
Abstract
The sex steroid hormone 17β-estradiol (E2) upregulates the levels of neuroglobin (NGB), a new neuroprotectant globin, to elicit its neuroprotective effect against H2O2-induced apoptosis. Several mechanisms could be proposed to justify the NGB involvement in E2 prevention of stress-induced apoptotic cell death. Here, we evaluate the ability of E2 to modulate the intracellular NGB localization and the NGB interaction with mitochondrial cytochrome c following the H2O2-induced toxicity. Present results demonstrate that NGB is expressed in the nuclei, mitochondria, and cytosol of human neuroblastoma SK-N-BE cells. E2, but not H2O2 treatment of SK-N-BE cells, reallocates NGB mainly at the mitochondria and contemporarily reduces the number of apoptotic nuclei and the levels of cleaved caspase-3. Remarkably, the E2 treatment strongly increases NGB–cytochrome c association into mitochondria and reduces the levels of cytochrome c into the cytosol of SK-N-BE cells. Although both estrogen receptors (ERα and ERβ) are expressed in the nucleus, mitochondria, and cytosol of SK-N-BE cells, this E2 effect specifically requires the mitochondrial ERβ activity. As a whole, these data demonstrate that the interception of the intrinsic apoptotic pathway into mitochondria (i.e., the prevention of cytochrome c release) is one of the pivotal mechanisms underlying E2-dependent NGB neuroprotection against H2O2 toxicity.
Similar content being viewed by others

Main
Neuroglobin (NGB), a globin family member, is a monomeric hexa-coordinated heme protein of 17 kDa, predominantly expressed in neurons of the central and peripheral nervous system. However, NGB mRNA and/or protein expression was also recognized in non-neuronal tissues.1, 2, 3 As a globin, NGB reversibly binds gaseous ligands, such as O2, CO, and NO,4, 5 facilitates scavenging of reactive nitrogen species (RNS) and reactive oxygen species (ROS),6 and catalyzes the conversion of nitrite to NO under hypoxic conditions.7 Remarkably, NGB reactivity is facilitated allosterically by phosphorylation and 14-3-3 binding.8
During the past decade, several experimental works suggested that NGB overexpression is protective against hypoxic/ischemic injury in the brain.1, 9 In human neuroblastoma cell lines, SH-SY5Y, NGB overexpression enhanced cell survival under the condition of anoxia or glucose deprivation.6, 10 Moreover, in transgenic rodents, increased levels of NGB significantly protect the brain tissues from hypoxic insult, whereas decreased NGB levels lead to the exacerbation of the tissue death.11, 12, 13 As a whole these data sustain a neuroprotective role of high NGB levels in neural tissues and neural-derived cell lines. Nevertheless, the underlying molecular mechanism(s) for the neuroprotective effect of NGB has remained controversial. Several NGB roles have been proposed: (i) NGB could exert a Mioglobin-like role, having an oxygen buffering or transport role in cells; (ii) NGB could scavenge the damage induced by either RNS or ROS; (iii) NGB could detoxify harmful excess of NO to nitrate at normoxia or produce NO for signaling functions from nitrite at hypoxia; and (iv) NGB could be involved in a signal transduction pathway, for example, by inhibiting the dissociation of guanosine diphosphate from G proteinα.1, 8, 9, 14 All these processes are at least in part associated with the prevention of apoptotic cell death and could be related to a possible interaction between NGB and cytochrome c,15 and thus a direct involvement of this globin in the mitochondrial (intrinsic) apoptotic pathway.15, 16, 17, 18
During ischemic episodes, cells can enter in the intrinsic apoptotic pathway via the partial release of ferric mitochondrial cytochrome c into the cytoplasm. Ferric cytochrome c is a required component of the caspase-cascade activating apoptosome.15, 19 Recently, based on the well-known redox reactions of NGB, it has been hypothesized that NGB could react with the oxidized cytochrome c released from the mitochondria.15, 17, 19 The first direct experimental evidence that NGB can interrupt the process of apoptosis at the level of cytochrome c reduction derived from a model system consisting of a cell lysate, free of mitochondria, where exogenous cytochrome c was added in the absence and in presence of NGB. In this system it was shown by computer modeling that NGB in the presence of ferric cytochrome c drastically lowers the enzymatic activity of caspase-9.18 However, if this process occurs the in cytosol or in the mitochondria, where NGB could prevent the onset of apoptosis directly sequestering cytochrome c, could not be ruled out.
Recently, we demonstrated that the sex steroid hormone 17β-estradiol (E2) increases up to 300% NGB levels acting as an endogenous modulator of NGB levels in the SK-N-BE human neuroblastoma cell line and in mouse primary hippocampal neurons. The E2-induced NGB upregulation is pivotal for the neuroprotective effect of E2 against H2O2-induced toxicity.20, 21 These results open the possibility that the protective role played by the E2-induced NGB could arise from NGB intervention in the mitochondrial apoptotic pathway and, in particular, from the association of NGB with cytochrome c in neuronal cells. Therefore, the aim of this work is to evaluate this hypothesis by assessing the interaction between NGB and cytochrome c in the presence of H2O2 as pro-apoptotic factor and E2 as a survival agent in SK-N-BE neuroblastoma cell line.
Results
E2, via NGB, protects SK-N-BE cells from H2O2-induced apoptosis
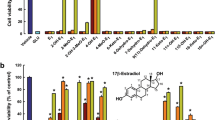
As expected, 24 h after 50 μM H2O2 insult, the SK-N-BE cell number is reduced (data not shown20) with the parallel increase of apoptotic nuclei (Figures 1a and a’) and cleaved caspase-3 (i.e., 17 kDa band) levels (Figure 1b). One nanomole of E2 strongly reduces the number of apoptotic nuclei and caspase-3 cleavage (Figure 1). In NGB-silenced SK-N-BE cells (Figure 1a”), E2 was unable to counteract the H2O2-induced increase of apoptotic nuclei and the cleavage of caspase-3, confirming the protective role of E2-induced NGB in preventing the apoptosis-induced by H2O2 toxicity.20

E2 effect on SK-N-BE human neuroblastoma cell line viability. (a) Nuclear morphology of SK-N-BE cells 24 h after H2O2 injury (50 μM). Cells, transfected with control plasmid or with SiNGB, were pretreated 24 h before the addition of H2O2 with either vehicle or E2 (1 nM). Fixed and permeabilized cells were stained with DAPI. Apoptotic nuclei were detected as condensed nuclei (arrow heads) (fluorescence microscopy, original magnification × 40). (a’) Data are means±S.D. of five independent experiments carried out in duplicate. Significant differences (P<0.001) were determined with ANOVA followed by Tukey–Kramer post-test: *significant difference versus vehicle—H2O2, °versus vehicle+H2O2. (a’’) Western blot analysis of NGB levels in cell transfected with control plasmid (MOCK) or with SiNGB. The figure represents a typical Western blot of three independent experiments. (b) Western blot analyses of caspase-3 activation were performed on cells stimulated with either the vehicle or pretreated with E2 (1 nM) for 24 h in the presence or absence of H2O2 50 μM treatment (24 h of stimulation). The amount of proteins was normalized to tubulin levels. Representative Western blots from three independent experiments are shown
E2 affects NGB intracellular localization
In SK-N-BE cells, NGB appears to be distributed in both nucleus and cytosol (Figure 2a). This localization has been confirmed in the HeLa cells transiently transfected with pcDNA-flag-NGB plasmid, showing a widespread distribution of fluorescent signal (Figure 2b). Furthermore, SK-N-BE cell fractionation into the three main cell compartments (i.e., nuclei, mitochondria, and cytosol) showed that NGB is principally localized into the cell nuclei and in mitochondria than in the cytosolic fraction. The nuclear Poly ADP-ribose polymerase (PARP), the mitochondrial cytochrome c oxidase-4 (COX-4; Complex IV), and the cytosolic protein phosphatase-2A (PP2A) have been used as the purity markers of cell fractions (Figures 2c and c’). Intriguingly, 1 nM E2 is able to reduce the nuclear localization of NGB. The E2 effect is already evident just 1 h after hormone stimulation and it is persistent and even more evident 24 h after stimulation (Figure 2d).

Localization of NGB in SK-N-BE cells and in flag-NGB transfected HeLa cells. (a) Fluorescence analysis of SK-N-BE cells. Cells were fixed and permeabilized, and stained with anti-NGB antibody (green, right panel) and costained with DAPI (left panel) (original magnification × 40). (b) Fluorescence analysis of Hela cells non transfected (NT, left panel) or transfected with pcDNA-flag-NGB plasmid (flag-NGB, right panel). Cells were fixed and permeabilized, then were stained with anti-flag M2 antibody (red) (original magnification × 40). Representative images from five different experiments are shown. (c) Western blot analysis (left panel) of NGB expression in nuclear, cytosolic, and mitochondrial fractions of SK-N-BE cells. The purity of fractions was assessed with PARP, cytochrome c oxidase-4 (Cox-4), and PP2A with respect to nucleus, mitochondria, and cytosol, respectively. The figure represents a typical Western blot of five independent experiments. (c’) Densitometric analysis of NGB distribution rate in the three fractions was calculated respect to whole protein amount. Data are±S.D. of five different experiments. (d) Confocal miscroscopy showing NGB distribution in SK-N-BE cells treated either with vehicle, or E2 (1 nM) for 1 h, or E2 (1 nM) for 24 h (original magnification × 63). Representative images from three different experiments are shown
The E2-induced (i.e., 1 nM for 24 h) reduction of NGB level into SK-N-BE cell nuclei is paralleled by the NGB increase in the mitochondrial and cytosolic fractions as assessed by the subcellular fractionation (Figures 3a and a’). This effect is specific for E2, in that only a significant increase of the NGB level in the mitochondrial fraction is reported after cell stimulation for 24 h with 50 μM H2O2 (Figures 3b and b’).

Effect of E2 and H2O2 on NGB intracellular localization in SK-N-BE cells. Western blot analysis of NGB expression in nuclear, cytosolic, and mitochondrial fractions of SK-N-BE cells treated either with vehicle or E2 (1 nM for 24 h) (a) or H2O2 (50 μM for 24 h) (b). The purity of fractions was assessed with PARP, Cox-4, and PP2A with respect to nucleus, mitochondria, and cytosol, respectively. The amount of protein was normalized by comparison with subcellular marker levels. The panels represent typical Western blots of five independent experiments. (a’ and b’), Densitometric analyses of NGB distribution in the three fractions was calculated respect to subcellular marker levels. Data are means±S.D. of five independent experiments carried out in duplicate. P<0.001 was calculated with ANOVA followed by Tukey–Kramer post-test: *significant versus vehicle (mitochondria), °versus vehicle (nuclei), §versus vehicle (cytosol)
E2 promotes NGB–cytochrome c association
This result prompted us to evaluate whether the association between NGB and cytochrome c occurs. In SK-N-BE mitochondrial fraction, a slight association between NGB and cytochrome c occurs (Figures 4a and a’). Upon E2 administration, NGB–cytocrome c association is increased in the mitochondria, whereas no association between proteins takes place in other subcellular compartments (data not shown). However, H2O2 insult (i.e., 50 μM for 24 h) increases mitochondrial NGB and cytochrome c association (Figures 4a and a’), which is further increased after the E2 treatment (i.e., 1 nM for 24 h) (Figures 4a and a’). Contemporarily upon H2O2 injury, the level of cytosolic cytochrome c increases with the parallel decrease into mitochondrial fraction (Figures 4b and b’). The E2 treatment reduces the amount of cytosolic cytochrome c in non-injured cells, and, most importantly, E2, administrated before H2O2, decreases the amount of cytochrome c present in the cytosolic fraction with the parallel increase of the cytochrome c amount in the mitochondrial fraction (Figures 4b and b’).

Effect of E2 and H2O2 on NGB association with cytochrome c (Cyt-c) in the mitochondrial fraction. SK-N-BE cells were grown for 24 h in the presence of either vehicle or E2 (1 nM). After 24 h, cells were stimulated with 50 μM H2O2 (24 h of stimulation), and then were fractionated and only the mitochondrial fraction was subjected to Cyt-c immunoprecipitation (IP), followed by Western blot (WB) with anti-NGB or anti-Cyt-c antibodies. (a) Representative Western blot of five independent experiments. (a’) Densitometric analysis of NGB association with Cyt-c. Data are means±S.D. of five independent experiments carried out in duplicate. P<0.001 was calculated with ANOVA followed by Tukey–Kramer post-test: *significant versus vehicle -H2 O2, °versus vehicle +H2 O2. (b) Analysis of NGB and Cyt-c levels in the mitochondria (left panel) and cytosol (right panel). Cox-4 and PP2A were used to normalize the total amount of protein in mitochondrial and cytosolic fraction, respectively. Cells were grown for 24 h in the presence of either vehicle or E2 (1 nM). After 24 h, cells were stimulated with 50 μM H2O2 (24 h of stimulation) and then fractionated. (b’) Densitometric analysis of Cyt-c levels in mitochondrial and cytosolic fraction. Data are mean±S.D. of five independent experiments. P<0.001 was calculated with ANOVA followed by Tukey–Kramer post-test: *significant versus vehicle−H2O2, °versus vehicle+H2O2
The E2-mediated increased association of NGB with cytochrome c in the mitochondrial fraction requires ERs. In fact, the cell pretreatment with ICI 182,780 (i.e., 1 μM 30 min before E2 administration), the pure antagonist of both ERs, prevents the E2 effect on mitochondrial NGB–cytochrome c association in the presence of H2O2 (Figure 5a). However, as in other cell models,22, 23 both ER subtypes are expressed in the nuclei, mitochondria, and cytosol of SK-N-BE cells in a different ratio (Figures 5b and b’). Nonetheless, the ERβ inhibitor THC ((R,R)-5,11-diethyl-5,6,11,12-tetrahydro-2,8-chrysenediol) pretreatment (i.e., 1 μM 30 min before E2 administration) impairs the E2 promotion of NGB–cytochrome c association, indicating that ERβ mediates the effect of E2 in inducing NGB–cytocrome c interactions (Figure 5c).

Estrogen receptors involvement in E2 effect on NGB association with cytochrome c (Cyt-c) in the mitochondrial fraction. (a) Effect of E2 (1 nM) and of ER antagonist ICI 182 780 (ICI, 1 μM) on NGB association with cytochrome c (Cyt-c) under H2O2 stimulus in the mitochondria. SK-N-BE cells were grown for 24 h in the presence of vehicle or E2 and/or ICI 182 780 (ICI, 1 μM). After 24 h, cells were stimulated with 50 μM H2O2 (24 h of stimulation) and then fractionated. Mitochondrial fraction was subjected to Cyt-c immunoprecipitation (IP), followed by Western blot (WB) with anti-NGB or anti-Cyt-c antibodies. Representative Western blots of five independent experiments. (b) Localization of ERs in SK-N-BE cell fractions. Western blot analysis of ERα and ERβ expression in nuclear, mitochondrial and cytosolic fractions of SK-N-BE cells. The purity of the nuclear, mitochondrial, and cytosolic fractions was assessed with PARP, cytochrome c oxidase-4 (Cox-4), and PP2A, respectively. Representative Western blots from five independent experiments are shown. (b’) ERα/ERβ ratio in subcellular fractions was calculated on densitometric analysis of Western blots reported in b. Data are means±S.D. of five independent experiments carried out in duplicate. (c) Effect of E2 (1 nM) and of the ERβ antagonist THC (1 μM) on NGB association with cytochrome c (Cyt-c) under H2O2 stimulus in the mitochondria. SK-N-BE cells were grown for 24 h in the presence of vehicle or E2 and/or THC. After 24 h, cells were stimulated with H2O2 50 μM (24 h of stimulation), and then were fractionated. Mitochondrial fraction was subjected to Cyt-c immunoprecipitation (IP), followed by Western blot (WB) with anti-NGB or anti-Cyt-c antibodies. Representative Western blots of five independent experiments
Discussion
In mitochondria, cytochrome c is located between the inner and the outer membrane having a pivotal role in transferring electrons from ubiquinone–cytochrome c reductase (Complex III) to COX-4 (Complex IV). Moreover, cytochrome c scavenges RNS and ROS, thus preventing cell oxidative stress and displays a central apoptotic role.19, 24 At least 15% of mitochondrial cytochrome c is bound to cardiolipin, an unusual lipid largely confined to the inner mitochondrial membrane. Remarkably, cardiolipin binding induces allosteric modifications in cytochrome c tertiary structure switching cytochrome c function(s) from mitochondrial respiration to apoptosis.19 Once in the cytosol and in the presence of ATP (and more efficiently in the presence of deoxyATP), ferric cytochrome c mediates the allosteric activation and hepta-oligomerization of the adapter molecule apoptosis-protease activating factor-1 (Apaf-1), generating the complex known as apoptosome.25 Each apoptosome can recruit seven dimers of caspase-9 favoring proteinase activation. These events allow the catalytic clevage of caspase-3 and other caspases, which eventually mediate the biochemical and morphological features of apoptosis (Ascenzi et al.19 and literature therein).
Bioinformatical sequence evaluations predicted NGB to be cytoplasmic.26 Moreover, the transfection of N- and C-terminal gene fusion constructs between green fluorescent protein and NGB into eukaryotic cell lines also did not produce evidence for any kind of attachment or import of this globin (in)to distinct intracellular structures.26 Thus, the cytoplasmic localization of NGB was undisputed and when firstly hypothesized, the interaction between cytochrome c and NGB was considered possible only when the ferric cytochrome c was released into the cytosol.27 Present results, obtained by immunofluorescent staining of endogenous and transfected NGB and by biochemical fractionaction, clearly demonstrate that NGB is expressed in the nucleus and mitochondria, and is scattered in the cytosol of SK-N-BE cell lines, confirming the widespread subcellular distribution of this globin. Similar NGB subcellular localization28, 29 and the interaction of NGB with intra-mitochondrial proteins has been recently reported in other cell lines.30, 31
These results lead us to hypothesize that NGB could directly interact with cytochrome c into mitochondria to prevent stress signals-induced cytochrome c release into the cytosol. To verify this hypothesis, we utilized two different stimuli which activate (i.e., H2O2) or inhibit (i.e., E2) neuronal apoptosis. Moreover, we recently20 demonstrated that the preventive effect of E2 against neuronal apoptosis requires NGB upregulation.20 Upon physiological E2 stimulation (i.e., 1 nM) or H2O2 challenge (50 μM for 24 h), NGB is reallocated in the mitochondrial fraction where its association with cytochrome c increases. However, during H2O2 insult the amount of mitochondrial NGB (i.e., about 2.0±0.2 fold) seems not to be sufficient to maintain cytochrome c into mitochondria, as a consequence a high level of this protein is still present into cytosol and the protection against H2O2-induced apoptosis could not occur. On the other hand, E2 stimulation in the absence of other stimuli leads to a greater NGB–cytochrome c association into mitochondria (i.e., about 3.3±0.15-fold), thereby reducing the amount of cytosolic cytochrome c.
From the functional viewpoint, the role of NGB appears to be important in the E2 regulation of neuronal function. A number of reports point out a relation between E2 and oxidative stress in several tissues.32, 33 However, in normal conditions, the E2-induced ROS increase does not trigger an apoptotic effect due to the upregulation of antioxidant mechanisms (e.g., manganese superoxide dismutase).32, 34 Regardless of compelling evidences, the mechanism by which estrogen protects cells from oxidative stress is not fully understood. Our result indicates for the first time that E2 induces mitochondrial NGB increase, thereby preventing oxidized cytochrome c release and the activation of apoptotic cascade. Thus, under physiological conditions, the E2-induced NGB–cytochrome c interaction can be considered as an E2 self-regulatory mechanism. Remarkably, the E2 effect on the enhancement of NGB–cytochrome c association increases under oxidative stress conditions (i.e., E2 1 nM 24 h before H2O2 administration) rather than under basal conditions. In agreement with these results, as E2 promotes NGB–cytochrome c association in the mitochondria, especially in the presence of an oxidative insult, the total amount of cytosolic cytochrome c appears to be reduced in SK-N-BE pretreated with E2 before H2O2 administration. As a whole this mechanism strengthens the idea that NGB could represent the intracellular mediator of the E2 anti-apoptotic effect in neuronal-derived cell lines, acting directly in mitochondria that represent the core of intrinsic apoptotic pathway start-up. The mechanisms underlying the NGB-based inhibition of the intrinsic apoptotic cascade can be related to the observation that ferrous NGB rapidly complexes with ferric cytochrome c released from cardiolipin thereby reducing rapidly it to the ferrous form, which in turn is no longer pro-apoptotic.17, 35, 36
E2 actions are mediated by its binding to two ER subtypes, which mediate different and somewhat opposite E2 functions. Recently, it has been demonstrated in mammary cancer cells and in the brain that the E2 effect on oxidative stress depends on the amount of ERs and more importantly on the ERα/ERβ ratio.32, 37 Our results indicate that in neuroblastoma cell line ERα/ERβ ratio differs in dependence on cell compartment being about 1.00±0.04 in the mitochondria. However, the E2 effect on the promotion of NGB–cytochrome c interaction under oxidative conditions requires ERβ activity, being impaired by both the pretreatment with the ERα and ERβ pure antagonist ICI, and in particular by the ERβ-specific antagonist, THC. Although still debated, most of the recent studies on E2 neuroprotective mechanisms are emphasizing the pivotal role of ERs localized in the mitochondria.23, 32, 38, 39 Present data further highlight the role of mitochondrial ERβ signaling in the prevention of apoptosis.
In conclusion, our findings suggest that the principal role played by NGB in the brain could be the reduction of neuronal death by resetting the trigger level of apoptosis leading to the onset of physiological response to stress. In this scenario, the E2 function is to accelerate the protective cell response to external insults21 by inducing a rapid and strong increase and reallocation of endogenous NGB, which in turn is pivotal to impair the activation of the intrinsic apoptotic pathway by direct interaction with cytochrome c in the mitochondria. However, further in vivo studies that will take into account the important role of E2 in NGB induction are required to better understand how this new mechanism of NGB endogenous modulation can be utilized to enhance the brain protective mechanisms.
Materials and Methods
Reagents
E2, Pen-Strep solution, H2O2, RPMI-1640 media without phenol red, Dulbecco’s modified Eagle medium (DMEM) without phenol red, charcoal-stripped fetal calf serum, protease inhibitor cocktail, bovine serum albumin fraction V (BSA), and mouse monoclonal anti-FlagM2 antibody were purchased from Sigma-Aldrich (St. Louis, MO, USA). Optimem was purchased from Gibco-BRL (Gaithersburg, MD, USA). The E2 antagonist fulvestrant (ICI 182,780, ICI), and the ERβ-selective antagonist THC were obtained from Tocris (Ballwin, MO, USA). Bradford protein assay was obtained from Bio-Rad Laboratories (Hercules, CA, USA). Human recombinant ERα and ERβ were obtained from Pan-Vera (Madison, WI, USA). Anti-PARP and anti-PP2A were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Monoclonal anti-human NGB (13C8) antibody was purchased from Abcam (Cambridge, UK). Anti-β-tubulin antibody was purchased from MP Biomedical (Solon, OH, USA). Polyclonal anti-cytochrome c and monoclonal anti-cytochrome c oxidase (COX-4) antibodies were purchased from Clontech Laboratories (Mountain View, CA, USA). Chemiluminescence reagent for Western blot ECL was obtained from GE Healthcare (Little Chalfont, UK). All the other products were from Sigma-Aldrich. Analytical or reagent grade products were used without further purification.
Cells
The human SK-N-BE neuroblastoma cell line was routinely grown in air containing 5% CO2 in modified, phenol red-free, RPMI-1640 medium containing 10% (v/v) charcoal-stripped fetal calf serum, L-glutamine (2.0 mM), Pen-Strep solution (penicillin 100 U/ml and streptomycin (100 mg/ml). Cells were passaged every 2 days. Cells were grown to ∼70% confluence in six-well plates before stimulation. Human cervix epithelioid carcinoma cells (HeLa) were routinely grown in air containing 5% CO2 in modified, phenol red-free, DMEM medium, containing 10% (v/v) charcoal-stripped fetal calf serum, L-glutamine (2 mM), gentamicin (0.1 mg/ml), and penicillin (100 U/ml). Cells were passaged every 2 days and media changed every 2 days.
Cell stimulation
Cells were simultaneously treated with vehicle (ethanol/PBS 1 : 10, v/v) or E2 (1 nM). H2O2 (50 μM) was administrated 24 h after E2 or vehicle treatment. When indicated, the anti-estrogen ICI (1 μM) and the ERβ inhibitor THC (1 μM) were added 30 min before E2 administration.
Plasmid flag-NGB and transfection
The pcDNA-flag-NGB (flag-NGB) was obtained by subcloning the NGB ORF from the NGBN1-pEGFP plasmid40 into the pcDNA-flag 3.1C. HeLa cells were grown to ∼70% confluence and then transfected with pcDNA-flag-NGB plasmid using lipofectamine reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Six hours after transfection, the medium was changed and 24 h after the cells were processed for confocal microscopy analysis.
DAPI staining
In some experiments, SK-N-BE cells were stimulated with E2 1 nM and/or 50 μM H2O2 for 24 h. Cells were processed in chamber slides and rinsed with PBS, pH 7.4, followed by fixation in 100% (v/v) cold methanol for 15 min. The slides were cover-slipped using Prolong Gold anti-fade reagent with 1 mg/ml DAPI (Invitrogen). Slides were viewed on an Olympus BX51 fluorescence microscope. Images were captured ( × 40 magnification) with Leica DFC 420 camera (Leica Microsystems, Wetzlar, Germany).
Confocal microscopy analysis
SK-N-BE cells and flag-NGB-transfected HeLa cells were stained with anti-NGB (1 : 50) and anti-FlagM2 (1 : 10 000) antibodies, respectively. Cells were processed in chamber slides and rinsed with PBS, pH 7.4, followed by fixation in formaldehyde 4% (v/v) for 1 h, and permeabilization with cold acetone 95% (v/v) for 3 min. Cells were rinsed in PBS and saturated with BSA 2% (w/v) for 1 h and then incubated with primary antibody at 4 °C o/n (anti-NGB) or 1 h at RT (anti-FlagM2). After that cells were rinsed three times in PBS for 5 min and incubated with Alexa Fluor 488 and 578 donkey anti-mouse secondary antibodies (Invitrogen) (1 : 400). The slides were cover-slipped using Prolong Gold anti-fade reagent. Confocal analysis ( × 63 magnification) was performed using LCS (Leica Microsystems).
Transfection of short interfering RNA
SK-N-BE cells, reaching 40–60% confluence, were transfected in serum-free condition with Stealth RNAi NGB-silencing RNA (NGB-silencing RNA (SiNGB); Invitrogen) according to the manufacturer’s instructions, using Oligofectamin (Invitrogen) as the transfection reagent. The sequence used for NGB oligonucleotides was 5′-CGUGAUUGAUGCUGCAGUGACCAAU-3′. The mismatch sequences used as a control were 5′-UGUGAUUUAUGGUGCAGUAACCAAC-3′. Briefly, oligofectamin and oligonucleotides (400 pM) were mixed with Optimem. The mixture was incubated for 20 min at room temperature, diluted with Optimem, and added to the cell medium for 4 h at 37 °C. The medium was added to cells to reach the growing conditions (i.e., 10% (v/v) serum). To evaluate the effective silencing of NGB, total proteins from cells transfected with MOCK (control) and with NGB were extracted 48 h after transfection, and NGB expression was tested by Western blot analysis using anti-NGB antibody.
Cell fractionation
The SK-N-BE cell fractionation was performed using the ApoAlert Cell Fractionation kit (Clontech) according to the manufacturer’s instructions. After stimulation, cells were washed in ice-cold PBS, collected with trypsin (1%, v/v), resuspended with complete medium, and centrifuged at 600 × g for 5 min. The pellet was resuspended in Fractionation Buffer Mix containing DTT 1 mM and homogenized in a Dounce tissue grinder. The homogenate was centrifuged at 700 × g for 10 min. The pellet was resuspended in the sample buffer containing 0.125 M Tris, pH 6.8, and 10% (w/v) SDS (nuclear fraction); the supernatant was centrifuged again at 10 000 × g for 25 min. The supernatant was collected (cytosolic fraction) and the pellet resuspended in the Fractionation Buffer Mix (mitochondrial fraction). The protein concentration of each fraction was determined using the Bradford protein assay. The lysate of each fraction was then processed for Western blot or used for immunoprecipitation assay.
True-blot coimmunoprecipitation
After stimulation, SK-N-BE cells were washed in ice-cold PBS, collected with trypsin (1%, v/v), and lysed in 50 μl lysis buffer 10 mM Tris, pH 7.5, 1 mM EDTA, 0.5 mM EGTA, 10 mM NaCl, 1% (v/v) Triton X-100, and 1% (w/v) sodium cholate, containing the protease inhibitor cocktail. Cell lysates were clarified by centrifugation and immunoprecipitated with TrueBlot (eBioscience, San Diego, CA, USA), which preferentially detects the native disulfide form of mouse or rabbit IgG, reducing interference by the ∼55 kDa heavy and ∼23 kDa light chains of the immunoprecipitating antibody. Briefly, after stimulation equal amounts of soluble cell extracts were incubated with 10 μg of anti-cytochrome c antibody. The lysates and antibodies were incubated at 4 °C for 1 h, then 20 μl of anti-Mouse IgG Beads (eBioscience) were added and samples incubated for 1 h on a rocking platform at 4 °C. Samples were centrifuged at 10 000 × g for 10 min, the supernatant was removed completely and beads (pelleted) were washed three times with 100 μl of lysis buffer. The SDS-reducing sample buffer (20 μl, containing 50 mM DTT) was added and samples were boiled at 100 °C for 5 min. Proteins (pelleted and supernatant) were resolved using 15% SDS-PAGE at 100 V for 1 h and then electrophoretically transferred to nitrocellulose for 45 min at 100 V and 4 °C. The nitrocellulose was treated with 5% (w/v) non-fat dry milk in 150 mM NaCl, 50 mM Tris HCl (pH 8.0), 0.1% (w/v) Tween-20, and then probed at 4 °C overnight with anti-NGB antibody (1 : 1000). The antibody reaction was visualized with the ECL chemiluminescence reagent for Western blot. The nitrocellulose was stripped with the Restore Western Blot Stripping Buffer for 10 min at room temperature and then probed with anti-cytochrome c antibody (final dilution 1 : 1000) to normalize the immunoprecipitate.
Western blot assays
After treatments, cells were lysed and solubilized in 0.125 M Tris, pH 6.8, containing 10% (w/v) SDS and protease inhibitor cocktail, and finally boiled for 2 min. Total proteins were quantified using the Bradford protein assay. Solubilized proteins (20 μg) were resolved by 7 or 15% SDS-PAGE at 100 V for 1 h at 25 °C and then electrophoretically transferred to nitrocellulose for 45 min at 100 V and 4 °C. The nitrocellulose was treated with 3% (w/v) BSA in 138 mM NaCl, 25 mM Tris, pH 8.0, and 0.1% (w/v) Tween-20 at 25 °C for 1 h and then probed overnight at 4 °C with either anti-NGB (final dilution 1 : 1000), anti-ERα MC-20 (final dilution 1 : 500), anti-ERβ H-150 (final dilution 1 : 3000), anti-PARP (final dilution 1 : 500), anti-COX-4 (final dilution 1 : 1000) or anti-PP2A (final dilution 1 : 1000). The nitrocellulose was stripped by the Restore Western Blot Stripping Buffer (Pierce Chemical, Rockford, IL, USA) for 10 min at room temperature and then probed with anti-β-tubulin antibody (final dilution 1 : 1000) to normalize total lysate. To evidence ERα and ERβ levels, electrophoresis was performed in the presence of 5 ng of recombinant ERα and ERβ; the antibody reaction was visualized with ECL chemiluminescence (GE Healthcare, Little Chalfont, UK). Densitometric analyses were performed by ImageJ software for Microsoft Windows. The densitometry quantification of protein was normalized to tubulin.
Statistical analysis
The statistical analysis was performed using ANOVA followed by Tukey–Kramer post-test with the GraphPad InStat3 software system (GraphPad software, La Jolla, CA, USA) for Windows. In all cases, P<0.05 was considered significant.
Abbreviations
- Apaf-1:
-
adapter molecule apoptosis-protease activating factor-1
- BSA:
-
bovine serum albumin
- COX-4:
-
cytochrome c oxidase-4
- DMEM:
-
Dulbecco’s modified Eagle medium
- E2:
-
17β-estradiol
- ERα:
-
estrogen receptor-α
- ERβ:
-
estrogen receptor-β
- ICI:
-
ICI 182,780 (fulvestrant)
- NGB:
-
neuroglobin
- PARP:
-
poly ADP-ribose polymerase
- PP2A:
-
phosphatase-2A
- RNS:
-
reactive nitrogen species
- ROS:
-
reactive oxygen species
- SiNGB:
-
neuroglobin-silencing RNA
- THC:
-
(R,R)-5,11-diethyl-5,6,11,12-tetrahydro-2,8-chrysenediol
References
Burmester T, Hankeln T . What is the function of neuroglobin? J Exp Biol 2009; 212: 1423–1428.
Burmester T, Weich B, Reinhardt S, Hankeln T . A vertebrate globin expressed in the brain. Nature 2000; 407: 520–523.
Emara M, Turner AR, Allalunis-Turner J . Hypoxic regulation of cytoglobin and neuroglobin expression in human normal and tumor tissues. Cancer Cell Int 2010; 10: 33.
Dewilde S, Kiger L, Burmester T, Hankeln T, Baudin-Creuza V, Aerts T et al. Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. J Biol Chem 2001; 276: 38949–38955.
Trent JT, Watts RA, Hargrove MS . Human neuroglobin, a hexacoordinate hemoglobin that reversibly binds oxygen. J Biol Chem 2001; 276: 30106–30110.
Fordel E, Thijs L, Martinet W, Schrijvers D, Moens L, Dewilde S . Anoxia or oxygen and glucose deprivation in SH-SY5Y cells: a step closer to the unraveling of neuroglobin and cytoglobin functions. Gene 2007; 398: 114–122.
Brunori M, Giuffre A, Nienhaus K, Nienhaus GU, Scandurra FM, B Vallone . Neuroglobin nitric oxide, and oxygen: functional pathways and conformational changes. Proc Natl Acad Sci USA 2005; 102: 8483–8488.
Jayaraman T, Tejero J, Chen BB, Blood AB, Frizzell S, Shapiro C et al. 14-3-3 binding and phosphorylation of neuroglobin during hypoxia modulate six-to-five heme pocket coordination and rate of nitrite reduction to nitric oxide. J Biol Chem 2011; 286: 42679–42689.
Greenberg DA, Jin K, Khan AA . Neuroglobin: an endogenous neuroprotectant. Curr Opin Pharmacol 2008; 8: 20–24.
Shao G, Gong KR, Li J, Xu XJ, Gao CY, Zeng XZ et al. Antihypoxic effects of neuroglobin in hypoxia-preconditioned mice and SH-SY5Y cells. Neurosignals 2009; 17: 196–202.
Khan AA, Wang Y, Sun Y, Mao XO, Xie L, Miles E et al. Neuroglobin-overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc Natl Acad Sci USA 2006; 103: 17944–17948.
Sun Y, Jin K, Mao XO, Zhu Y, Greenberg DA . Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc Natl Acad Sci USA 2001; 98: 15306–15311.
Sun Y, Jin K, Peel A, Mao XO, Xie L, Greenberg DA . Neuroglobin protects the brain from experimental stroke in vivo. Proc Natl Acad Sci USA 2003; 100: 3497–3500.
Yu Z, Fan X, Lo EH, Wang X . Neuroprotective roles and mechanisms of neuroglobin. Neurol Res 2009; 31: 122–127.
Brittain T, Skommer J, Henty K, Birch N, Raychaudhuri S . A role for human neuroglobin in apoptosis. IUBMB Life 2010; 62: 878–885.
Brittain T, Skommer J, Raychaudhuri S, Birch N . An antiapoptotic neuroprotective role for neuroglobin. Int J Mol Sci 2010; 11: 2306–2321.
Fago A, Mathews AJ, Brittain T . A role for neuroglobin: resetting the trigger level for apoptosis in neuronal and retinal cells. IUBMB Life 2008; 60: 398–401.
Raychaudhuri S, Skommer J, Henty K, Birch N, Brittain T . Neuroglobin protects nerve cells from apoptosis by inhibiting the intrinsic pathway of cell death. Apoptosis 2010; 15: 401–411.
Ascenzi P, Polticelli F, Marino M, Santucci R, Coletta M . Cardiolipin drives cytochrome c proapoptotic and antiapoptotic actions. IUBMB Life 2011; 63: 160–165.
De Marinis E, Ascenzi P, Pellegrini M, Galluzzo P, Bulzomi P, Arevalo MA et al. 17Beta-estradiol--a new modulator of neuroglobin levels in neurons: role in neuroprotection against HO-induced toxicity. Neurosignals 2010; 18: 223–235.
De Marinis E, Marino M, P Ascenzi . Neuroglobin estrogens, and neuroprotection. IUBMB Life 2011; 63: 140–145.
Arnold S, Beyer C . Neuroprotection by estrogen in the brain: the mitochondrial compartment as presumed therapeutic target. J Neurochem 2009; 110: 1–11.
Simpkins JW, Yang SH, Sarkar SN, Pearce V . Estrogen actions on mitochondria-physiological and pathological implications. Mol Cell Endocrinol 2008; 290: 51–59.
Ow YP, Green DR, Hao Z, Mak TW . Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol 2008; 9: 532–542.
Purring-Koch C, McLendon G . Cytochrome c binding to Apaf-1: the effects of dATP and ionic strength. Proc Natl Acad Sci USA 2000; 97: 11928–11931.
Hankeln T, Wystub S, Laufs T, Schmidt M, Gerlach F, Saaler-Reinhardt S et al. The cellular and subcellular localization of neuroglobin and cytoglobin -- a clue to their function? IUBMB Life 2004; 56: 671–679.
Brittain T, Skommer J . Does a redox cycle provide a mechanism for setting the capacity of neuroglobin to protect cells from apoptosis? IUBMB Life 2012; 64: 419–422.
Hundahl CA, Allen GC, Hannibal J, Kjaer K, Rehfeld JF, Dewilde S et al. Anatomical characterization of cytoglobin and neuroglobin mRNA and protein expression in the mouse brain. Brain Res 2010; 1331: 58–73.
Hundahl CA, Luuk H, Ilmjarv S, Falktoft B, Raida Z, Vikesaa J et al. Neuroglobin-deficiency exacerbates Hif1A and c-FOS response, but does not affect neuronal survival during severe hypoxia in vivo. PLoS One 2011; 6: e28160.
Haines B, Demaria M, Mao X, Xie L, Campisi J, Jin K et al. Hypoxia-inducible factor-1 and neuroglobin expression. Neurosci Lett 2012; 51: 137–1340.
Yu Z, Liu N, Wang Y, Li X, Wang X . Identification of neuroglobin-interacting proteins using yeast two-hybrid screening. Neuroscience 2012; 200: 99–105.
Arnold S, Victor MB, Beyer C . Estrogen and the regulation of mitochondrial structure and function in the brain. J Steroid Biochem Mol Biol 2012; 131: 2–9.
Okoh V, Deoraj A, Roy D . Estrogen-induced reactive oxygen species-mediated signalings contribute to breast cancer. Biochim Biophys Acta 2011; 1815: 115–133.
Borras C, Gambini J, Lopez-Grueso R, Pallardo FV, Vina J . Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim Biophys Acta 2010; 1802: 205–211.
Bonding SH, Henty K, Dingley AJ, Brittain T . The binding of cytochrome c to neuroglobin: a docking and surface plasmon resonance study. Int J Biol Macromol 2008; 43: 295–299.
Fago A, Mathews AJ, Dewilde S, Moens L, Brittain T . The reactions of neuroglobin with CO: evidence for two forms of the ferrous protein. J Inorg Biochem 2006; 100: 1339–1343.
Nadal-Serrano M, Sastre-Serra J, Pons DG, Miro AM, Oliver J, Roca P . The ERalpha/ERbeta ratio determines oxidative stress in breast cancer cell lines in response to 17beta-estradiol. J Cell Biochem 2012; 113: 3178–3185.
Flynn JM, Dimitrijevich SD, Younes M, Skliris G, Murphy LC, Cammarata PR . Role of wild-type estrogen receptor-beta in mitochondrial cytoprotection of cultured normal male and female human lens epithelial cells. Am J Physiol Endocrinol Metab 2008; 295: E637–E647.
Yang SH, Sarkar SN, Liu R, Perez EJ, Wang X, Wen Y et al. Estrogen receptor beta as a mitochondrial vulnerability factor. J Biol Chem 2009; 284: 9540–9548.
Fordel E, Thijs L, Martinet W, Lenjou M, Laufs T, Van Bockstaele D et al. Neuroglobin and cytoglobin overexpression protects human SH-SY5Y neuroblastoma cells against oxidative stress-induced cell death. Neurosci Lett 2006; 410: 146–151.
Acknowledgements
The generous gift of NGBN1-pEGFP plasmid from Professor Sylvia Dewilde (University of Antwerp, Belgium) is gratefully acknowledged. This work was partially supported by grants from the Italian Ministry of Education, University, and Research (PRIN 2010-2011 20109MXHMR_01 to P.A.) and from University Roma Tre to P.A. and M.M.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Raschellá
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
De Marinis, E., Fiocchetti, M., Acconcia, F. et al. Neuroglobin upregulation induced by 17β-estradiol sequesters cytocrome c in the mitochondria preventing H2O2-induced apoptosis of neuroblastoma cells. Cell Death Dis 4, e508 (2013). https://doi.org/10.1038/cddis.2013.30
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.30
Keywords
This article is cited by
-
Identification of novel prognostic risk signature of breast cancer based on ferroptosis-related genes
Scientific Reports (2022)
-
Estrogen-induced downregulation of TASK-1 expression through estrogen receptor β in N2A cells
Molecular Biology Reports (2022)
-
Auditory function and dysfunction: estrogen makes a difference
Cellular and Molecular Life Sciences (2020)
-
Mn-TAT PTD-Ngb attenuates oxidative injury by an enhanced ROS scavenging ability and the regulation of redox signaling pathway
Scientific Reports (2019)
-
Mitochondrial Neuroglobin Is Necessary for Protection Induced by Conditioned Medium from Human Adipose-Derived Mesenchymal Stem Cells in Astrocytic Cells Subjected to Scratch and Metabolic Injury
Molecular Neurobiology (2019)
