model and the adoptive transfer model 101. Administration of HU-308, with a selectivity of ∼500× for CB2 vs CB1 102, improved EAE symptoms and reduced spinal cord lesions and microglial activation 97. Unlike CB1, CB2 activation is not associated with psychoactive effects. Therefore, targeting CB2 with selective agonists might be a more attractive way to treat MS.
It is also interesting to notice that the key components of the endocannabinoid system are all altered in MS patients. AEA and palmitoylethanolamide (PEA) were found to be higher in RRMS samples compared to controls. AEA, PEA, and oleoylethanolamide were also increased in the plasma of SPMS patients; PPMS patients had higher AEA plasma levels compared to controls. mRNA level of fatty acid amide hydrolase, the enzyme responsible for the degradation of endocannabinoids, was decreased in SPMS but not in RRMS or PPMS blood. CB1 and CB2 mRNAs were increased in the PPMS patients 103,104. The fact that all these alterations will lead to the activation of the endocannabinoid system suggests that the body might employ these as a mechanism to compensate for the over-activation of immune responses.
Chemokine receptors
Chemokines are cytokines initially characterized to be associated with leukocyte chemotaxis and inflammatory responses. Chemokines are classified on the basis of their structural properties, regarding the number and position of the conserved cysteine residues at the amino-terminal, into two major (CXC and CC) and two minor (C and CX3C) subfamilies 105,106. Chemokines were the first members of cytokine family to be shown to interact with GPCRs. Chemokine receptors comprise 10 CCR family members, 7 CXCR family members and other receptors including XCR1, CCRL1 and 2, and CX3CR1. Three decoy receptors, D6, DARC, and CCX-CKR (Chemocentryx-chemokine receptor), which bind chemokines with high affinity but do not elicit signal transduction, have also been reported 107. Many chemokines bind multiple receptors and most receptors bind multiple chemokines, suggesting the possibility of functional redundancy, which is also likely to be modulated by both the spatial and temporal control of expression. Chemokine receptors signal through heterotrimeric G-proteins, which in turn regulate diverse signal transduction pathways, including intracellular calcium, mitogen-activated protein kinases, PLCβ, PI3K, Ras, and Rho GTPases pathways, etc. 108. These signal mechanisms are believed to be responsible for cell movement beyond immune cell trafficking, as they also regulate other processes, such as hematopoiesis 109, angiogenesis 110, and organogenesis, including CNS formation 111.
The infiltration of leukocytes into the CNS is an essential step in the neuro-pathogenesis of MS. Leukocyte extravasation from the bloodstream is a multi-step process that depends on fluid dynamics within the vasculature and molecular interactions between circulating leukocytes and the vascular endothelium. An important step in this cascade is the binding of chemokines displayed on the vascular endothelial cell surface to chemokine receptors on circulating leukocytes, initiating intracellular signaling that leads to integrin activation, leukocyte arrest, and extravasation 112. Indeed, during the pathogenesis of MS or EAE, the expression of many chemokines and/or their receptors has been found to be altered significantly in the demyelinating plaques or the periphery immune tissues (summarized in Table 1).
Modulating immune cell migration into the CNS may represent an ideal way of combating neuro-inflammation, and accurately determining which processes or physiological roles may be regulated by a given chemokine or its receptors is crucial. The best means of investigating the actual functions of chemokines and their receptors are probably those using gene-manipulated (transgenic or knockout) mice and specific pharmacological blockers of the receptors (summarized in Table 2). Even though the attempt to translate knowledge from the animal models to the human situation is criticized, these models have rapidly led to an understanding of chemokine signaling pathways and have provided the basis for their use as therapeutic targets.
The recent failures of CCR1 antagonists BX471 (Berlex/Schering), MLN 3701 and MLN 3897 (Millennium) and CCR2 antagonist MK-0812 (Merck), which showed promising results in animal models 113, in treating MS in phase II clinical trials highlighted the difficulty of this animal-to-human translation 114. Functional redundancy of chemokines and their receptors might contribute to this problem. Moreover, the redundancy could vary between species, making it difficult to predict what the outcome of an antagonist tested in animal models will be in humans. Finally, the heterogeneity of MS 114,115 might further complicate the problem. Despite these difficulties, pharmaceutical companies still consider chemokine receptors as promising therapeutic targets. Almost every major company has a list of potential blockers in clinical development for different indications, including MS (summarized in Table 3).
Leukotriene receptors
Leukotrienes are potent pro-inflammatory mediators derived from arachidonic acid via the sequential actions of cytosolic phospholipase A2α (cPLA2α), 5-lipoxygenase (5-LO), and LTA4 hydrolase (LTA4H) for LTB4, or LTC4 synthase (LTC4S) for cysteinyl leukotrienes (CysLTs, including LTC4, LTD4, and LTE4) 116. There have been contradictory reports regarding the change of leukotriene levels in MS patients and animal models. Some studies found that the LTB4 and LTC4 levels are significantly increased in the cerebrospinal fluid (CSF) of MS patients compared with the controls 117,118. But no significant difference exists in LTC4 production between MS and control peripheral blood monocytes and macrophages 118,119. These results were also verified in the animal model of EAE 120. Other studies, however, found normal CSF concentrations of leukotrienes 121. Such discrepancies have been attributed to the difficulties in measuring leukotrienes accurately in body fluids.
However, the studies of the key enzymes of the leukotriene biosynthesis pathways revealed important roles of leukotrienes in the pathogenesis of MS. The biosynthesis of leukotrienes in inflammatory cells begins with the cleavage of arachidonic acid from nuclear membrane glycerophospholipids by cPLA2α. Marusic et al. 122,123 reported that cPLA2α-deficient mice are resistant to EAE and blocking cPLA2α with specific inhibitors prevents EAE development and greatly reduces antigen-induced production of Th1-type cytokines and IL-17. Another key enzyme, 5-LO, which catalyses the conversion of arachidonic acid to 5-hydroperoxyeicosatetraenoic acid and subsequently the unstable precursor LTA4, was found to be upregulated in both MS lesions and EAE brains by microarray analysis 124. 5-LO-specific inhibitors-treated guinea pigs showed significantly lower histological inflammation and better clinical outcome compared with controls upon EAE induction 120,125,126.
The biological effects of LTB4 are mediated via two GPCRs, BLT1 and BLT2 127. CysLTs also activate two GPCRs, namely CysLT1 and CysLT2 128. Inhibition of LTB4 receptors by antagonists or gene knockout alleviates disease pathology in EAE. An early study found that LTB4 receptor antagonist treatment significantly reduced, but did not completely inhibit the cachectic response in a guinea pig EAE model 129. Similar results were also observed in a murine model of EAE upon antagonist treatment 130. A recent study with BLT1-knockout mice found that BLT1 deficiency led to delayed onset and less severe symptoms of EAE, and BLT1−/− lymphocytes showed impaired proliferation ability and decreased cytokine production 127. Our recent study with two anti-asthmatic drugs (montelukast and zafirlukast) targeting CysLT1 indicated that blocking CysLT1 could alleviate CNS inflammatory cell infiltration and pathogenesis of EAE by reducing the permeability of the blood brain barrier (BBB) and the chemotaxis of pathogenic T cells 131.
Opioid receptors
Opioid compounds such as morphine modulate nociceptive pathways in the nervous system and produce powerful analgesia, and are used to treat various types of pain. It has also been noticed for a long time that opioids can alter the immune responses. Acute or chronic administration of opioids is known to have inhibitory effects on humoral and cellular immune responses, including antibody production, natural killer-lymphocyte activity, cytokine expression, and phagocytic activity 132,133,134. Increased morbidity and mortality due to artificial infection and faster cancer progression have been well documented in animal studies with morphine treatment 134,135. However, from other studies it emerges that not all opioids induce the same immuno-suppressive effects 136,137.
The endogenous opioid system is comprised of native opioid peptides and four opioid receptors: delta, mu, kappa receptors, and opioid receptor-like 1 138. Preclinical investigations utilizing animal models, as well as clinical observations with MS patients, suggested alteration of endogenous opioid systems in the disease. In the TMEV model of MS, mRNA levels of the mu, delta, and kappa opioid receptors were significantly decreased in the spinal cord at days 90, 150, and 180 post infection 139. The loss of opioid receptors might partially explain the common central neuropathic pain in MS patients 140. Pregnant woman usually have higher levels of endogenous opioids 141. They experience remission of MS and have fewer relapses during their pregnancy. However, these women exhibit a marked increase in relapse rate 3 months after delivery, when endogenous opioid levels are decreased 142,143. These findings suggest that one or more of the elevated opioids are acting with relevant receptors to attenuate the pathogenesis of MS. However, Gironi et al. 144 have reported a reduction of β-endorphin levels in PBMCs from patients with clinically inactive MS, but demonstrated an increase of β-endorphin in PBMCs from patients experiencing a relapse. The same group also found that β-endorphin level varies in different forms of MS. The lowest PBMC β-endorphin level was observed in primary and secondary progressive forms of MS, while the highest level was found in patients with benign and relapsing remitting forms of MS 145. Treatment with IFN-β seems to induce an increase of this opioid in MS patients 144. The increase of β-endorphin concentration during a clinical relapse may represent a possible control mechanism aimed at downregulating the inflammatory process.
Opioids have been used to treat EAE or MS. Met-enkephalin, an endogenous opioid, inhibited the onset and progression of EAE 146. A preliminary clinical trial showed that intrathecally given met-enkephalin exerted a beneficial effect on 13 patients with chronic severe progressive MS 147. Rats treated with MR 2034, a kappa opioid receptor agonist, showed a pronounced suppression of EAE clinical signs, CNS histological lesions, and anti-myelin basic protein antibody production 148. However, other reports have shown that naltrexone (NTX), a non-selective opioid receptor antagonist, has protective effects on both EAE animals and MS patients. Zagon et al. 149 demonstrated that low-dose NTX (LDN, 0.1 mg/kg) markedly reduced the severity and disease index of the treated mice, and over 33% of the MOG-treated animals receiving LDN treatment did not exhibit behavioral signs of disease. On the other hand, high-dose NTX (HDN, 10 mg/kg) displayed no beneficial effect. A 6-month phase II multi-center pilot trial with LDN has been carried out in 40 patients with PPMS. A significant reduction of spasticity was observed at the end of the trial 150. The therapeutic effect of LDN might be explained by the elevated endogenous opioids and opioid receptors due to the temporary blockade of the opioid receptors 151,152,153.
These results suggest that endogenous opioids and their receptors may play important roles in the development of EAE and MS, though the exact ligand, receptor or their mechanisms remain to be elucidated. Nevertheless, LDN, which has been successful in the treatment of Crohn's disease 154, would represent a safe, non-toxic, and generically available agent for attenuating MS and possibly other autoimmune diseases.
Sphingosine-1-phosphate receptors
Sphingolipids were first identified in the ethanolic brain extracts in the 1870s and were named after the Greek mythological creature, Sphinx, because of their enigmatic nature 155. Sphingosine-1-phosphate (S1P) represents a minor constituent of total sphingolipids. It is found abundantly in vertebrate blood and lymph. With the discovery of five S1P receptors 156,157, designated S1P1-5, S1P is recognized as an important extracellular lipid mediator.
S1P receptors are ubiquitously but differentially expressed on all cells, including a wide range of cells that are involved in the development of MS. Genetic deletion of S1P1 in mice demonstrated that this receptor plays key roles in angiogenesis and vascular maturation 158, immune cell trafficking 159, endothelial barrier function 160, and vascular tone 110. Like S1P1, S1P2 and S1P3 are also widely expressed. S1P4 has a more restricted expression pattern and is detectable predominantly within immune compartments and leukocytes 161, and may play a role in regulating T-cell cytokine production 162. S1P5 is expressed primarily in the white matter of the CNS, but its precise role remains to be clarified.
MS is generally believed to be caused by the invasion of autoreactive T cells into the CNS, which leads to demyelination and axonal damage. The S1P1 receptor has been shown to regulate the recirculation of lymphocytes 163,164,165 and their egress from secondary lymphatic organs 159. Therefore, targeting S1P1 to reduce circulating T cells might be an effective treatment for MS and other autoimmune diseases.
Recently, fingolimod (FTY720), a non-selective S1P receptor modulator, has been approved by the US FDA as the first oral, first-line treatment for RRMS. It outperformed the established first-line therapy IFN-β1a in a 1-year, double-blind, and double-dummy phase III study (known as TRANSFORMS) 166. In animal studies, prophylactic administration of fingolimod completely prevented development of EAE features, whereas therapeutic administration significantly reduced the clinical severity of EAE 167,168. As a structural analog of sphingosine 169, fingolimod is phosphorylated in vivo by sphingosine kinase 2 170 to produce fingolimod-phosphate, which binds to four of the five S1P receptors (S1P1 and S1P3-5) with high affinity. Fingolimod-phosphate initially activates lymphocyte S1P1 but subsequently induces S1P1 internalization and downregulation, which prevents lymphocyte egress from lymphoid tissues, therefore reducing pathogenic lymphocyte infiltration into the CNS 163,171. Fingolimod is also reported to ameliorate EAE by suppressing both cellular and humoral immune responses 172.
Fingolimod is able to cross the BBB 173, and may therefore have direct CNS effects, which is unique compared to other immunologically targeted MS therapies. S1P receptors are also expressed in many CNS cells and have been shown to influence cell proliferation, morphology, and migration 174,175. S1P and S1P1 have been implicated to mediate the migration of neural stem cells towards sites of injury in the spinal cord 175. Recent studies in EAE also suggested a key role of the neuronal S1P1 in disease progression 176. Re-myelination has been documented in human MS lesions and animal models 177,178. S1P1 and S1P5 are both expressed on oligodendrocytes and may be involved in the re-myelination process. Fingolimod can increase the number of both progenitor and mature oligodendrocytes in vitro. It can protect oligodendrocytes from cell death induced by cytokines or the withdrawal of growth factors, and modulate process outgrowth 179,180. High levels of S1P1 and S1P3 are also found in astrocytes 181, a glial cell type that might act like immune cells to enhance the immune responses and inhibit myelin repair. S1P induces activation and proliferation of astrocytes in vitro, while injection of S1P into the striata of mouse brains induced astrogliosis 182. In a recent study with conditional knockout mice, EAE was attenuated and fingolimod efficacy was lost in mutants lacking S1P1 on GFAP-expressing astrocytes but not on neurons 183. Receptor rescue and pharmacological experiments supported the loss of S1P1 on astrocytes via functional antagonism by fingolimod-phosphate as a primary mechanism of fingolimod. S1P1 is also expressed and plays important physiological roles in neurons. Genetic deletion of S1P1 resulted in defective neuronal development 184. Fingolimod has been shown to display neuroprotective effect in both in vitro and EAE animal models 185.
Fingolimod thus represents a new generation of medicines for MS treatment with the advantages of oral administration, and beneficially affecting not only the immune system to reduce inflammatory damage but also the CNS to promote neuroprotection and repair. Fingolimod is a non-selective sphingosine receptor modulator. Targeting other receptors, especially the S1P3 receptor, has been reported to induce certain cardiovascular side effects 186. Pharmaceutical companies are currently developing more specific S1P1 agonists to avoid such potential problems. For example, ACT-128800, an orally available S1P1 receptor agonist ∼650-fold more selective for human S1P1 over S1P3 than the natural ligand, is currently under phase II clinical investigation to treat RRMS 187,188,189.
Other GPCRs
Many other GPCRs have also been reported to be involved in the pathogenesis of EAE. For example, blocking dopamine D2-like-receptors (including D2, D3, and D4) with antagonist L750667 promoted DC-mediated Th17 differentiation 190. This is consistent with a previous report that D2 receptor agonist bromocriptine displayed therapeutic effect on acute and relapsing EAE models 191. In contrast, SCH23390, a D1-like-receptor antagonist, inhibited DC-mediated Th17 differentiation and prevented EAE in mice 190.
Histamine also plays a key regulatory role in EAE and exerts its effect through four GPCRs designated H1, H2, H3, and H4 receptors. Histidine decarboxylase is the necessary enzyme to make histamine. The histidine decarboxylase-deficient mice are genetically unable to make histamine. EAE was found to be significantly more severe in these animals 192. H3R knockout mice also developed a more severe EAE and neuro-inflammation compared with the wild-type animals 193, indicating that the immunosuppressive effect of histamine might be mediated via H3R. However, H1R-deficient mice have been found to be more resistant to EAE induction than wild-type controls 194. Treatment with pyrilamine or hydroxyzine, the H1R antagonists, led to reduced clinical signs of EAE and brain mast cell activation 195,196,197. Results from a pilot open-label clinical trial also indicated that hydroxyzine can partially inhibit brain mast cell activation and reduce MS symptoms 198.
Increasing evidence suggests that the platelet-activating factor (PAF) and its receptor (PAFR) are involved in EAE and MS pathogenesis. PAF was upregulated in peripheral-blood leukocytes during EAE induction, and the receptor antagonist treatment or gene knockout led to alleviation of clinical signs 195,196,199,200. Polymorphism of PAFR gene has also been identified to be correlated with the susceptibility to MS in human 201.
Apart from leukotrienes, prostaglandins form another family of important signaling molecules derived from arachidonic acid. Prostaglandin E2 (PGE2) was found to be increased in the CSF of MS patients 118,202. Among the four PGE2 receptors, EP1-EP4, only the EP4-knockout significantly suppressed EAE induction. This was mimicked in wild-type mice and to a greater extent, in EP2-knockout mice by administration of the EP4 antagonist ONO-AE3-208 during the immunization or preclinical phase 203,204. But ONO-AE3-208 administration at EAE onset had little effect on disease severity. In contrast, administration of the EP4 agonist ONO-AE1-329 at EAE onset delayed and suppressed disease progression as well as inhibited the associated increase in permeability of the BBB 204. Thus, PGE2 exerts dual functions in EAE, facilitating Th1 and Th17 cell generation redundantly through EP4 and EP2 during immunization and attenuating invasion of these cells into the brain by protecting the BBB integrity through EP4 203,204.
Other GPCRs that have been reported to be involved in EAE or MS pathogenesis, such as serotonin receptors, GPR30, and a list of peptide receptors, are summarized in Table 4.
Conclusion
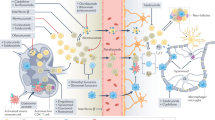
As summarized herein, a large body of evidence from both in vivo and in vitro studies suggests that GPCRs mediate important physiological or pathological functions in the development of MS (Figure 1). Targeted blockade or activation of GPCR-mediated signaling may provide novel approaches to treating MS. Given the current lack of effective pharmacological targets for the treatment of MS, the continued identification and study of GPCRs in MS pathogenesis may eventually lead to major breakthroughs and new pharmacological strategies. One of the best examples of targeting GPCRs to treat MS is the case of fingolimod (FTY720), an S1P1 receptor modulator. Application of this drug significantly reduced the relapse rates, the risk of disability progression, and MRI measures of disease activity in MS patients, as compared with IFN-β1a or placebo. More interestingly, as a wide variety of drugs or compounds targeting GPCRs have already been developed for the treatment of other human diseases, repositioning of these agents might greatly facilitate the development of novel therapies for MS or other autoimmune diseases. We anticipate an exciting future for the discovery of new drugs for MS by targeting GPCRs.

Pathogenesis of multiple sclerosis and critical roles of GPCRs. Studies with genetic manipulations and/or pharmacological interventions suggest that GPCRs mediate important processes in the development of EAE or MS: (I) T-cell activation; (II) T-cell egress from lymphoid tissues; (III) migration and infiltration of inflammatory cells from the periphery to the CNS; (IV) BBB integrity maintenance; (V) astrocyte activation; (VI) microglia activation; (VII) demyelination and neurotoxicity.
References
McFarlin DE, McFarland HF . Multiple sclerosis (first of two parts). N Engl J Med 1982; 307:1183–1188.
McFarlin DE, McFarland HF . Multiple sclerosis (second of two parts). N Engl J Med 1982; 307:1246–1251.
Rosati G . The prevalence of multiple sclerosis in the world: an update. Neurol Sci 2001; 22:117–139.
Compston A, Coles A . Multiple sclerosis. Lancet 2002; 359:1221–1231.
Frohman EM, Racke MK, Raine CS . Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med 2006; 354:942–955.
Pettinelli CB, McFarlin DE . Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J Immunol 1981; 127:1420–1423.
Sospedra M, Martin R . Immunology of multiple sclerosis. Annu Rev Immunol 2005; 23:683–747.
Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233–240.
Ivanov, II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006; 126:1121–1133.
Tzartos JS, Friese MA, Craner MJ, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 2008; 172:146–155.
McFarland HF . Correlation between MR and clinical findings of disease activity in multiple sclerosis. AJNR Am J Neuroradiol 1999; 20:1777–1778.
Raine CS, Scheinberg LC . On the immunopathology of plaque development and repair in multiple sclerosis. J Neuroimmunol 1988; 20:189–201.
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H . Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000; 47:707–717.
Compston A, Coles A . Multiple sclerosis. Lancet 2008; 372:1502–1517.
Baranzini SE, Mudge J, van Velkinburgh JC, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 2010; 464:1351–1356.
Marinissen MJ, Gutkind JS . G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 2001; 22:368–376.
Allen JA, Roth BL . Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol 2011; 51:117–144.
Bourne HR, Sanders DA, McCormick F . The GTPase superfamily: conserved structure and molecular mechanism. Nature 1991; 349:117–127.
Dorsam RT, Gutkind JS . G-protein-coupled receptors and cancer. Nat Rev Cancer 2007; 7:79–94.
Lappano R, Maggiolini M . G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov 2011; 10:47–60.
Mayne M, Shepel PN, Jiang Y, Geiger JD, Power C . Dysregulation of adenosine A1 receptor-mediated cytokine expression in peripheral blood mononuclear cells from multiple sclerosis patients. Ann Neurol 1999; 45:633–639.
Johnston JB, Silva C, Gonzalez G, et al. Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann Neurol 2001; 49:650–658.
Tsutsui S, Schnermann J, Noorbakhsh F, et al. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J Neurosci 2004; 24:1521–1529.
Mills JH, Thompson LF, Mueller C, et al. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2008; 105:9325–9330.
Chen GQ, Chen YY, Wang XS, et al. Chronic caffeine treatment attenuates experimental autoimmune encephalomyelitis induced by guinea pig spinal cord homogenates in Wistar rats. Brain Res 2010; 1309:116–125.
Milne GR, Palmer TM . Anti-inflammatory and immunosuppressive effects of the A2A adenosine receptor. ScientificWorldJournal 2011; 11:320–339.
Hasko G, Linden J, Cronstein B, Pacher P . Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 2008; 7:759–770.
Montesinos MC, Gadangi P, Longaker M, et al. Wound healing is accelerated by agonists of adenosine A2 (G alpha s-linked) receptors. J Exp Med 1997; 186:1615–1620.
Luijk B, van den Berge M, Kerstjens HA, et al. Effect of an inhaled adenosine A2A agonist on the allergen-induced late asthmatic response. Allergy 2008; 63:75–80.
Ledent C, Vaugeois JM, Schiffmann SN, et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 1997; 388:674–678.
Successful and effective: Montelukast in clinical practice. Internist 1999; 40(12 Suppl Montelukas):1–4.
Fredholm BB, Irenius E, Kull B, Schulte G . Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol 2001; 61:443–448.
Fredholm BB . Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 2007; 14:1315–1323.
Hasko G, Csoka B, Nemeth ZH, Vizi ES, Pacher P . A(2B) adenosine receptors in immunity and inflammation. Trends Immunol 2009; 30:263–270.
Kolachala V, Asamoah V, Wang L, et al. TNF-alpha upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: a basis for A2bR overexpression in colitis. Cell Mol Life Sci 2005; 62:2647–2657.
Kolachala V, Ruble B, Vijay-Kumar M, et al. Blockade of adenosine A2B receptors ameliorates murine colitis. Br J Pharmacol 2008; 155:127–137.
Kolachala VL, Vijay-Kumar M, Dalmasso G, et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology 2008; 135:861–870.
Lee JY, Jhun BS, Oh YT, et al. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/Akt and NF-kappaB activation in murine BV2 microglial cells. Neurosci Lett 2006; 396:1–6.
Levy O, Coughlin M, Cronstein BN, Roy RM, Desai A, Wessels MR . The adenosine system selectively inhibits TLR-mediated TNF-alpha production in the human newborn. J Immunol 2006; 177:1956–1966.
Silva LC, Ortigosa LC, Benard G . Anti-TNF-alpha agents in the treatment of immune-mediated inflammatory diseases: mechanisms of action and pitfalls. Immunotherapy 2010; 2:817–833.
Sicotte NL, Voskuhl RR . Onset of multiple sclerosis associated with anti-TNF therapy. Neurology 2001; 57:1885–1888.
van Oosten BW, Barkhof F, Truyen L, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 1996; 47:1531–1534.
TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999; 53:457–465.
Varani K, Maniero S, Vincenzi F, et al. A receptors are overexpressed in pleura from patients with mesothelioma and reduce cell growth via Akt/nuclear factor-kappaB pathway. Am J Respir Crit Care Med 2011; 183:522–530.
Bar-Yehuda S, Silverman MH, Kerns WD, Ochaion A, Cohen S, Fishman P . The anti-inflammatory effect of A3 adenosine receptor agonists: a novel targeted therapy for rheumatoid arthritis. Expert Opin Investig Drugs 2007; 16:1601–1613.
Webster JI, Tonelli L, Sternberg EM . Neuroendocrine regulation of immunity. Annu Rev Immunol 2002; 20:125–163.
Straub RH . Complexity of the bi-directional neuroimmune junction in the spleen. Trends Pharmacol Sci 2004; 25:640–646.
Mackenzie FJ, Leonard JP, Cuzner ML . Changes in lymphocyte beta-adrenergic receptor density and noradrenaline content of the spleen are early indicators of immune reactivity in acute experimental allergic encephalomyelitis in the Lewis rat. J Neuroimmunol 1989; 23:93–100.
Breneman SM, Moynihan JA, Grota LJ, Felten DL, Felten SY . Splenic norepinephrine is decreased in MRL-lpr/lpr mice. Brain Behav Immun 1993; 7:135–143.
Zoukos Y, Leonard JP, Thomaides T, Thompson AJ, Cuzner ML . beta-Adrenergic receptor density and function of peripheral blood mononuclear cells are increased in multiple sclerosis: a regulatory role for cortisol and interleukin-1. Ann Neurol 1992; 31:657–662.
Zoukos Y, Kidd D, Woodroofe MN, Kendall BE, Thompson AJ, Cuzner ML . Increased expression of high affinity IL-2 receptors and beta-adrenoceptors on peripheral blood mononuclear cells is associated with clinical and MRI activity in multiple sclerosis. Brain 1994; 117(Pt 2):307–315.
Zoukos Y, Thomaides T, Mathias CJ, Cuzner ML . High beta-adrenoceptor density on peripheral blood mononuclear cells in progressive multiple sclerosis: a manifestation of autonomic dysfunction? Acta Neurol Scand 1994; 90:382–387.
Zoukos Y, Thomaides TN, Kidd D, Cuzner ML, Thompson A . Expression of beta2 adrenoreceptors on peripheral blood mononuclear cells in patients with primary and secondary progressive multiple sclerosis: a longitudinal six month study. J Neurol Neurosurg Psychiatry 2003; 74:197–202.
Karaszewski JW, Reder AT, Maselli R, Brown M, Arnason BG . Sympathetic skin responses are decreased and lymphocyte beta-adrenergic receptors are increased in progressive multiple sclerosis. Ann Neurol 1990; 27:366–372.
Karaszewski JW, Reder AT, Anlar B, Kim WC, Arnason BG . Increased lymphocyte beta-adrenergic receptor density in progressive multiple sclerosis is specific for the CD8+, CD28− suppressor cell. Ann Neurol 1991; 30:42–47.
Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE . Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol 1997; 158:4200–4210.
Seiffert K, Hosoi J, Torii H, et al. Catecholamines inhibit the antigen-presenting capability of epidermal Langerhans cells. J Immunol 2002; 168:6128–6135.
Maestroni GJ . Short exposure of maturing, bone marrow-derived dendritic cells to norepinephrine: impact on kinetics of cytokine production and Th development. J Neuroimmunol 2002; 129:106–114.
Maestroni GJ, Mazzola P . Langerhans cells beta 2-adrenoceptors: role in migration, cytokine production, Th priming and contact hypersensitivity. J Neuroimmunol 2003; 144:91–99.
Kim BJ, Jones HP . Epinephrine-primed murine bone marrow-derived dendritic cells facilitate production of IL-17A and IL-4 but not IFN-gamma by CD4+ T cells. Brain Behav Immun 2010; 24:1126–1136.
De Keyser J, Wilczak N, Leta R, Streetland C . Astrocytes in multiple sclerosis lack beta-2 adrenergic receptors. Neurology 1999; 53:1628–1633.
Mantyh PW, Rogers SD, Allen CJ, et al. Beta 2-adrenergic receptors are expressed by glia in vivo in the normal and injured central nervous system in the rat, rabbit, and human. J Neurosci 1995; 15:152–164.
Cohen Z, Molinatti G, Hamel E . Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab 1997; 17:894–904.
Zeinstra E, Wilczak N, De Keyser J . [3H]dihydroalprenolol binding to beta adrenergic receptors in multiple sclerosis brain. Neurosci Lett 2000; 289:75–77.
Nikcevich KM, Gordon KB, Tan L, et al. IFN-gamma-activated primary murine astrocytes express B7 costimulatory molecules and prime naive antigen-specific T cells. J Immunol 1997; 158:614–621.
Soos JM, Morrow J, Ashley TA, Szente BE, Bikoff EK, Zamvil SS . Astrocytes express elements of the class II endocytic pathway and process central nervous system autoantigen for presentation to encephalitogenic T cells. J Immunol 1998; 161:5959–5966.
Frohman EM, Vayuvegula B, Gupta S, van den Noort S . Norepinephrine inhibits gamma-interferon-induced major histocompatibility class II (Ia) antigen expression on cultured astrocytes via beta-2-adrenergic signal transduction mechanisms. Proc Natl Acad Sci USA 1988; 85:1292–1296.
Frohman EM, Vayuvegula B, van den Noort S, Gupta S . Norepinephrine inhibits gamma-interferon-induced MHC class II (Ia) antigen expression on cultured brain astrocytes. J Neuroimmunol 1988; 17:89–101.
Gavrilyuk V, Horvath P, Weinberg G, Feinstein DL . A 27-bp region of the inducible nitric oxide synthase promoter regulates expression in glial cells. J Neurochem 2001; 78:129–140.
Gavrilyuk V, Dello Russo C, Heneka MT, Pelligrino D, Weinberg G, Feinstein DL . Norepinephrine increases I kappa B alpha expression in astrocytes. J Biol Chem 2002; 277:29662–29668.
De Keyser J, Laureys G, Demol F, Wilczak N, Mostert J, Clinckers R . Astrocytes as potential targets to suppress inflammatory demyelinating lesions in multiple sclerosis. Neurochem Int 2010, 57:446–450.
Wiegmann K, Muthyala S, Kim DH, Arnason BG, Chelmicka-Schorr E . Beta-adrenergic agonists suppress chronic/relapsing experimental allergic encephalomyelitis (CREAE) in Lewis rats. J Neuroimmunol 1995; 56:201–206.
Makhlouf K, Weiner HL, Khoury SJ . Potential of beta2-adrenoceptor agonists as add-on therapy for multiple sclerosis: focus on salbutamol (albuterol). CNS Drugs 2002; 16:1–8.
De Keyser J, Zeinstra E, Mostert J, Wilczak N . Beta 2-adrenoceptor involvement in inflammatory demyelination and axonal degeneration in multiple sclerosis. Trends Pharmacol Sci 2004; 25:67–71.
De Keyser J, Zeinstra E, Wilczak N . Astrocytic beta2-adrenergic receptors and multiple sclerosis. Neurobiol Dis 2004; 15:331–339.
Khoury SJ, Healy BC, Kivisakk P, et al. A randomized controlled double-masked trial of albuterol add-on therapy in patients with multiple sclerosis. Arch Neurol 2010; 67:1055–1061.
Brosnan CF, Goldmuntz EA, Cammer W, Factor SM, Bloom BR, Norton WT . Prazosin, an alpha 1-adrenergic receptor antagonist, suppresses experimental autoimmune encephalomyelitis in the Lewis rat. Proc Natl Acad Sci USA 1985; 82:5915–5919.
Goldmuntz EA, Brosnan CF, Chiu FC, Norton WT . Astrocytic reactivity and intermediate filament metabolism in experimental autoimmune encephalomyelitis: the effect of suppression with prazosin. Brain Res 1986; 397:16–26.
Brosnan CF, Sacks HJ, Goldschmidt RC, Goldmuntz EA, Norton WT . Prazosin treatment during the effector stage of disease suppresses experimental autoimmune encephalomyelitis in the Lewis rat. J Immunol 1986; 137:3451–3456.
Malanga G, Reiter RD, Garay E . Update on tizanidine for muscle spasticity and emerging indications. Expert Opin Pharmacother 2008; 9:2209–2215.
Kamen L . Henney HR 3rd, Runyan JD . A practical overview of tizanidine use for spasticity secondary to multiple sclerosis, stroke, and spinal cord injury. Curr Med Res Opin 2008; 24:425–439.
Pacher P, Batkai S, Kunos G . The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 2006; 58:389–462.
Brown AJ . Novel cannabinoid receptors. Br J Pharmacol 2007; 152:567–575.
Ryberg E, Larsson N, Sjogren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 2007; 152:1092–1101.
Pacher P, Mechoulam R . Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 2011; 50:193–211.
Demuth DG, Molleman A . Cannabinoid signalling. Life Sci 2006; 78:549–563.
Arevalo-Martin A, Vela JM, Molina-Holgado E, Borrell J, Guaza C . Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci 2003; 23:2511–2516.
Ortega-Gutierrez S, Molina-Holgado E, Arevalo-Martin A, et al. Activation of the endocannabinoid system as therapeutic approach in a murine model of multiple sclerosis. FASEB J 2005; 19:1338–1340.
Cabranes A, Venderova K, de Lago E, et al. Decreased endocannabinoid levels in the brain and beneficial effects of agents activating cannabinoid and/or vanilloid receptors in a rat model of multiple sclerosis. Neurobiol Dis 2005; 20:207–217.
Sanchez AJ, Gonzalez-Perez P, Galve-Roperh I, Garcia-Merino A . R-(+)-[2,3-Dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo-[1,2,3-de]-1,4 -benzoxazin-6-yl]-1-naphtalenylmethanone (WIN-2) ameliorates experimental autoimmune encephalomyelitis and induces encephalitogenic T cell apoptosis: partial involvement of the CB(2) receptor. Biochem Pharmacol 2006; 72:1697–1706.
Lyman WD, Sonett JR, Brosnan CF, Elkin R, Bornstein MB . Delta 9-tetrahydrocannabinol: a novel treatment for experimental autoimmune encephalomyelitis. J Neuroimmunol 1989; 23:73–81.
Wirguin I, Mechoulam R, Breuer A, Schezen E, Weidenfeld J, Brenner T . Suppression of experimental autoimmune encephalomyelitis by cannabinoids. Immunopharmacology 1994; 28:209–214.
Baker D, Pryce G, Croxford JL, et al. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature 2000; 404:84–87.
Bifulco M, Laezza C, Malfitano AM . From anecdotal evidence of cannabinoids in multiple sclerosis to emerging new therapeutical approaches. Multiple Sclerosis 2007; 13:133–134.
Killestein J, Hoogervorst EL, Reif M, et al. Safety, tolerability, and efficacy of orally administered cannabinoids in MS. Neurology 2002; 58:1404–1407.
Maresz K, Pryce G, Ponomarev ED, et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med 2007; 13:492–497.
Palazuelos J, Davoust N, Julien B, et al. The CB(2) cannabinoid receptor controls myeloid progenitor trafficking: involvement in the pathogenesis of an animal model of multiple sclerosis. J Biol Chem 2008; 283:13320–13329.
Ni X, Geller EB, Eppihimer MJ, Eisenstein TK, Adler MW, Tuma RF . Win 55212-2, a cannabinoid receptor agonist, attenuates leukocyte/endothelial interactions in an experimental autoimmune encephalomyelitis model. Mult Scler 2004; 10:158–164.
Ehrhart J, Obregon D, Mori T, et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation 2005; 2:29.
Wiley JL, Beletskaya ID, Ng EW, et al. Resorcinol derivatives: a novel template for the development of cannabinoid CB(1)/CB(2) and CB(2)-selective agonists. J Pharmacol Exp Ther 2002; 301:679–689.
Zhang M, Martin BR, Adler MW, et al. Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J Neuroimmune Pharmacol 2009; 4:249–259.
Hanus L, Breuer A, Tchilibon S, et al. HU-308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci USA 1999; 96:14228–14233.
Jean-Gilles L, Feng S, Tench CR, et al. Plasma endocannabinoid levels in multiple sclerosis. J Neurol Sci 2009; 287:212–215.
Benito C, Romero JP, Tolon RM, et al. Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis. J Neurosci 2007; 27:2396–2402.
Ali S, Lazennec G . Chemokines: novel targets for breast cancer metastasis. Cancer Metastasis Rev 2007; 26:401–420.
Vindrieux D, Escobar P, Lazennec G . Emerging roles of chemokines in prostate cancer. Endocr Relat Cancer 2009; 16:663–673.
Mantovani A, Bonecchi R, Locati M . Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol 2006; 6:907–918.
Thelen M, Stein JV . How chemokines invite leukocytes to dance. Nat Immunol 2008; 9:953–959.
Broxmeyer HE . Chemokines in hematopoiesis. Curr Opin Hematol 2008; 15:49–58.
Benelli R, Lorusso G, Albini A, Noonan DM . Cytokines and chemokines as regulators of angiogenesis in health and disease. Curr Pharm Des 2006; 12:3101–3115.
Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G . Chemokines and their receptors in the central nervous system. Front Neuroendocrinol 2001; 22:147–184.
Holman DW, Klein RS, Ransohoff RM . The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta; 1812:220–230.
Ubogu EE . Chemokine receptors as specific anti-inflammatory targets in peripheral nerves. Endocr Metab Immune Disord Drug Targets; 11:141–153.
Zipp F, Hartung HP, Hillert J, et al. Blockade of chemokine signaling in patients with multiple sclerosis. Neurology 2006; 67:1880–1883.
Infante-Duarte C, Waiczies S, Wuerfel J, Zipp F . New developments in understanding and treating neuroinflammation. J Mol Med 2008; 86:975–985.
Tantisira KG, Drazen JM . Genetics and pharmacogenetics of the leukotriene pathway. J Allergy Clin Immunol 2009; 124:422–427.
Neu IS, Metzger G, Zschocke J, Zelezny R, Mayatepek E . Leukotrienes in patients with clinically active multiple sclerosis. Acta Neurol Scand 2002; 105:63–66.
Dore-Duffy P, Ho SY, Donovan C . Cerebrospinal fluid eicosanoid levels: endogenous PGD2 and LTC4 synthesis by antigen-presenting cells that migrate to the central nervous system. Neurology 1991; 41:322–324.
Merrill JE, Strom SR, Ellison GW, Myers LW . In vitro study of mediators of inflammation in multiple sclerosis. J Clin Immunol 1989; 9:84–96.
Prosiegel M, Neu I, Mallinger J, et al. Suppression of experimental autoimmune encephalomyelitis by dual cyclo-oxygenase and 5-lipoxygenase inhibition. Acta Neurol Scand 1989; 79:223–226.
Haupts M, Smektala K, Finkbeiner T, Simmet T, Gehlen W . Immunoreactive leukotriene C4 levels in CSF of MS patients. Acta Neurol Scand 1992; 85:365–367.
Marusic S, Leach MW, Pelker JW, et al. Cytosolic phospholipase A2 alpha-deficient mice are resistant to experimental autoimmune encephalomyelitis. J Exp Med 2005; 202:841–851.
Marusic S, Thakker P, Pelker JW, et al. Blockade of cytosolic phospholipase A2 alpha prevents experimental autoimmune encephalomyelitis and diminishes development of Th1 and Th17 responses. J Neuroimmunol 2008; 204:29–37.
Whitney LW, Ludwin SK, McFarland HF, Biddison WE . Microarray analysis of gene expression in multiple sclerosis and EAE identifies 5-lipoxygenase as a component of inflammatory lesions. J Neuroimmunol 2001; 121:40–48.
Prosiegel M, Neu I, Vogl S, Hoffmann G, Wildfeuer A, Ruhenstroth-Bauer G . Suppression of experimental autoimmune encephalomyelitis by sulfasalazine. Acta Neurol Scand 1990; 81:237–238.
Neu IS, Mallinger J, Wildfeuer A, Mehlber L . Suppression of experimental autoimmune encephalomyelitis by a new specific leukotriene biosynthesis inhibitor. J Neurol 1992; 239:470–471.
Kihara Y, Yokomizo T, Kunita A, et al. The leukotriene B4 receptor, BLT1, is required for the induction of experimental autoimmune encephalomyelitis. Biochem Biophys Res Commun 2010; 394:673–678.
Jiang Y, Borrelli LA, Kanaoka Y, Bacskai BJ, Boyce JA . CysLT2 receptors interact with CysLT1 receptors and down-modulate cysteinyl leukotriene dependent mitogenic responses of mast cells. Blood 2007; 110:3263–3270.
Fretland DJ, Widomski DL, Shone RL, Levin S, Gaginella TS . Effect of the leukotriene B4 receptor antagonist, SC-41930, on experimental allergic encephalomyelitis (EAE) in the guinea pig. Agents Actions 1991; 34:172–174.
Gladue RP, Carroll LA, Milici AJ, et al. Inhibition of leukotriene B4-receptor interaction suppresses eosinophil infiltration and disease pathology in a murine model of experimental allergic encephalomyelitis. J Exp Med 1996; 183:1893–1898.
Wang L, Du C, Lv J, Wei W, Cui Y, Xie X . Antiasthmatic drugs targeting the cysteinyl leukotriene receptor 1 alleviate central nervous system inflammatory cell infiltration and pathogenesis of experimental autoimmune encephalomyelitis. J Immunol 2011; 87:2336–2345.
Vallejo R, de Leon-Casasola O, Benyamin R . Opioid therapy and immunosuppression: a review. Am J Ther 2004; 11:354–365.
Brack A, Rittner HL, Stein C . Immunosuppressive effects of opioids-clinical relevance. J Neuroimmune Pharmacol 2011; 6:490–450.
Sacerdote P . Opioids and the immune system. Palliat Med 2006; 20(Suppl 1):s9–s15.
Risdahl JM, Khanna KV, Peterson PK, Molitor TW . Opiates and infection. J Neuroimmunol 1998; 83:4–18.
Sacerdote P, Limiroli E, Gaspani L . Experimental evidence for immunomodulatory effects of opioids. Adv Exp Med Biol 2003; 521:106–116.
Sacerdote P, Manfredi B, Mantegazza P, Panerai AE . Antinociceptive and immunosuppressive effects of opiate drugs: a structure-related activity study. Br J Pharmacol 1997; 121:834–840.
Waldhoer M, Bartlett SE, Whistler JL . Opioid receptors. Annu Rev Biochem 2004; 73:953–990.
Lynch JL, Alley JF, Wellman L, Beitz AJ . Decreased spinal cord opioid receptor mRNA expression and antinociception in a Theiler's murine encephalomyelitis virus model of multiple sclerosis. Brain Res 2008; 1191:180–191.
Baron R . Mechanisms of disease: neuropathic pain--a clinical perspective. Nat Clin Pract Neurol 2006; 2:95–106.
Akil H, Watson SJ, Young E, Lewis ME, Khachaturian H, Walker JM . Endogenous opioids: biology and function. Annu Rev Neurosci 1984; 7:223–255.
Lutton JD, Winston R, Rodman TC . Multiple sclerosis: etiological mechanisms and future directions. Exp Biol Med (Maywood) 2004; 229:12–20.
Vukusic S, Hutchinson M, Hours M, et al. Pregnancy and multiple sclerosis (the PRIMS study): clinical predictors of post-partum relapse. Brain 2004; 127:1353–1360.
Gironi M, Martinelli V, Brambilla E, et al. Beta-endorphin concentrations in peripheral blood mononuclear cells of patients with multiple sclerosis: effects of treatment with interferon beta. Arch Neurol 2000; 57:1178–1181.
Gironi M, Furlan R, Rovaris M, et al. Beta endorphin concentrations in PBMC of patients with different clinical phenotypes of multiple sclerosis. J Neurol Neurosurg Psychiatry 2003; 74:495–497.
Zagon IS, Rahn KA, Bonneau RH, Turel AP, McLaughlin PJ . Opioid growth factor suppresses expression of experimental autoimmune encephalomyelitis. Brain Res 2010; 1310:154–161.
Jankovic BD . Enkephalins and immune inflammatory reactions. Acta Neurol 1991; 13:433–441.
Radulovic J, Djergovic D, Miljevic C, Jankovic BD . kappa-Opioid receptor functions: possible relevance to experimental allergic encephalomyelitis. Neuroimmunomodulation 1994; 1:236–241.
Zagon IS, Rahn KA, Turel AP, McLaughlin PJ . Endogenous opioids regulate expression of experimental autoimmune encephalomyelitis: a new paradigm for the treatment of multiple sclerosis. Exp Biol Med 2009; 234:1383–1392.
Gironi M, Martinelli-Boneschi F, Sacerdote P, et al. A pilot trial of low-dose naltrexone in primary progressive multiple sclerosis. Mult Scler 2008; 14:1076–1083.
Misra AL . Current status of preclinical research on disposition, pharmacokinetics, and metabolism of naltrexone. NIDA Res Monogr 1981; 28:132–146.
Zagon IS, McLaughlin PJ . Naltrexone modulates tumor response in mice with neuroblastoma. Science 1983; 221:671–673.
Zagon IS, McLaughlin PJ . Duration of opiate receptor blockade determines tumorigenic response in mice with neuroblastoma: a role for endogenous opioid systems in cancer. Life Sci 1984; 35:409–416.
Smith JP, Stock H, Bingaman S, Mauger D, Rogosnitzky M, Zagon IS . Low-dose naltrexone therapy improves active Crohn's disease. Am J Gastroenterol 2007; 102:820–828.
Spiegel S, Milstien S . Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 2003; 4:397–407.
Spiegel S, Milstien S . Functions of a new family of sphingosine-1-phosphate receptors. Biochim Biophys Acta 2000; 1484:107–116.
Chun J, Goetzl EJ, Hla T, et al. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol Rev 2002; 54:265–269.
Liu Y, Wada R, Yamashita T, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest 2000; 106:951–961.
Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004; 427:355–360.
Singleton PA, Dudek SM, Chiang ET, Garcia JG . Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J 2005; 19:1646–1656.
Graler MH, Bernhardt G, Lipp M . EDG6, a novel G-protein-coupled receptor related to receptors for bioactive lysophospholipids, is specifically expressed in lymphoid tissue. Genomics 1998; 53:164–169.
Wang W, Graeler MH, Goetzl EJ . Type 4 sphingosine 1-phosphate G protein-coupled receptor (S1P4) transduces S1P effects on T cell proliferation and cytokine secretion without signaling migration. FASEB J 2005; 19:1731–1733.
Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 2002; 277:21453–21457.
Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002; 296:346–349.
Xie JH, Nomura N, Koprak SL, Quackenbush EJ, Forrest MJ, Rosen H . Sphingosine-1-phosphate receptor agonism impairs the efficiency of the local immune response by altering trafficking of naive and antigen-activated CD4+ T cells. J Immunol 2003; 170:3662–3670.
Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov; 9:883–897.
Webb M, Tham CS, Lin FF, et al. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J Neuroimmunol 2004; 153:108–121.
Kataoka H, Sugahara K, Shimano K, et al. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol 2005; 2:439–448.
Nofer JR, Bot M, Brodde M, et al. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2007; 115:501–508.
Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T . Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem 2003; 278:47408–47415.
Chiba K . FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissues and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol Ther 2005; 108:308–319.
Papadopoulos D, Rundle J, Patel R, et al. FTY720 ameliorates MOG-induced experimental autoimmune encephalomyelitis by suppressing both cellular and humoral immune responses. J Neurosci Res 2010; 88:346–359.
Foster CA, Howard LM, Schweitzer A, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther 2007; 323:469–475.
Chun J, Weiner JA, Fukushima N, et al. Neurobiology of receptor-mediated lysophospholipid signaling. From the first lysophospholipid receptor to roles in nervous system function and development. Ann N Y Acad Sci 2000; 905:110–117.
Kimura A, Ohmori T, Ohkawa R, et al. Essential roles of sphingosine 1-phosphate/S1P1 receptor axis in the migration of neural stem cells toward a site of spinal cord injury. Stem Cells 2007; 25:115–124.
Lee CW, Choi JW, Chun J . Neurological S1P signaling as an emerging mechanism of action of oral FTY720 (fingolimod) in multiple sclerosis. Arch Pharm Res 2010; 33:1567–1574.
Patrikios P, Stadelmann C, Kutzelnigg A, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006; 129:3165–3172.
Lassmann H, Bruck W, Lucchinetti C, Rodriguez M . Remyelination in multiple sclerosis. Mult Scler 1997; 3:133–136.
Miron VE, Jung CG, Kim HJ, Kennedy TE, Soliven B, Antel JP . FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol 2008; 63:61–71.
Coelho RP, Payne SG, Bittman R, Spiegel S, Sato-Bigbee C . The immunomodulator FTY720 has a direct cytoprotective effect in oligodendrocyte progenitors. J Pharmacol Exp Ther 2007; 323:626–635.
Rao TS, Lariosa-Willingham KD, Lin FF, et al. Pharmacological characterization of lysophospholipid receptor signal transduction pathways in rat cerebrocortical astrocytes. Brain Res 2003; 990:182–194.
Sorensen SD, Nicole O, Peavy RD, et al. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol Pharmacol 2003; 64:1199–1209.
Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci USA 2011; 108:751–756.
Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL . Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol 2005; 25:11113–11121.
Balatoni B, Storch MK, Swoboda EM, et al. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res Bull 2007; 74:307–316.
Alewijnse AE, Peters SL . Sphingolipid signalling in the cardiovascular system: good, bad or both? Eur J Pharmacol 2008; 585:292–302.
Piali L, Froidevaux S, Hess P, et al. The selective sphingosine 1-phosphate receptor 1 agonist ponesimod protects against lymphocyte-mediated tissue inflammation. J Pharmacol Exp Ther 2012; 337:547–556.
ClinicalTrials.gov Identifier: NCT01006265. http://clinicaltrials.gov/. 2009.
ClinicalTrials.gov Identifier: NCT01093326. http://clinicaltrials.gov/. 2010.
Nakano K, Higashi T, Hashimoto K, Takagi R, Tanaka Y, Matsushita S . Antagonizing dopamine D1-like receptor inhibits Th17 cell differentiation: preventive and therapeutic effects on experimental autoimmune encephalomyelitis. Biochem Biophys Res Commun 2008; 373:286–291.
Dijkstra CD, van der Voort ER, De Groot CJ, et al. Therapeutic effect of the D2-dopamine agonist bromocriptine on acute and relapsing experimental allergic encephalomyelitis. Psychoneuroendocrinology 1994; 19:135–142.
Musio S, Gallo B, Scabeni S, et al. A key regulatory role for histamine in experimental autoimmune encephalomyelitis: disease exacerbation in histidine decarboxylase-deficient mice. J Immunol 2006; 176:17–26.
Teuscher C, Subramanian M . Noubade R, et al. Central histamine H3 receptor signaling negatively regulates susceptibility to autoimmune inflammatory disease of the CNS. Proc Natl Acad Sci USA 2007; 104:10146–10151.
Noubade R, Milligan G, Zachary JF, et al. Histamine receptor H1 is required for TCR-mediated p38 MAPK activation and optimal IFN-gamma production in mice. J Clin Invest 2007; 117:3507–3518.
El Behi M, Zephir H, Lefranc D, et al. Changes in self-reactive IgG antibody repertoire after treatment of experimental autoimmune encephalomyelitis with anti-allergic drugs. J Neuroimmunol 2007; 182:80–88.
Pedotti R, DeVoss JJ, Youssef S, et al. Multiple elements of the allergic arm of the immune response modulate autoimmune demyelination. Proc Natl Acad Sci USA 2003; 100:1867–1872.
Dimitriadou V, Pang X, Theoharides TC . Hydroxyzine inhibits experimental allergic encephalomyelitis (EAE) and associated brain mast cell activation. Int J Immunopharmacol 2000; 22:673–684.
Logothetis L, Mylonas IA, Baloyannis S, et al. A pilot, open label, clinical trial using hydroxyzine in multiple sclerosis. Int J Immunopathol Pharmacol 2005; 18:771–778.
Koehler NK, Roebbert M, Dehghani K, et al. Up-regulation of platelet-derived growth factor by peripheral-blood leukocytes during experimental allergic encephalomyelitis. J Neurosci Res 2008; 86:392–402.
Kihara Y, Ishii S, Kita Y, Toda A, Shimada A, Shimizu T . Dual phase regulation of experimental allergic encephalomyelitis by platelet-activating factor. J Exp Med 2005; 202:853–863.
Osoegawa M, Miyagishi R, Ochi H, et al. Platelet-activating factor receptor gene polymorphism in Japanese patients with multiple sclerosis. J Neuroimmunol 2005; 161:195–198.
Bolton C, Turner AM, Turk JL . Prostaglandin levels in cerebrospinal fluid from multiple sclerosis patients in remission and relapse. J Neuroimmunol 1984; 6:151–159.
Yao C, Sakata D, Esaki Y, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med 2009; 15:633–640.
Esaki Y, Li Y, Sakata D, et al. Dual roles of PGE2-EP4 signaling in mouse experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2010; 107:12233–12238.
Sunnemark D, Eltayeb S, Wallstrom E, et al. Differential expression of the chemokine receptors CX3CR1 and CCR1 by microglia and macrophages in myelin-oligodendrocyte-glycoprotein-induced experimental autoimmune encephalomyelitis. Brain Pathol 2003; 13:617–629.
Trebst C, Sorensen TL, Kivisakk P, et al. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am J Pathol 2001; 159:1701–1710.
Glabinski AR, Bielecki B, O'Bryant S, Selmaj K, Ransohoff RM . Experimental autoimmune encephalomyelitis: CC chemokine receptor expression by trafficking cells. J Autoimmun 2002; 19:175–181.
Eltayeb S, Berg AL, Lassmann H, et al. Temporal expression and cellular origin of CC chemokine receptors CCR1, CCR2 and CCR5 in the central nervous system: insight into mechanisms of MOG-induced EAE. J Neuroinflammation 2007; 4:14.
Simpson J, Rezaie P, Newcombe J, Cuzner ML, Male D, Woodroofe MN . Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol 2000; 108:192–200.
Sorensen TL, Sellebjerg F . Distinct chemokine receptor and cytokine expression profile in secondary progressive MS. Neurology 2001; 57:1371–1376.
Mahad DJ, Ransohoff RM . The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 2003; 15:23–32.
Jiang Y, Salafranca MN, Adhikari S, et al. Chemokine receptor expression in cultured glia and rat experimental allergic encephalomyelitis. J Neuroimmunol 1998; 86:1–12.
Jee Y, Yoon WK, Okura Y, Tanuma N, Matsumoto Y . Upregulation of monocyte chemotactic protein-1 and CC chemokine receptor 2 in the central nervous system is closely associated with relapse of autoimmune encephalomyelitis in Lewis rats. J Neuroimmunol 2002; 128:49–57.
Misu T, Onodera H, Fujihara K, et al. Chemokine receptor expression on T cells in blood and cerebrospinal fluid at relapse and remission of multiple sclerosis: imbalance of Th1/Th2-associated chemokine signaling. J Neuroimmunol 2001; 114:207–212.
Columba-Cabezas S, Serafini B, Ambrosini E, et al. Induction of macrophage-derived chemokine/CCL22 expression in experimental autoimmune encephalomyelitis and cultured microglia: implications for disease regulation. J Neuroimmunol 2002; 130:10–21.
Balashov KE, Rottman JB, Weiner HL, Hancock WW . CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci USA 1999; 96:6873–6878.
Martinez-Caceres EM, Espejo C, Brieva L, et al. Expression of chemokine receptors in the different clinical forms of multiple sclerosis. Mult Scler 2002; 8:390–395.
Trebst C, Konig F, Ransohoff R, Bruck W, Stangel M . CCR5 expression on macrophages/microglia is associated with early remyelination in multiple sclerosis lesions. Mult Scler 2008; 14:728–733.
Kivisakk P, Mahad DJ, Callahan MK, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol 2004; 55:627–638.
Bielecki B, Mazurek A, Wolinski P, Glabinski A . Expression of chemokine receptors CCR7 and CCR8 in the CNS during ChREAE. Scand J Immunol 2007; 66:383–392.
Trebst C, Staugaitis SM, Kivisakk P, et al. CC chemokine receptor 8 in the central nervous system is associated with phagocytic macrophages. Am J Pathol 2003; 162:427–438.
Infante-Duarte C, Weber A, Kratzschmar J, et al. Frequency of blood CX3CR1-positive natural killer cells correlates with disease activity in multiple sclerosis patients. FASEB J 2005; 19:1902–1904.
Sunnemark D, Eltayeb S, Nilsson M, et al. CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: kinetics and cellular origin. J Neuroinflammation 2005; 2:17.
Omari KM, John GR, Sealfon SC, Raine CS . CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain 2005; 128:1003–1015.
Omari KM, John G, Lango R, Raine CS . Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia 2006; 53:24–31.
Filipovic R, Jakovcevski I, Zecevic N . GRO-alpha and CXCR2 in the human fetal brain and multiple sclerosis lesions. Dev Neurosci 2003; 25:279–290.
Glabinski AR, O'Bryant S, Selmaj K, Ransohoff RM . CXC chemokine receptors expression during chronic relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci 2000; 917:135–144.
Sindern E, Patzold T, Ossege LM, Gisevius A, Malin JP . Expression of chemokine receptor CXCR3 on cerebrospinal fluid T-cells is related to active MRI lesion appearance in patients with relapsing-remitting multiple sclerosis. J Neuroimmunol 2002; 131:186–190.
Sorensen TL, Trebst C, Kivisakk P, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol 2002; 127:59–68.
Glabinski AR, Bielecki B, Ransohoff RM . Chemokine upregulation follows cytokine expression in chronic relapsing experimental autoimmune encephalomyelitis. Scand J Immunol 2003; 58:81–88.
Nakajima H, Fukuda K, Doi Y, et al. Expression of TH1/TH2-related chemokine receptors on peripheral T cells and correlation with clinical disease activity in patients with multiple sclerosis. Eur Neurol 2004; 52:162–168.
Godiska R, Chantry D, Dietsch GN, Gray PW . Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol 1995; 58:167–176.
Mahad D, Callahan MK, Williams KA, et al. Modulating CCR2 and CCL2 at the blood-brain barrier: relevance for multiple sclerosis pathogenesis. Brain 2006; 129:212–223.
Mahad DJ, Howell SJ, Woodroofe MN . Expression of chemokines in the CSF and correlation with clinical disease activity in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry 2002; 72:498–502.
Glabinski AR, Tuohy VK, Ransohoff RM . Expression of chemokines RANTES, MIP-1alpha and GRO-alpha correlates with inflammation in acute experimental autoimmune encephalomyelitis. Neuroimmunomodulation 1998; 5:166–171.
Kennedy KJ, Strieter RM, Kunkel SL, Lukacs NW, Karpus WJ . Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1alpha and monocyte chemotactic protein-1. J Neuroimmunol 1998; 92:98–108.
Sellebjerg F, Bornsen L, Khademi M, et al. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology 2009; 73:2003–2010.
Narikawa K, Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y . CSF chemokine levels in relapsing neuromyelitis optica and multiple sclerosis. J Neuroimmunol 2004; 149:182–186.
Krumbholz M, Theil D, Steinmeyer F, et al. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J Neuroimmunol 2007; 190:72–79.
Columba-Cabezas S, Serafini B, Ambrosini E, Aloisi F . Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the maintenance of chronic neuroinflammation. Brain Pathol 2003; 13:38–51.
Alt C, Laschinger M, Engelhardt B . Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood-brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur J Immunol 2002; 32:2133–2144.
Kastenbauer S, Koedel U, Wick M, Kieseier BC, Hartung HP, Pfister HW . CSF and serum levels of soluble fractalkine (CX3CL1) in inflammatory diseases of the nervous system. J Neuroimmunol 2003; 137:210–217.
Carter SL, Muller M, Manders PM, Campbell IL . Induction of the genes for Cxcl9 and Cxcl10 is dependent on IFN-gamma but shows differential cellular expression in experimental autoimmune encephalomyelitis and by astrocytes and microglia in vitro. Glia 2007; 55:1728–1739.
Szczucinski A, Kalinowska A, Losy J . CXCL11 (Interferon-inducible T-cell alpha chemoattractant) and interleukin-18 in relapsing-remitting multiple sclerosis patients treated with methylprednisolone. Eur Neurol 2007; 58:228–232.
Calderon TM, Eugenin EA, Lopez L, et al. A role for CXCL12 (SDF-1alpha) in the pathogenesis of multiple sclerosis: regulation of CXCL12 expression in astrocytes by soluble myelin basic protein. J Neuroimmunol 2006; 177:27–39.
McCandless EE, Piccio L, Woerner BM, et al. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol 2008; 172:799–808.
Krumbholz M, Theil D, Cepok S, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006; 129:200–211.
Rottman JB, Slavin AJ, Silva R, Weiner HL, Gerard CG, Hancock WW . Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur J Immunol 2000; 30:2372–2377.
Eltayeb S, Sunnemark D, Berg AL, et al. Effector stage CC chemokine receptor-1 selective antagonism reduces multiple sclerosis-like rat disease. J Neuroimmunol 2003; 142:75–85.
Liang M, Mallari C, Rosser M, et al. Identification and characterization of a potent, selective, and orally active antagonist of the CC chemokine receptor-1. J Biol Chem 2000; 275:19000–19008.
Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ . CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 2000; 192:899–905.
Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD . Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med 2000; 192:1075–1080.
Gaupp S, Pitt D, Kuziel WA, Cannella B, Raine CS . Experimental autoimmune encephalomyelitis (EAE) in CCR2(−/−) mice: susceptibility in multiple strains. Am J Pathol 2003; 162:139–150.
Bennett JL, Elhofy A, Charo I, Miller SD, Dal Canto MC, Karpus WJ . CCR2 regulates development of Theiler's murine encephalomyelitis virus-induced demyelinating disease. Viral Immunol 2007; 20:19–33.
Tran EH, Kuziel WA, Owens T . Induction of experimental autoimmune encephalomyelitis in C57BL/6 mice deficient in either the chemokine macrophage inflammatory protein-1alpha or its CCR5 receptor. Eur J Immunol 2000; 30:1410–1415.
Ni J, Zhu YN, Zhong XG, et al. The chemokine receptor antagonist, TAK-779, decreased experimental autoimmune encephalomyelitis by reducing inflammatory cell migration into the central nervous system, without affecting T cell function. Br J Pharmacol 2009; 158:2046–2056.
Villares R, Cadenas V, Lozano M, et al. CCR6 regulates EAE pathogenesis by controlling regulatory CD4+ T-cell recruitment to target tissues. Eur J Immunol 2009; 39:1671–1681.
Elhofy A, Depaolo RW, Lira SA, Lukacs NW, Karpus WJ . Mice deficient for CCR6 fail to control chronic experimental autoimmune encephalomyelitis. J Neuroimmunol 2009; 213:91–99.
Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 2009; 10:514–523.
Kim JV, Jiang N, Tadokoro CE, et al. Two-photon laser scanning microscopy imaging of intact spinal cord and cerebral cortex reveals requirement for CXCR6 and neuroinflammation in immune cell infiltration of cortical injury sites. J Immunol Methods 2010; 352:89–100.
Kuwabara T, Ishikawa F, Yasuda T, et al. CCR7 ligands are required for development of experimental autoimmune encephalomyelitis through generating IL-23-dependent Th17 cells. J Immunol 2009; 183:2513–2521.
Graham KL, Zabel BA, Loghavi S, et al. Chemokine-like receptor-1 expression by central nervous system-infiltrating leukocytes and involvement in a model of autoimmune demyelinating disease. J Immunol 2009; 183:6717–6723.
Huang D, Shi FD, Jung S, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J 2006; 20:896–905.
Carlson T, Kroenke M, Rao P, Lane TE, Segal B . The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med 2008; 205:811–823.
Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH . Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp Neurol 2009; 220:44–56.
Liu L, Huang D, Matsui M, et al. Severe disease, unaltered leukocyte migration, and reduced IFN-gamma production in CXCR3−/− mice with experimental autoimmune encephalomyelitis. J Immunol 2006; 176:4399–4409.
Muller M, Carter SL, Hofer MJ, et al. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol 2007; 179:2774–2786.
Kohler RE, Comerford I, Townley S, Haylock-Jacobs S, Clark-Lewis I, McColl SR . Antagonism of the chemokine receptors CXCR3 and CXCR4 reduces the pathology of experimental autoimmune encephalomyelitis. Brain Pathol 2008; 18:504–516.
McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS . CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol 2006; 177:8053–8064.
Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM . Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med 2001; 193:713–726.
Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD . An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol 1995; 155:5003–5010.
Elhofy A, Wang J, Tani M, et al. Transgenic expression of CCL2 in the central nervous system prevents experimental autoimmune encephalomyelitis. J Leukoc Biol 2005; 77:229–237.
Fife BT, Kennedy KJ, Paniagua MC, et al. CXCL10 (IFN-gamma-inducible protein-10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol 2001; 166:7617–7624.
Narumi S, Kaburaki T, Yoneyama H, Iwamura H, Kobayashi Y, Matsushima K . Neutralization of IFN-inducible protein 10/CXCL10 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol 2002; 32:1784–1791.
Klein RS, Izikson L, Means T, et al. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol 2004; 172:550–559.
Bagaeva LV, Rao P, Powers JM, Segal BM . CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol 2006; 176:7676–7685.
Hamann I, Zipp F, Infante-Duarte C . Therapeutic targeting of chemokine signaling in multiple sclerosis. J Neurol Sci 2008; 274:31–38.
ClinicalTrials.gov Identifier: NCT01199640. http://clinicaltrials.gov/. 2010.
Platten M, Youssef S, Hur EM, et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc Natl Acad Sci USA 2009; 106:14948–14953.
Stegbauer J, Lee DH, Seubert S, et al. Role of the renin-angiotensin system in autoimmune inflammation of the central nervous system. Proc Natl Acad Sci USA 2009; 106:14942–14947.
Schulze-Topphoff U, Prat A, Prozorovski T, et al. Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat Med 2009; 15:788–793.
Dos Santos AC, Roffe E, Arantes RM, et al. Kinin B2 receptor regulates chemokines CCL2 and CCL5 expression and modulates leukocyte recruitment and pathology in experimental autoimmune encephalomyelitis (EAE) in mice. J Neuroinflammation 2008; 5:49.
Shin T, Kang B, Tanuma N, et al. Intrathecal administration of endothelin-1 receptor antagonist ameliorates autoimmune encephalomyelitis in Lewis rats. Neuroreport 2001; 12:1465–1468.
Osmers I, Smith SS, Parks BW, et al. Deletion of the G2A receptor fails to attenuate experimental autoimmune encephalomyelitis. J Neuroimmunol 2009; 207:18–23.
Wraith DC, Pope R, Butzkueven H, et al. A role for galanin in human and experimental inflammatory demyelination. Proc Natl Acad Sci USA 2009; 106:15466–15471.
Constantinescu CS, Hilliard B, Ventura E, Rostami A . Luzindole, a melatonin receptor antagonist, suppresses experimental autoimmune encephalomyelitis. Pathobiology 1997; 65:190–194.
Freire-Garabal M, Nunez MJ, Balboa J, et al. Administration of the 5-hydroxytryptamine(1A) receptor antagonist WAY100635 suppresses acute experimental allergic encephalomyelitis in Lewis rats. Neurosci Lett 2003; 342:33–36.
Nessler S, Stadelmann C, Bittner A, et al. Suppression of autoimmune encephalomyelitis by a neurokinin-1 receptor antagonist--a putative role for substance P in CNS inflammation. J Neuroimmunol 2006; 179:1–8.
Bedoui S, Miyake S, Lin Y, et al. Neuropeptide Y (NPY) suppresses experimental autoimmune encephalomyelitis: NPY1 receptor-specific inhibition of autoreactive Th1 responses in vivo. J Immunol 2003; 171:3451–3458.
Reinke EK, Johnson MJ, Ling C, et al. Substance P receptor mediated maintenance of chronic inflammation in EAE. J Neuroimmunol 2006; 180:117–125.
Tan YV, Abad C, Lopez R, et al. Pituitary adenylyl cyclase-activating polypeptide is an intrinsic regulator of Treg abundance and protects against experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2009; 106:2012–2017.
Acknowledgements
We thank Dr Ru Zhang, Mr Wei Wei and Ms Jie Lv for discussion and critical reading of the manuscript. This work was supported by grants from the National Natural Science Foundation of China (31000399, 31171348, 90713047, and 31071227), and the Ministry of Science and Technology of China (2012CB910404, 2008DFB30150 and 2009ZX09302-001).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Du, C., Xie, X. G protein-coupled receptors as therapeutic targets for multiple sclerosis. Cell Res 22, 1108–1128 (2012). https://doi.org/10.1038/cr.2012.87
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2012.87
Keywords
This article is cited by
-
Diphtheria toxin induced but not CSF1R inhibitor mediated microglia ablation model leads to the loss of CSF/ventricular spaces in vivo that is independent of cytokine upregulation
Journal of Neuroinflammation (2022)
-
Cannabinoid and endocannabinoid system: a promising therapeutic intervention for multiple sclerosis
Molecular Biology Reports (2022)
-
WNT16 Requires Gα Subunits as Intracellular Partners for Both Its Canonical and Non-Canonical WNT Signalling Activity in Osteoblasts
Calcified Tissue International (2020)
-
Adenosine Promotes the Recovery of Mice from the Cuprizone-Induced Behavioral and Morphological Changes while Effecting on Microglia and Inflammatory Cytokines in the Brain
Journal of Neuroimmune Pharmacology (2018)
-
Deficiency of the G protein Gαq ameliorates experimental autoimmune encephalomyelitis with impaired DC-derived IL-6 production and Th17 differentiation
Cellular & Molecular Immunology (2017)
