
Abstract
HIF-1 is associated with poor prognoses and therapeutic resistance in cancer patients. We previously developed a novel hypoxia-inducible factor (HIF)-1 inhibitor, IDF-11774, a clinical candidate for cancer therapy. We also reported that IDF-1174 inhibited HSP70 chaperone activity and suppressed accumulation of HIF-1α. In this study, IDF-11774 inhibited the accumulation of HIF-1α in vitro and in vivo in colorectal carcinoma HCT116 cells under hypoxic conditions. Moreover, IDF-11774 treatment suppressed angiogenesis of cancer cells by reducing the expression of HIF-1 target genes, reduced glucose uptake, thereby sensitizing cells to growth under low glucose conditions, and decreased the extracellular acidification rate (ECAR) and oxygen consumption rate of cancer cells. Metabolic profiling of IDF-11774-treated cells revealed low levels of NAD+, NADP+, and lactate, as well as of intermediates in glycolysis and the tricarboxylic acid cycle. In addition, we observed elevated AMP and diminished ATP levels, resulting in a high AMP/ATP ratio. The level of AMP-activated protein kinase phosphorylation also increased, leading to inhibition of mTOR signaling in treated cells. In vivo xenograft assays demonstrated that IDF-11774 exhibited substantial anticancer efficacy in mouse models containing KRAS, PTEN, or VHL mutations, which often occur in malignant cancers. Collectively, our data indicate that IDF-11774 suppressed hypoxia-induced HIF-1α accumulation and repressed tumor growth by targeting energy production-related cancer metabolism.
Similar content being viewed by others

Main
Most cancer cells produce energy by glycolysis rather than mitochondrial oxidative phosphorylation, regardless of oxygen availability; this phenomenon is termed the Warburg effect.1 Specifically, this metabolic phenotype of cancer is regulated by the HIF-1, PI3K, p53, MYC, and AMP-activated protein kinase (AMPK)-liver kinase B1 pathways. Although HIF-1α is rapidly degraded under normoxic conditions, this protein is stabilized and dimerizes with the HIF-1β subunit in the nucleus under conditions of hypoxia.2, 3 These HIF-1α/β heterodimers subsequently bind to hypoxia-response elements (HREs) (5′-RCGTG-3′, where R is A or G) in the promoters of target genes involved in angiogenesis, metastasis, and resistance to apoptosis, thereby activating their transcription.4 In addition, HIF-1 activates glycolysis by facilitating the transcription of metabolic genes such as glucose transporters (GLUTs), hexokinase, pyruvate kinase M2, and lactate dehydrogenase A, leading to the reprograming of cancer cell metabolism.4, 5 Therefore, inhibition of HIF-1 could impair the metabolic adaptability of cancer cells and render them sensitive to cancer therapy.6
Although many efforts have been made to develop HIF-1 inhibitors, only few have reached clinical trials.7, 8 In particular, BAY 87-2243, a mitochondrial complex I inhibitor, was shown to reduce hypoxia-induced HIF-1α accumulation and suppress tumor growth in an H460 xenograft model;9 PX-478 was found to prevent hypoxia-mediated HIF-1 signaling by inhibiting HIF-1 translation,10 and to exert antitumor activity in various human cancer cell xenograft models;11 and KCN-1, a benzopyran analog, suppresses HIF-1 activity by disrupting the interaction of HIF-1α with the transcriptional coactivator p300 in glioma cells.12 In addition, NSC-134754 was found to reduce HIF-1 activity and tumor growth in a prostate cancer xenograft model.13
In previous studies, we reported the establishment of HIF-1 inhibitors based on the aryloxyacetylamino benzoic acid scaffold.14, 15, 16 From lead optimization studies, we recently developed an orally administered HIF-1 inhibitor, IDF-11774, which has been approved as a clinical candidate for a phase I study by the Korea Food and Drug Administration.17 Using chemical probes, we previously demonstrated that IDF-11774 inhibits HSP70 chaperone activity by binding to its allosteric pocket, rather than the ATP-binding site in its nucleotide-binding domain.18 The HSP70 family is reported to be associated with malignancy, clinical cancer stage, and poor prognosis of various cancers.19, 20
Here, we further analyze the anticancer efficacy of IDF-11774 in vitro and in vivo, as well as its mode of action. Our data demonstrate that IDF-11774 reduced cancer cell growth through the regulation of cancer glycolytic metabolism and energy production. IDF-11774 showed significant antitumor efficacy in xenograft assays of various cancer models bearing drug resistance mutations, suggesting that IDF-11774 may be used by itself or in combination with other agents in cancer therapeutics.
Results
IDF-11774 inhibits HIF-1α accumulation and suppresses angiogenesis
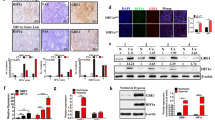
Previously, we screened a focused library of aryloxyacetylamino benzoic acid scaffolds and performed lead optimization for the identification of an HIF-1α inhibitor. We found that IDF-11774 reduced the HRE-luciferase activity of HIF-1α (IC50=3.65 μM) and blocked HIF-1α accumulation under hypoxic conditions in HCT116 human colon cancer cells (Figure 1a and Supplementary Figure S1a). We next evaluated the effect of IDF-11774 on HIF-1α accumulation in vivo using a bioluminescence imaging assay. Luciferase activity and HIF-1α accumulation were strongly suppressed in the tumors of mice treated by oral administration of IDF-11774, compared with the control (Figure 1b and Supplementary Figure S1b).

IDF-11774 inhibits HIF-1α accumulation in HCT116 cells. (a) Structure of IDF-11774 and its effect on HIF-1α accumulation, as determined by western blot analysis. (b) In vivo bioluminescence imaging of HIF-1 activity. The relative luminescence in live tumors and HIF-1α levels in tumor tissue were measured after treatment for 4 days. Data are presented as the means and standard deviations of the results from three independent experiments; *P<0.05, compared with vehicle control. (c) Quantitative real-time PCR analysis of the mRNA expression levels of HIF-1α target genes in HCT116 cells treated with IDF-11774 for 18 h. *P<0.05 and **P<0.01, compared with the untreated hypoxia control. (d) In vitro tube formation: HUVECs were treated with DMSO, IDF-11774, or sunitinib under 1% O2 for 24 h. (e) CAM assay: in situ inhibition of angiogenesis in the chicken embryo by treatment with IDF-11774 (20 μg per egg) or sunitinib (10 μg per egg)
Subsequently, we found that IDF-11774 inhibited the expression of the HIF-1 target genes VEGF and EPO, which are involved in angiogenesis, but not of the gene encoding HIF-1α itself (Figure 1c). Therefore, we investigated the effects of the inhibitor on angiogenesis using both in vitro and in vivo assays. In the in vitro tube formation assay, human umbilical vascular endothelial cells (HUVECs) treated with IDF-11774 showed reduced capillary network formation on Matrigel, similar to that observed with sunitinib, the positive control (Figure 1d). This IDF-11774-mediated inhibition of in vitro tube formation was rescued by the addition of VEGF (Supplementary Figure S1c). Similarly, chick embryo chicken chorioallantoic membrane (CAM) analyses revealed treatment with IDF-11774 and sunitinib resulted in reduced vessel formation within the CAM in vivo, compared with that observed in the negative control (Figure 1e). These results strongly indicate that IDF-11774 potently represses angiogenesis.
IDF-11774 inhibits glucose-dependent cancer metabolism
We next revealed that IDF-11774 treatment resulted in reduced mRNA expression of GLUT1 and pyruvate dehydrogenase kinase 1 (PDK1), which are targets of HIF-1 (Figure 2a). Therefore, we assessed whether IDF-11774 affected the metabolism of HCT116 cells. Glucose uptake assay analysis demonstrated that IDF-11774 markedly suppressed the cellular uptake of [3H]2-deoxyglucose (2DG) (Figure 2b). In addition, intracellular ATP levels were significantly reduced in the presence of IDF-11774 and were affected to a greater degree under low glucose conditions (5.5 mM) (Figure 2c). Indeed, cells treated with IDF-11774 in low glucose medium exhibited stronger growth inhibition than cells treated in high glucose (25 mM) (Figure 2d). Conversely, 5-fluorouracil (5-FU) and sorafenib exerted no glucose-related inhibitory effects on the growth of cancer cells (Figure 2d and Supplementary Figure S2).

Effect of IDF-11774 on glycolytic energy metabolism and cell growth. (a) Effect of IDF-11774 on mRNA expression after treatment of HCT116 cells with IDF-11774 for 6 h; *P<0.05 and **P<0.01, compared with the untreated hypoxia control. RPL13A was used as a control. (b) Effect of IDF-11774 on glucose transport using 2DG. (c) Levels of glucose-dependent ATP production in HCT116 cells cultivated in the presence of IDF-11774 for 12 h. (d) Effect of IDF-11774 on glucose-dependent cell growth. The growth of cells treated with IDF-11774 or 5-FU for 72 h was compared in media containing 25 or 5.5 mM glucose. Data are presented as the means and standard deviations of the results from three independent experiments; *P<0.05, **P<0.01, and ***P<0.001, compared with 25 mM glucose. (e and f) Effects of IDF-11774 on (e) glycolysis and (f) mitochondrial respiration
Subsequently, the effect of IDF-11774 on the ECAR, which is an indicator of glycolysis activity, was determined using an XF-24 Extracellular Flux Analyzer (Seahorse Biosciences, North Billerica, MA, USA). Treatment of cells with IDF-11774 inhibited both glucose-mediated basal and oligomycin-mediated ECAR in a dose-dependent manner (Figure 2e), suggesting that IDF-11774 inhibits glycolysis in HCT116 cells. We further investigated the effects of IDF-11774 on mitochondrial respiration by measuring the oxygen consumption rate (OCR). IDF-11774 significantly inhibited OCR in a concentration-dependent manner (Figure 2f), demonstrating that this compound also blocks mitochondrial respiration.
Metabolic profiling of cells treated with IDF-11774
To confirm that IDF-11774 affects the metabolism of HCT116 cells, we examined the metabolite profiles of cells treated with IDF-11774 under hypoxic condition via 1H-NMR spectroscopy. Treatment of cells with IDF-11774 for 12 h did not affect HCT116 cell growth (Supplementary Figure S3). A total of 49 metabolites, including amino acids, carbohydrates, organic acids, and nucleotides, in HCT116 cell extracts were identified and quantified under the test conditions (Figure 3a). Notably, IDF-11774 treatment resulted in reductions in the levels of many glycolysis and tricarboxylic acid (TCA) cycle metabolites. In particular, IDF-11774 significantly reduced the levels of intracellular lactate, an end product of glycolysis (Figure 3b), and NAD+ and NADP+, which are metabolites related to nicotinamide metabolism (Figure 3c). In addition, the levels of TCA cycle intermediates such as fumarate, malate, and succinate were apparently reduced in HCT116 cells treated with IDF-11774, compared with the control (Figure 3d). Furthermore, IDF-11774 treatment resulted in increased cellular AMP levels and decreased ATP levels, resulting in an elevated AMP/ATP ratio (Figure 3e). These data suggest that growth inhibition in HCT116 cells treated with IDF-11774 under hypoxic conditions was the result of inhibition of energy production metabolism.

Metabolic profile of cells treated with IDF-11774 under hypoxic conditions. (a) Heatmaps of quantified metabolites in HCT116 cells treated with or without 10 μM IDF-11774 for 12 h. (b–e) Levels of (b) lactate, (c) NAD+ and NADP+, (d) TCA cycle intermediate metabolites, and (e) AMP and ATP in HCT116 cells treated with or without 10 μM IDF-11774 for 12 h. Data are presented as the means and standard deviations of the results from three independent experiments; *P<0.05; **P<0.01 and ***P<0.001, compared with the untreated control
Finally, we performed western blot analysis to determine the degree of IDF-11774-mediated activation of AMPK signaling consequent to the elevation of the AMP/ATP ratio. IDF-11774 treatment resulted in increased phosphorylation of AMPK, which inactivates ACC and suppressed the phosphorylation of mTOR and 4EBP1, thereby ultimately attenuating the translation of HIF-1α (Figure 4a). These results suggest that IDF-11774 inhibits glucose-dependent cancer metabolism through positive feedback regulation of HIF-1α inhibition (Figure 4b).

Effect of IDF-11774 on energy metabolism. (a) Western blot analysis of the expression levels of proteins involved in AMPK activation in the presence or absence of IDF-11774. (b) Model depicting the mode of action of IDF-11774: IDF-11774 suppresses HIF-1α accumulation, thereby inhibiting glucose-dependent energy metabolism and mitochondrial respiration in cancer cells. HIF-1α accumulation was reduced by the enhanced degradation of HIF-1α consequent to increased local oxygen concentrations and the translational attenuation of HIF-1α by mTOR inactivation
IDF-11774 suppresses tumor growth in various cancer xenograft models
To evaluate the in vivo antitumor efficacy of IDF-11774, we performed a xenograft assay using HCT116 cells (Figures 5a–d and Supplementary Table S1). When IDF-11774 was orally administered daily for two weeks, significant dose-dependent tumor regression was observed in the mouse model (Figure 5a). We then tested whether IDF-11774 could be used for combination therapy with other agents to enhance its anticancer efficacy in an HCT116 xenograft model. The combination of IDF-11774 with the multikinase inhibitor sunitinib resulted in a significant increase in anticancer efficacy, compared with individual treatment with either agent (Figure 5b). When intravenous treatment of IDF-11774 (intravenously, twice a week) was combined with sunitinib (per oral, once a day), a similar enhancement of antitumor efficacy was obtained, compared with each treatment alone (Figure 5c). In addition, the combined treatment of IDF-11774 with the multikinase inhibitors sorafenib or lapatinib also showed enhancement in anticancer efficacy (Figure 5d). Notably, however, no significant weight loss or side effects, such as skin ulcers or other severe symptoms, were observed in any of the treated mice.

Antitumor efficacy of IDF-11774 in HCT116 xenograft models. Graphic depictions of tumor size in xenograft model mice in the presence or absence of (a) IDF-11774 (per oral (p.o.), 10, 30, or 60 mg/kg, once a day (q.d.)), (b) IDF-11774 and sunitinib (p.o., 30 mg/kg each, q.d.), (c) IDF-11774 (intranvenously (i.v.), 2/wk; 30 mg/kg, q.d.) and sunitinib (p.o., 30 mg/kg, q.d.), or (d) IDF-11774 (p.o., 60 mg/kg, q.d.) and sorafenib or lapatinib (p.o., 30 mg/kg each, q.d.). Data are presented as the means and standard deviations of the results from five independent experiments; ***P<0.001, compared with vehicle control
To investigate additional applications of IDF-11774, the effects of IDF-11774 on other cancer cells were investigated. IDF-11774 inhibited HIF-1α accumulation and the growth of various cancer cells (Supplementary Figures S4a and S4b). Furthermore, we investigated the in vivo antitumor efficacy of IDF-11774 in various xenograft models (Table 1 and Supplementary Figure S5). IDF-11774 exhibited strong efficacy in inhibiting the growth of A549 cell tumor, which contain a KRAS mutation. IDF-11774 also inhibited the tumor growth of NCI-H1975 cells bearing an EGFR T790M mutation and wild-type KRAS. Notably, IDF-11774 demonstrated good response in both PC-3 prostate cancer cells, which are PTEN-null, as well as MIA-PaCa-2 pancreatic cancer cells. Last, IDF-11774 significantly suppressed the tumor growth of Caki-1 renal cancer cells containing wild-type VHL and of 786-O cells with truncated VHL. Collectively, IDF-11774 showed significant antitumor efficacy in xenograft models of tumors harboring various mutations.
Discussion
Cancer cells show alterations in cellular metabolism, such as aerobic glycolysis, high fatty acid synthesis, and rapid glutamine metabolism, which is linked to therapeutic resistance.1, 21, 22 Accordingly, mutations and alterations in several metabolism-related enzymes, including IDH1, IDH2, SDH, FH, and PKM2, have been discovered in various cancers.23 Therefore, it has been suggested that targeting of metabolic pathways likely represents an efficient strategy to improve antitumor efficacy and overcome therapeutic resistance in the development of anticancer drugs.24 In particular, metabolic inhibitors targeting GLUTs, PDK1, and glycolytic enzymes have been shown to enhance anticancer efficacy and mitigate drug resistance.24
We have developed an orally administered anticancer agent, IDF-11774, which reduced HIF-1α accumulation both in vitro and in vivo. IDF-11774 suppressed in vitro tube formation and vascularization in an in vivo CAM assay, indicating an inhibition of angiogenesis. In a previous report, IDF-11774 inhibited HSP70 chaperone activity, resulting in suppression of HIF-1α refolding.25 Moreover, IDF-11774 reduced the OCR and ATP production, thereby increasing intracellular oxygen tension to stimulate HIF-1α degradation. Furthermore, IDF-11774 suppressed GLUT1 expression and ECAR, resulting in sensitization of cells to growth under low glucose conditions, and significantly inhibited mitochondrial respiration, resulting in increased local oxygen tension to promote the proteasomal degradation of HIF-1α.25 It has been reported that HIF-1 inhibition increases mitochondria-mediated oxygen consumption due to reduction of PDK1 expression.26 Therefore, our observation of decrease in OCR in the presence of IDF-11774 is likely the effect of HSP70 inhibition, which is HIF-1α -independent.
In a cell culture system, short exposure to hypoxic conditions resulted in increased HIF-1α accumulation, while prolonged exposure to hypoxia resulted in reduced accumulation of this protein.27 In our study, to understand the effects of IDF-11774 on HIF-1α expression, we evaluated the levels of HIF-1α production in HCT116 cells after exposure to hypoxia for 6 h, at which peak levels of HIF-1α were previously observed. Indeed, Luo et al. suggest that HIF-1α is degraded by CHIP/HSP70-mediated proteasomal degradation in HEK293 cells during prolonged exposure to hypoxic conditions.28 It appears that coordinated positive and negative regulatory mechanisms control the transcription and degradation of HIF-1α to maintain cell homeostasis. Consistent with this conclusion, several reports have described distinct regulatory mechanisms underlying the degradation of HIF-1α in cancer cells.29, 30, 31 In addition, complex relationships between transcriptional activation of HSP70 family members by HIF-1α have been reported.32, 33
Next, to confirm the effect of IDF-11774 on cancer metabolism, we examined the metabolic profile of cells treated with IDF-11774. IDF-11774 significantly reduced the intracellular lactate level, indicating a decrease in glycolysis. In addition, there was a significant decrease in the levels of intermediates in glycolysis and the TCA cycle, as well as in amino acids, which are necessary for biosynthesis of the building blocks of cell growth. Furthermore, cellular concentrations of NAD+ and NADP+ decreased in the presence of IDF-11774, compared with the control. NAD+ is converted to NADH by coupling with the glycolytic pathway, β-oxidation, and the TCA cycle, whereas NADP+ to NADPH conversion is accomplished by the pentose phosphate pathway. NADH is used for ATP production in the mitochondria via the electron transport chain, while NADPH, a crucial antioxidant, is used for fatty acid biosynthesis, which provides lipids for membrane biogenesis and confers a survival advantage to cancer cells. As expected, IDF-11774 treatment yielded reduced ATP levels, resulting in an elevated AMP/ATP ratio. The activation of AMPK and suppression of mTOR phosphorylation were also observed in the presence of IDF-11774. It is further likely that IDF-11774 inhibited energy production-related metabolism of cancer cells, because IDF-11774 caused significant glucose-dependent cell death and the suppression of mitochondrial oxidative phosphorylation.
Notably, we revealed that IDF-11774 suppressed the growth of various cancer cells in vitro and in vivo. In particular, IDF-11774 significantly suppressed tumor growth in both Caki-1 and 786-O renal cancer cells regardless of the presence of a VHL mutation, which is HIF-1α independent and differs from the effects of other HIF-1 inhibitors that function only in cancer cells containing wild-type VHL.9 Additionally, it was previously reported that combination treatment using the HIF inhibitor BAY 87-2243 and the BRAF inhibitor vemurafenib potentially yielded enhanced tumor growth inhibition in nude mice bearing a BRAF mutant melanoma xenograft.34 Similarly, IDF-11774 provided substantial inhibition of tumor growth when combined with sunitinib, sorafenib, or the HER2 inhibitor lapatinib. Therefore, we suggest that IDF-11774 represents a metabolic inhibitor suitable for use in cancer therapy to cure cancers containing mutations in KRAS, PTEN, EGFR T790M, p53, PI3K, and VHL, alone or in combination with other anticancer drugs or radiotherapy.
In summary, we report a novel HIF-1 inhibitor, IDF-11774, which suppressed HIF-1α accumulation in colorectal cancer cells in vitro and in vivo. Additionally, IDF-11774 inhibited glucose uptake, ECAR, and OCR, leading to decreased glucose-dependent energy metabolism. Metabolic analysis of cells treated with IDF-11774 detected significant changes in the levels of glycolysis and TCA cycle metabolites. Furthermore, IDF-11774 exhibited significant efficacy in inhibiting the growth of tumors bearing mutations regulating cancer metabolism. Taken together, our results demonstrate that IDF-11774 targets cancer metabolism to suppress the growth of cancer cells.
Materials and Methods
Materials
IDF-11774 were synthesized as described previously.18 Sunitinib and 5-FU were obtained from Sigma-Aldrich (St. Louis, MO, USA). Sorafenib and lapatinib were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Stock solutions of compounds were prepared in DMSO at 10 mM and stored at −20 °C. The following antibodies were used: anti-β-actin (sc-47778), anti-ACCα (sc-30212), and anti-AMPKα (sc-25792) from Santa Cruz Biotechnology; anti-HER2 (2242), anti-mTOR (2983), anti-p-mTOR (2971), anti-pACC (3661), anti-4EBP1 (9644), anti-p-4EBP1 (9459), and anti-p-AMPK (2531) from Cell Signaling Technology (Beverly, MA, USA); anti-HIF-1α (610958) and anti-cyclin D1 (554180) from BD Biosciences (San Diego, CA, USA); anti-β-tubulin (ab15568) from Abcam (Cambridge, MA, USA).
Cell culture
The following human cancer cell lines were obtained from the Bioevaluation Center at the Korea Research Institute of Bioscience and Biotechnology (KRIBB): HCT116 colon cancer cells, non-small-cell lung cancer adenocarcinoma A549 and NCI-H1975, pancreatic cancer MIA-PaCa-2, prostate cancer PC-3, and renal cancer Caki-1 and 786-O. Cells were cultured in a 5% CO2 atmosphere at 37 °C in Dulbecco’s modified Eagle’s medium (Gibco, Carlsbad, CA, USA) or RPMI (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco). Hypoxic conditions were achieved by placing the cells in a 1% O2, 94% N2, and 5% CO2 multigas incubator (Sanyo, Osaka, Japan).
Western blot analysis
Western blot analysis was performed as described previously.35 Cells were lysed with 1 × RIPA buffer (Millipore, Billerica, MA, USA) containing 1 mM Na3VO4, 1 mM sodium fluoride, 1 mM PMSF, and a protease inhibitor cocktail (Roche, Basel, Switzerland), and the protein concentrations of the resulting lysates were quantified using a BCA Assay Kit (Bio-Rad, Hercules, CA, USA). Western blot signals were detected using an Enhanced Chemiluminescence (ECL) Kit (Millipore).
Quantitative real-time PCR
Real-time PCR was performed using RT2 SYBR Green qPCR Mastermix (Qiagen, Venlo, The Netherlands). Data were analyzed using the Rotor-Gene 6000 Series Software (Corbett Research, Cambridge, UK). VEGF, EPO, GLUT1, PDK1, and HIF-1α primers were purchased from Bioneer (Daejeon, Korea).
In vivo/in vitro HIF-1 reporter assay
HRE-luciferase activity was measured as described previously.36 For bioluminescence imaging assays, IDF-11774 (50 mg/kg) was administered orally to Balb/c nude mice bearing tumors (100 mm3) formed from HCT116 cells (1 × 107) expressing HRE-luciferase by subcutaneous injection. After intraperitoneal injection of d-luciferin, bioluminescence images of each tumor were analyzed using the IVIS Lumina II Luminescence Imaging System (Caliper Life Science, Alameda, CA, USA) and Living Image Software (Caliper Life Science).
In vitro tube formation assay
In vitro tube formation assays were performed using HUVECs as described previously.16 The tubule branches were observed under a microscope and photographed.
Chicken CAM assay
The CAM assay was performed as described previously.37 IDF-11774 (20 μg per egg)- or Sunitinib (10 μg per egg)-loaded thermanox coverslips (Nalge NUNC, Rochester, NY, USA) were laid on the CAM surface.
Measurement of glucose uptake
Glucose uptake was measured as described previously.38 HCT116 cells in Krebs-Ringer phosphate buffer were incubated with 2DG (0.5 μCi/ml), and tritium activity was measured by Tri-Carb Liquid Scintillation Counting (Perkin-Elmer, Wellesley, MA, USA).
Measurement of glycolytic flux and mitochondrial respiration
The ECAR was measured using a XF-24 extracellular flux analyzer (Seahorse Biosciences), according to the manufacturer’s instructions. The OCR was measured as described previously.25
1H-NMR analysis of metabolites
Polar metabolites were extracted from cells with solvent composed of methanol, water, and chloroform. 1H-NMR spectra were acquired on a Bruker Avance III HD 800 MHz FT-NMR Spectrometer (Bruker BioSpin Co., Billerica, MA, USA) at 298 K using a 5-mm triple-resonance inverse cryoprobe with Z-gradients. The 1D NOESY pulse sequence was applied to suppress the residual water signal. For each sample, 256 transients were collected into 64 000 data points using a spectral width of 16 393.4. All NMR spectra were phased and baseline corrected using Chenomx NMR suite version 7.1 (Chenomx Inc., Edmonton, AB, Canada). Metabolite identities were confirmed based on total correlation spectroscopy (2D 1H-1H TOCSY) and spiking experiments. Quantification was achieved using the 800 MHz library from Chenomx NMR Suite version 7.1. The amount of each metabolite was calculated according to the number of cells present in the additional sets of sample dishes prepared using the same procedures.
Xenograft model
All animal experimental protocols were approved by the bioethics committee of the Korea Research Institute of Bioscience and Biotechnology. Cancer cells were injected subcutaneously into 4- to 6-week-old female Balb/c nude mice to generate tumors (5 mice per group). When the tumors grew to 100 mm3, IDF-11774 was administered orally (per oral) or intravenously for 15 days. Tumor volumes (V) were determined using the following equation: V (mm3)=(length × width × height) × 0.5. Percentage tumor growth inhibition (%TGI) values were calculated for each treatment group (T) versus the control (C) using initial (i) and final (f) tumor volumes, according to the formula: %TGI=(1 – [Tf – Ti]/[Cf – Ci]) × 100.
Statistical analysis
Differences between results were analyzed using Student’s t-test for unpaired observations and Dunnett’s test for multiple comparisons.
References
Hsu PP, Sabatini DM . Cancer cell metabolism: Warburg and beyond. Cell 2008; 134: 703–707.
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2000; 2: 423–427.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292: 464–468.
Semenza GL . Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003; 3: 721–732.
Semenza GL . HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 2010; 20: 51–56.
Semenza GL . HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 2013; 123: 3664–3671.
Ban HS, Uto Y, Won M, Nakamura H . Hypoxia-inducible factor (HIF) inhibitors: a patent survey (2011-2015). Expert Opin Ther Patents 2016; 26: 309–322.
Xia Y, Choi HK, Lee K . Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem 2012; 49: 24–40.
Ellinghaus P, Heisler I, Unterschemmann K, Haerter M, Beck H, Greschat S et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med 2013; 2: 611–624.
Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL et al. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther 2008; 7: 90–100.
Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G . Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol Cancer Ther 2004; 3: 233–244.
Yin S, Kaluz S, Devi NS, Jabbar AA, de Noronha RG, Mun J et al. Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1alpha interaction with cofactors p300/CBP. Clin Cancer Res 2012; 18: 6623–6633.
Baker LC, Boult JK, Walker-Samuel S, Chung YL, Jamin Y, Ashcroft M et al. The HIF-pathway inhibitor NSC-134754 induces metabolic changes and anti-tumour activity while maintaining vascular function. Br J Cancer 2012; 106: 1638–1647.
Lee K, Lee JH, Boovanahalli SK, Jin Y, Lee M, Jin X et al. (Aryloxyacetylamino)benzoic acid analogues: a new class of hypoxia-inducible factor-1 inhibitors. J Med Chem 2007; 50: 1675–1684.
Lee K, Kang JE, Park SK, Jin Y, Chung KS, Kim HM et al. LW6, a novel HIF-1 inhibitor, promotes proteasomal degradation of HIF-1alpha via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol 2010; 80: 982–989.
Naik R, Won M, Kim BK, Xia Y, Choi HK, Jin G et al. Synthesis and structure–activity relationship of (E)-phenoxyacrylic amide derivatives as hypoxia-inducible factor (HIF) 1alpha inhibitors. J Med Chem 2012; 55: 10564–10571.
Lee K, Won M-S, Kim H-M, Park S-K, Lee K-H, Lee C-W et alinventorsPreparation of adamantyl heterocyclic compounds as HIF-1α inhibitors for treatment of cancer, diabetic retinopathy, and rheumatoid arthritis. Patent No. WO2013048164A1, 2013.
Ban HS, Naik R, Kim HM, Kim B-K, Lee H, Kim I et al. Identification of targets of the HIF-1 inhibitor IDF-11774 using alkyne-conjugated photoaffinity probes. Bioconjug Chem 2016; 27: 1911–1920.
Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, Jaattela M . Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev 2005; 19: 570–582.
Murphy ME . The HSP70 family and cancer. Carcinogenesis 2013; 34: 1181–1188.
Cairns RA, Harris IS, Mak TW . Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11: 85–95.
Vander Heiden MG, Cantley LC, Thompson CB . Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033.
Teicher BA, Linehan WM, Helman LJ . Targeting cancer metabolism. Clin Cancer Res 2012; 18: 5537–5545.
Zhao Y, Butler EB, Tan M . Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013; 4: e532.
Lee K, Ban HS, Naik R, Hong YS, Son S, Kim BK et al. Identification of malate dehydrogenase 2 as a target protein of the HIF-1 inhibitor LW6 using chemical probes. Angew Chem Int Ed 2013; 52: 10286–10289.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC . HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3: 187–197.
Lee Dong C, Sohn Hyun A, Park Z-Y, Oh S, Kang Yun K, Lee K-m et al. A lactate-induced response to hypoxia. Cell 2015; 161: 595–609.
Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but not HIF-2alpha. J Biol Chem 2010; 285: 3651–663..
Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M et al. OS-9 interacts with hypoxia-inducible factor 1α and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1α. Mol Cell 2005; 17: 503–512.
Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL . RACK1 competes with HSP90 for binding to HIF-1α and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1α. Mol Cell 2007; 25: 207–217.
Yang S-J, Park YS, Cho JH, Moon B, Ahn H-J, Lee JY et al. Regulation of hypoxia responses by flavin adenine dinucleotide-dependent modulation of HIF-1α protein stability. EMBO J 2017; 36: 1011–1028.
Huang W-J, Xia L-M, Zhu F, Huang B, Zhou C, Zhu H-F et al. Transcriptional upregulation of HSP70-2 by HIF-1 in cancer cells in response to hypoxia. Int J Cancer 2009; 124: 298–305.
Gogate SS, Fujita N, Skubutyte R, Shapiro IM, Risbud MV . Tonicity enhancer binding protein (TonEBP) and hypoxia-inducible factor (HIF) coordinate heat shock protein 70 (Hsp70) expression in hypoxic nucleus pulposus cells: Role of Hsp70 in HIF-1α degradation. J Bone Miner Res 2012; 27: 1106–1117.
Schöckel L, Glasauer A, Basit F, Bitschar K, Truong H, Erdmann G et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab 2015; 3: 11.
Im JY, Lee KW, Won KJ, Kim BK, Ban HS, Yoon SH et al. DNA damage-induced apoptosis suppressor (DDIAS), a novel target of NFATc1, is associated with cisplatin resistance in lung cancer. Biochim Biophys Acta 2016; 1863: 40–49.
Naik R, Won M, Ban HS, Bhattarai D, Xu X, Eo Y et al. Synthesis and structure-activity relationship study of chemical probes as hypoxia induced factor-1alpha/malate dehydrogenase 2 inhibitors. J Med Chem 2014; 57: 9522–9538.
Lee JH, Lee DH, Lee HS, Choi JS, Kim KW, Hong SS . Deguelin inhibits human hepatocellular carcinoma by antiangiogenesis and apoptosis. Oncol Rep 2008; 20: 129–134.
Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA 2012; 109: 13787–13792.
Acknowledgements
This work was supported by the KRIBB Initiative program, National Research Foundation (NRF) grants (2012M3A9C1053532, 2015M3A6A4065734 and 2015M3A9A8032460), Health Technology R&D grant (HI13C2162) from Ministry of Health and Welfare and Korea Basic Science Institute (C36705). This work was also supported by research program of Dongguk University, 2017.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Agostini
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ban, H., Kim, BK., Lee, H. et al. The novel hypoxia-inducible factor-1α inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis 8, e2843 (2017). https://doi.org/10.1038/cddis.2017.235
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2017.235
This article is cited by
-
Associations between HIFs and tumor immune checkpoints: mechanism and therapy
Discover Oncology (2024)
-
NF-κB signaling in neoplastic transition from epithelial to mesenchymal phenotype
Cell Communication and Signaling (2023)
-
AIMP2-DX2 provides therapeutic interface to control KRAS-driven tumorigenesis
Nature Communications (2022)
-
VHL suppresses RAPTOR and inhibits mTORC1 signaling in clear cell renal cell carcinoma
Scientific Reports (2021)
-
Hypoxia-induced alternative splicing: the 11th Hallmark of Cancer
Journal of Experimental & Clinical Cancer Research (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}