
Abstract
Abexinostat is a pan histone deacetylase inhibitor (HDACi) that demonstrates efficacy in malignancy treatment. Like other HDACi, this drug induces a profound thrombocytopenia whose mechanism is only partially understood. We have analyzed its effect at doses reached in patient plasma on in vitro megakaryopoiesis derived from human CD34+ cells. When added at day 0 in culture, abexinostat inhibited CFU-MK growth, megakaryocyte (MK) proliferation and differentiation. These effects required only a short incubation period. Decreased proliferation was due to induction of apoptosis and was not related to a defect in TPO/MPL/JAK2/STAT signaling. When added later (day 8), the compound induced a dose-dependent decrease (up to 10-fold) in proplatelet (PPT) formation. Gene profiling from MK revealed a silencing in the expression of DNA repair genes with a marked RAD51 decrease at protein level. DNA double-strand breaks were increased as attested by elevated γH2AX phosphorylation level. Moreover, ATM was phosphorylated leading to p53 stabilization and increased BAX and p21 expression. The use of a p53 shRNA rescued apoptosis, and only partially the defect in PPT formation. These results suggest that HDACi induces a thrombocytopenia by a p53-dependent mechanism along MK differentiation and a p53-dependent and -independent mechanism for PPT formation.
Similar content being viewed by others


Main
Acetylation/deacetylation of lysine residues is one of the post-translational modifications that modulate the function of proteins as histones and non-histone proteins such as the transcription factor GATA1, the chaperone HSP90, tubulin and p53.1, 2, 3 These modifications are regulated by histone acetyl transferases and histone deacetylases (HDAC).4, 5, 6, 7 The four classes of HDAC (I, IIa, IIb and IV)8 display common and specific functions and localizations, for example, class I HDACs are nuclear enzymes, whereas class II and IV HDACs are both in the nucleus and in the cytoplasm. Their main function is the co-repression of gene transcription by removing acetyl groups from lysine residues located in the NH2 terminal tail of histone 3 and 4. HDACs by this mechanism have a central role in the regulation of the DNA repair cell machinery.9, 10
The compounds known as HDAC inhibitors (HDACi) that induce a shift in the balance between acetylation and deacetylation of proteins represent a new class of anticancer agents.6 This has been initially demonstrated in acute promyelocytic leukemia cells, in which the recruitment of a HDAC by the PLZF-RAR fusion protein represses RAR target genes and HDACi restore the ability of retinoic acid to induce leukemia cell differentiation.11, 12 In this model, HDACi also promote the transcriptional activity of p53 through increasing its acetylation.2 These drugs have now been tested in a broad range of malignancies including T-cell lymphomas,13 myeloproliferative neoplasms (MPN), gliomas and colon cancers.14, 15 They can induce cell cycle arrest through p21 induction, and apoptosis through radical oxygen species overproduction.16
Although some HDACi are specific of a class of HDAC, for example, those derived from the bicyclic depsipeptide, such as Romidepsin, preferentially target class I HDAC,17 most of them, such as panobinostat and abexinostat (S78454, PCI-24781), derive from a hydroxamic acid structure and are pan HDAC inhibitors. Thrombocytopenia, which is a constantly observed side effect of these drugs, is limiting dose escalade and drug combinations.18, 19, 20, 21, 22 The molecular mechanisms of these thrombocytopenia remain a matter of speculation. Conditional knock-out of hdac1 and hdac2 genes in mice induces a thrombocytopenia by inducing megakaryocyte (MK) apoptosis,23 and both Panobinostat and Romidepsin induce a thrombocytopenia in mice. Noteworthy, this drug effect, probably involving actomyosin cytoskeleton, was rescued by thrombopoietin (TPO) mimetics.24 It has also been suggested that a very low dose of Panobinostat could inhibit proplatelet (PPT) formation through increasing the level of acetylated tubulin.25 This drug could also downregulates GATA1 expression at both transcriptional and post-transcriptional levels.26
By investigating the effects of pharmacologically relevant doses of the pan-HDACi abexinostat, on human megakaryopoiesis derived in vitro from CD34+ cells, we demonstrate here that the compound has two main effects. It inhibits MK differentiation by inducing progenitor and precursor apoptosis through silencing of several DNA repair genes, including RAD51, leading to the accumulation of DNA double-strand breaks (DSBs) and the induction of p53. In addition, a defect in PPT formation was found, which was mainly p53-independent suggesting that Abexinostat directly targets some effectors implicated in PPT formation.
Results
The pan-HDAC inhibitor Abexinostat inhibits hematopoietic colony formation
We explored the effects of drug doses ranging from 10 to 100 nM on the ex vivo growth of human hematopoietic progenitor cells because it has been shown that the peak plasma concentration of abexinostat (called also PCI-24781, S78454) ranges from 295 to 185 nM at 4 h post dose27 and these doses in ex vivo experiments were found to be toxic. When abexinostat was added to the CD34+ cells at the onset of the methylcellulose cultures, no significant effect was seen at 10 nM compared with control cultures. However, the total number of colonies, including mixed colonies derived from CFU-GEMM progenitors, was nearly 50% reduced in the presence of 50 nM abexinostat and more than 60% at 100 nM in adult CD34+ cells whatever their origin derived either from leukapheresis or bone marrow. BFU-E were more sensitive to abexinostat than CFU-GM, as the decrease in BFU-E-derived colonies reached 60% and 70% at 50 and 100 nM, respectively, versus 30% and 50% for CFU-GM-derived colonies (Figures 1a and b). The most dramatic dose-dependent effect was on the CFU-MK growth inhibition assessed in fibrin clots that reached 62.5% at 50 nM and more than 90% at 100 nM (Figure 1c).

The pan HDACi abexinostat (A) decreases hematopoietic progenitors growth. Representative experiments are shown to illustrate the number of progenitors generated in a semi-solid assay in the presence of indicated doses of abexinostat, n=3. In each experiment, CD34+ cells were seeded in triplicate. Results: mean±S.E.M., ***P<0.0001; **P<0.004; *P<0.05. (a and b) Number of BFU-E, CFU-GM CFU-GEMM and total colonies (total CFU) generated by plating 500 CD34+ cells from leukapheresis or bone marrow samples in methylcellulose assays in the presence of SCF+IL-3+EPO+G-CSF+ IL-6 as described in Materials and Methods. (a) Leukapheresis samples; (b) bone marrow exposed all along the culture or only 24 h to abexinostat. (c) Number of CFU-MK colonies generated by plating 1000 CD34+ cells in a fibrin clot assay in the presence of TPO+SCF+IL-6. (d and e) Number of CFU-MK colonies generated by plating CD34+ cells from leukapheresis (d) exposed 24 h to the indicated concentration of abexinostat, then washed before plating 500 CD34+ cells or from bone marrow samples exposed all along the culture or only 24 h to abexinostat (e)
Because abexinostat was added at the onset of the culture and all along, it was possible that the reduction in colony formation was due to an effect on the differentiation process. Thus, to test the effects of an exposure for a short period, CD34+ cells were seeded in liquid serum-free medium supplemented with cytokines and exposed during 24 h to abexinostat at 10, 50 or 100 nM. Then abexinostat-treated cells were washed and plated in methylcellulose or fibrin assay. A marked inhibition of mobilized or bone marrow CFU-MK growth was also found demonstrating that abexinostat acts at the level of CFU-MK (Figures 1d and e). Similar results were found for BFU-E and CFU-GM growth (Figure 1b) further demonstrating that abexinostat acts at the level of hematopoietic progenitors.
The pan-HDAC inhibitor abexinostat alters CD34+ cell proliferation and induces apoptosis
Because abexinostat decreased the cloning efficacy of CFU-MK, we tested the effects of increasing doses of abexinostat (1, 5, 10, 20, 50 and 100 nM added at the onset of culture and all along the culture) on the generation of MK from CD34+ cells in a serum-free liquid culture supplemented with TPO. Cell counts were performed at day 6 of culture. Abexinostat led to a dose-dependent decrease in alive cell number, with nearly 90% inhibition at 100 nM (Figure 2a). This decrease was associated with an increase in apoptotic cells as revealed by the increase of the Annexin+IP+ cell population (38.6%, 45.4% and 51.8%, respectively, for 20, 50 and 100 nM versus 34% for control, mean of three independent experiment, Figure 2b). A representative experiment is shown in Figure 2c. These results show that the drug-induced decrease in cell amplification correlates with an increase in apoptosis.

Abexinostat induces a decrease of cellular growth, an increase in apoptosis and a decrease of ploidy. (a) CD34+ cells were seeded in liquid culture in the presence of TPO±increasing dose of abexinostat, viable cells were counted using trypan blue exclusion at day 6 and results are the mean±S.E.M. (representative experiments of n=3;*P<0.05; **P<0.004; ***P<0.002). (b and c) CD34+ cells were seeded with TPO±increasing dose of abexinostat for 48 h then stained with allophycocyanin-Annexin V-propidium iodide. The percentage of apoptotic cells was determined by flow cytometry. (b) Mean of three independent experiments; (c) representative experiments. *P<0.04; **P<0.001, ***P<0.0001. (d and e) Mean ploidy value of MKs at day 10 of culture with indicated doses of abexinostat (d) mean of three independent experiments: control N: 3.82; 10 nM: 3.66; 50 nM: 2.59; 100 nM: 2.35. *P<0.03; (e) representative ploidy experiment. (f) Mean ploidy value at indicated times, with 100 nM abexinostat added at day 1 or 8. Control at day 8N: 3.80; control at day 10N: 4.7; day 10 analysis for 100 nM abexinostat added either at day 1 (N: 2.51) or at day 8 (N: 4.03)
To study the effects of abexinostat during MK differentiation, we added the HDACi at day 3 and analyzed the culture at day 10. As expected, apoptosis was elevated in control cultures as 50% of MKs were cells at the end of differentiation. Nevertheless, abexinostat treatment increased the percentage of cells in apoptosis as attested both by the subG1 peak (data not shown) and the presence of annexin V-positive cells (Supplementary Figure 1). The mean of three independent experiments showed a marked decrease in ploidy level in cultures treated with 50 or 100 nM abexinostat (2.59 and 2.35 N, respectively) compared with 3.82 N (control) or 3.66 N (10 nM) leading to a nearly complete absence of >4 N MK at 100 nM (Figure 2d and representative experiment in Figure 2e). To analyze whether the effects of abexinostat on polyploidization were either indirect by early apoptosis of MK progenitors or direct by affecting the ploidization process, abexinostat (100 nM) was added in culture either at day 1 or at day 8 and the ploidy was measured at both day 8 and 10 (Figure 2f). When abexinostat was added at day 1 of culture, the mean ploidy level measured at day 10 was decreased (2.51 N) compared with control cultures (4.7 N). In contrast, when added at day 8, abexinostat had no significant effect on ploidization (day 8 ploidy: 3.8 N, day 10 ploidy without abexinostat: 4.7 N and day 10 ploidy with abexinostat; 4.03 N) suggesting that abexinostat had minor direct effects on the ploidization process.
The pan-HDACi abexinostat prevents PPT formation
Addition of the pan-HDACi at day 8 of the culture was observed to decrease PPT formation quantified 4 days later, that is, the decrease in PPT formation was already 45% at 10 nM, reached 95% at 50 nM and was virtually complete at 100 nM (Figures 3a and b). Low concentrations (eg, 20 nM) modified the structure of the PPTs with less branching extensions (Figure 3a, iii), whereas PPT inhibition observed at higher concentrations was associated with an apoptotic MK morphology (Figure 3a, iv, v). To understand if this defect in PPT formation only required a short treatment, MKs from control cultures (day 8) were incubated with 100 nM of abexinostat for only 24 h and PPT formation was scored 4 days later (day 12). A similar inhibition of PPT formation was observed (Figures 3c and d), suggesting that HDACi directly affect the PPT formation process.

Abexinostat affects proplatelet formation. CD41+-sorted cells were seeded in triplicate in 96 wells with TPO and increasing doses of abexinostat (from 10 to 100 nM). Proplatelets were scored at day 12 under an inverted microscope. (a) Examples of proplatelet bearing MKs after treatment with indicated doses of the pan-HDACi. Proplatelet bearing MKs in control (i) and at 10 nM (ii) were similar; at 20 nM (iii), MK displayed less branching extensions compared with control MKs (i); at 50 nM (iv) and 100 nM (v) of abexinostat most MKs displayed apoptotic morphology and had shorter proplatelets. (b) Typical dose response of abexinostat on proplatelet formation. One representative experiment out of four experiments with increasing dose of abexinostat; ***P<0.0005. (c and d) Effects of a abexinostat short-time exposure on proplatelet formation. MKs were treated 24 h with 100 nM of abexinostat at day 8 (A-100 nM), then washed and seeded in triplicate in serum-free medium plus 10 ng/ml TPO. (d) Proplatelet formation was scored after 3 supplementary days of culture, n=2, P<0.0001. In c, even a short treatment (24 h) induces a proplatelet defect with a few extensions
Abexinostat had minor effects on the TPO/MPL/JAK2 signaling pathway
MK differentiation is controlled by JAK2 activation mediated by TPO binding to MPL.28 It has been underscored that HDAC inhibitors were able to inhibit the chaperone function of HSP90 leading to JAK2 degradation, especially in cells expressing the mutant JAK2V617F and subsequently to apoptosis.29 We thus studied whether alteration in the TPO signaling could be seen in MKs after abexinostat treatment. To this aim, CD41+ cells were sorted at day 7 of culture, seeded in serum-free medium with TPO with or without 50 nM abexinostat for 4 additional days. Then, MKs were TPO-deprived 12 h in the presence or absence of 50 nM abexinostat and then stimulated during 10 min, 1 h and 24 h with TPO and with or without 50 nM abexinostat (Figure 4a). The total JAK2 level was slightly modified in comparison to the control at 24 h. No changes were observed in the phosphorylation of STAT3 and ERK except a slight decrease for STAT5. Similar results were obtained when sorted day 7 MKs were treated only for 24 h with abexinostat (data not shown). This result suggests that although inducing a moderate decrease in JAK2, abexinostat did not significantly alter the MPL/TPO/JAK2/STAT signaling. However, it may alter other TPO-dependent or -independent pathways such as BMP4 signaling.30

Representative immunoblot blot analysis of the TPO/MPL/JAK2 signaling pathway in MKs treated or not with abexinostat. CD41+ cells were sorted at day 7 of culture, seeded in serum-free medium with TPO with or without 50 nM abexinostat for 4 additional days. Then, MKs were TPO-deprived 12 h in the presence or absence of 50 nM abexinostat and then stimulated for indicated times with or without 50 nM abexinostat. Phosphorylation of STAT5, STAT3 and ERK was evaluated by western blot as well as the JAK2 level. Actin was used as a loading control
Abexinostat treatment leads to a decrease of DNA repair and DNA DSBs
To investigate the mechanisms of abexinostat-induced inhibition of MK differentiation, we performed a transcriptional profiling on MKs sorted at day 7 and cultured for 24 h in the presence or absence of 100 nM abexinostat. A change in expression in a large number of genes was observed after abexinostat treatment (1012 genes upregulated and 476 genes downregulated with a P-value of 1 × 10−5 and an absolute fold change >2). Gene set enrichment analysis between the treated and untreated cells reveals that the great majority of pathways in MSigDB database was not affected. Indeed in the three subsets pathways KEGG, Biocarta and Reactome only 14/186, 0/217 and 18/430, respectively, appears differential. DNA repair, DNA DSB, DNA recombination and response to DNA damage were the most prominent signatures of abexinostat-treated cells (Figure 5a), underscoring the key role of HDAC on the DNA repair mechanism.9, 10 In contrast, genes implicated in DNA repair such as RAD51, AP1, Exo1, TIMELESS-interacting protein or BRCA1 were decreased by twofold to nearly threefold (Table 1). As RAD51 is one of main protein involved in DSB repair through homologous recombination, we studied its expression at the protein level and found a near complete disappearance of the protein (Figure 5b) as previously reported in the HCT116 colon cell line.31 The expression level of γH2AX detected by western blotting was used as a surrogate marker of DSB and a marked increase in γH2AX level was seen after abexinostat treatment (Figure 6a). Moreover, ATM phosphorylation responsible in turn of γH2AX was also observed after abexinostat treatment (Figure 6a).

Abexinostat modulates expression of genes involved in DNA repair. (a) Gene set enrichment analysis (GSEA) of CD41+ cells treated with 100 nM of abexinostat reveals that DNA repair, DNA double-strand break (DSB), DNA recombination and response to DNA damage were the most prominent signatures of abexinostat-treated cells. (b) RAD51 protein level by immunoblot analysis. Day 7 CD41+-sorted MK were cultured for 48 h with TPO with or without increasing the dose of abexinostat. HSC70 was used as loading control

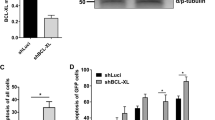
Activation of p53 in abexinostat-treated MK. CD41+ cells were sorted at day 7 of culture and seeded in serum-free medium with TPO with or without indicated doses of abexinostat for 48 h. (a) Immunoblot analysis of the phosphorylation status of ATM and γH2AX. (b) Immunoblot analysis of caspase 3 in sorted MK treated with or without abexinostat (50 and 100 nM). (c) Immunoblot analysis of the phosphorylated p53 on serine residue 15, BAX and BCL-xL. HSC70 used as loading charge. (d) qRT-PCR studies of p53 target genes. Data are mean±S.E.M., n=3, each experiments performed in triplicate, ***P<0.0007
DNA-damage accumulation is usually associated with a DNA-damage response characterized by a halt in proliferation and apoptosis through usually p53-dependent or -independent mechanisms. Thus, we studied whether p53 was activated after abexinostat treatment.
Abexinostat treatment induces p53 activation in MKs
To understand if the p53 pathway was induced, purified MKs were treated with increasing doses of abexinostat during 48 h and qRT-PCR and western blots were performed. Subsequently to ATM phosphorylation (Figure 6a), we found a p53 increase in MKs treated with abexinostat (data not shown) as well as its phosphorylated form on Serine 15 (Figure 6c). This was associated with an increased apoptosis as shown by the complete cleavage of caspase 3 at 100 nM abexinostat (Figure 6b). These results indicate that abexinostat may lead to caspase-mediated apoptosis through DSB induction that activates a p53-dependent pathway. Two p53 target genes, P21 and BAX, were induced after HDACi as shown by qRT-PCR, further demonstrating the functional activation of p53 (Figure 6d). BAX was also induced at protein level in a dose-dependent manner by abexinostat, whereas the anti-apoptotic protein BCL-xL was decreased by half at 50 nM and by more than 80% at 100 nM (Figure 6c).
p53 knockdown decreases apoptosis induced by abexinostat, but does not rescue PPT formation
To study whether p53 silencing could revert the apoptosis induced by abexinostat, MKs were transduced at day 5 of culture with either a lentivirus vector encoding a shRNAp53 (shp53) and GFP or the same vector containing an irrelevant sequence (SCR). MKs were sorted at day 7 on both CD41 and GFP expression, and cultured in the presence or absence of 10, 50 and 100 nM abexinostat for 2 days prior performing flow cytometry analysis. The shp53 led to a nearly complete inhibition of the p53 protein expression as shown in Figure 7a. Interestingly, this p53 knockdown totally prevented abexinostat to induce apoptosis whatever the concentration used, including the highest concentrations, as assessed by Annexin V staining (Figure 7b). In addition, in the absence of abexinostat, p53 knockdown decreased the number of apoptotic MKs compared with the shSCR (18.8 versus 10% and 32 versus 11.3 in two experiments). PPT formation was subsequently studied. The level of PPT formation was always lower in the controls transduced with the shSCR than in untransduced cells (compare Figures 3 and 7). However, the shp53 increased threefold PPT formation in comparison to the shSCR (Figure 7d), suggesting that p53 may negatively regulate PPT formation in vitro by a mechanism, which remains to be investigated. However, the p53 knockdown could not completely rescue the inhibition of PPT formation induced by abexinostat. Indeed, when shp53-transduced MKs were treated by 50 nM abexinostat, an increased PPT formation was seen (2.5% in shp53-transduced MKs compared with 0.6% in SCR-transduced MKs), but normal values (around 12% for the shp53 and 5% with the shSCR) were not reached (Figure 7d). Furthermore, in the presence of 100 nM abexinostat, no PPT formation by shp53-transduced MKs was seen despite the fact that they remained alive with a spherical appearance (Figure 7c).

p53 knockdown rescues MK apoptosis, but not proplatelet formation. (a) Immunoblot analysis of p53 performed on sorted MK transduced with shSCR or shp53. (b) Untreated or treated MKs were stained with Annexin V and analyzed by flow cytometry. Data represent the mean±S.E. of the % of Annexin V+ cells, n=2. In these two independent experiments, results show that the shp53 markedly reduced the apoptosis induced by abexinostat in comparison to the SCR. (c and d) Proplatelet formation derived from shp53-treated and non-treated MK. shp53 increased proplatelet formation by MKs in control cultures (P<0.0007), and in 50 nM abexinostat cultures (**P<0.002) (d). At 100 nM abexinostat, MKs kept their round shapes and remained alive but did not form proplatelets. ***P<0.0007
Altogether, these results show that abexinostat affects in vitro megakaryopoiesis by two mechanisms: one at the level of hematopoietic progenitors and during differentiation by induction of apoptosis through a p53 mechanism related to induction of DSB and at the level of PPT formation by p53-dependent and -independent mechanisms.
Discussion
HDAC inhibitors (HDACi) are currently tested in the treatment of a wide range of malignancies32 and transient but severe thrombocytopenia is their prominent, dose-limiting side effect. A thrombocytopenia is observed in up to 30% of the patients and can be either isolated or associated with other cytopenias.18, 19, 20 The molecular mechanisms of this thrombocytopenia remain a controversial issue. Here, we show in human primary MKs established in culture that the pan-HDACi abexinostat also called S78454 or PCI-24781, a pan-HDACi structurally related to hydroxyamic acid that is representative of this compound class, has two distinct effects at pharmacologically relevant doses: it promotes MK apoptosis in a p53-dependent manner, and it decreases PPT formation independently of p53.
HDACi have been shown to induce cell cycle arrest and/or apoptosis through different mechanisms, including downregulation of anti-apoptotic proteins such as BCL2, upregulation of apoptotic proteins such as BAX and BAK, and overproduction of radical oxygen species.16 These effects, initially related only to the acetylation of histone proteins that regulate gene transcription, can also be explained by the acetylation of non-histone proteins,8, 9 as recently underscored in JAK2V617F MPN.32, 33 In the mouse, hdac1 and hdac2 double knockout, which encode two class I HDACs, demonstrated that the global level of class I HDAC was critical for erythroid development and even more for the MK lineage.23 A complete deletion of hdac1 associated with hdac2 haploinsufficiency was sufficient to dramatically reduce the MK number in bone marrow through apoptosis induction and the platelet count in blood.23
At 100 nM that is the lower concentration compared with the peak plasma concentration of abexinostat also called PCI-24781 in humans,27 we show here that the pan-HDACi completely abolishes the growth of MK colonies, due to mainly apoptosis induction in both MK progenitors and precursors. This effect of abexinostat on progenitors was not specific of the MK lineage and was found on multipotent progenitors BFU-E and CFU-GM. However, CFU-MKs were more sensitive to abexinostat than the other progenitors and CFU-GEMMM and BFU-E more sensitive than CFU-GM.
In JAK2V617F MPN, HDAC inhibition promotes the kinase degradation through probably inhibition of HDAC6 and consequently by altering the chaperone effect of HSP90.34, 35 This mechanism preferentially targets the mutated version of JAK233 and does not appear to account for abexinostat toxicity toward human MKs, in which the JAK/STAT signaling remains operative. However, MK survival, proliferation and differentiation may also depend on the BMP4/receptor loop30 and on other signaling pathways such as the mTOR pathway. We cannot exclude that HDACi may operate on these regulatory pathways because a decrease in the level of mTOR transcripts was found in gene profiling (http://www.ebi.ac.uk/arrayexpress/) and was confirmed by qRT-PCR. This will require further investigations.
HDACi were also reported to affect the DNA repair machinery functioning.31, 36, 37, 38 In this line, we found that several DNA repair pathways, both in non-homologous end joining and homologous recombination, are altered in human MKs exposed to the pan-HDACi. The decreased expression of RAD51, which is the most important effector in homologous recombination, together with the increase in γH2AX level, argued for a DSB-processing defect similar to that observed in abexinostat-treated tumor cell lines exposed to radiation.30 We show that exposure of human MKs to the pan-HDACi induces ATM phosphorylation, stabilizes and phosphorylates p53 and promotes the induction of several p53 target genes, whereas p53 knockdown rescues the apoptotic phenotype.
Mice treated with Panobinostat or Romidepsin (class I HDACi)24 and Lewis rats treated with FR23522526 exhibit a decrease in their platelet counts without apoptosis of their bone marrow MKs, suggesting an alternative mechanism of toxicity. This alternative mechanism is presumably a defect in PPT formation,24 which may occur at a lower dose. Accordingly, we observed a defect in PPT formation starting at 10 nM abexinostat and this defect was only partially rescued by the downregulation of p53. The mechanisms that could account for the decreased formation of PPT upon inhibition of HDAC include the defective nuclear translocation of NF-κB, the downregulation of GATA1 and NF-E2,39, 40 the deregulation of GATA1/FOG1 interaction or the function by acetylation of GATA1,1, 41 as well as changes in the remodeling complex (NuRD) that recruits these transcription factors to their targets.42, 43 Cytoskeleton abnormalities have also been involved in HDACi-induced thrombocytopenia, as a consequence of altered expression of several Rho GTPases,24 which was not observed in our experiments in gene array analysis (http://www.ebi.ac.uk/arrayexpress/). The disrupted microtubule organization observed in HDACi-treated MK was also correlated to tubulin hyperacetylation.26 HDAC6, which is responsible for tubulin deacetylation, is a key regulator of the cytoskeleton,44 but thrombocytopenia had not been described in hdac6 knockout mice.45 In addition, class I HDACi such as Romidepsin, which induces a thrombocytopenia, have a minor effect on HDAC6. During the experiments reported here, we noticed marked changes in the mRNA expression level of myosin genes such as MYH10 or MYL9 (http://www.ebi.ac.uk/arrayexpress/). Interestingly, downregulation of MYL9 could provoke a marked decrease in PPT formation.46 The regulation of myosin light chains by HDAC and their role in HDACi-induced inhibition of PPT formation will therefore require further investigation.
Altogether, the present study uses the pan-HDACi abexinostat to demonstrate that thrombocytopenia classically induced by this class of anticancer drugs involves two complementary mechanisms. The first one involves the deregulation of the DNA repair machinery, leading to p53 activation and apoptosis induction. The second that could occur at a lower dose is the inhibition of PPT formation, which is both p53 dependent and independent and may be related in part to cytoskeleton modifications.
Materials and Methods
Isolation of CD34+ cells
CD34+ cells were obtained from leukapheresis samples after mobilization performed on patients or from bone marrow from patients undergoing hip surgery after informed consent in agreement with our Institute Ethic Committee (Assistance Publique des Hôpitaux de Paris) and in accordance with the Declaration of Helsinki. CD34+ cells were isolated by a positive selection using an immunomagnetic cell sorting system (AutoMacs; Miltenyi Biotec, Bergisch Gladbach, Germany) and cultured in serum-free medium in the presence of recombinant human (rhu) TPO (10 ng/ml; Kirin Brewery, Tokyo, Japan).
Quantification of clonogenic progenitors in semi-solid cultures
CD34+ cells were plated either in 1% methylcellulose assay (StemCells technologies, Vancouver, Canada) to quantify Erythroid (BFU-E) and granulocytic (CFU-GM) progenitors or in serum-free fibrin clot assay46 for quantification of MK progenitors (CFU-MK) in the absence or presence of various concentration of abexinostat. Cultures in methylcellulose were stimulated by addition of rhu growth factors: G-CSF (20 ng/ml), IL-3 (100 U/ml) SCF (50 ng/ml, Biovitrum AB, Stockholm, Sweden), IL-6 (100 U/ml) and human EPO (1 U/ml). Cultures in fibrin clot were stimulated with 10 ng/ml TPO (Kirin Brewery, Tokyo, Japan) and 50 ng/ml SCF. They were incubated at 37 °C in a fully humidified atmosphere containing 5% CO2 in air and scored after 12–14 days for BFU-E-, CFU-GM and CFU-GEMM-derived colonies using an inverted microscope. MK colonies were enumerated at day 12 after labeling by an indirect immuno-alkaline phosphatase staining technique using an anti-CD41a monoclonal antibody (MoAb; Becton Dickinson, le Pont de Claix, France; clone HIP8), as previously described.47 Culture dishes were entirely scanned under an inverted microscope at × 4 or × 10 magnification.
Liquid serum-free medium and purification of the CD41+ cell population
CD34+ cells were cultured in serum-free liquid medium in the presence of TPO (10 ng/ml). Ingredients used to prepare the serum-free medium were as previously described.48 To purify the CD41+ cell population, cells in suspension culture were collected at days 6–7, stained with an anti-CD41a-phycoerythrin (PE) monoclonal antibody (MoAb; PharMingen, San Diego, CA, USA) and selected by cell sorting (Influx, Becton Dickinson). The purified CD41+ cell population was subsequently grown in serum-free liquid medium without or with TPO and in the presence or absence of abexinostat for an additional 24 h. When stated, CD41+ cells were starved overnight from TPO and then TPO stimulated for different time periods without or with abexinostat and cells were recovered in dry pellet for subsequent analysis.
PPT formation
For PPT formation studies, CD41+-sorted cells were seeded in 96-well plates in serum-free liquid medium supplemented with 10 ng/ml TPO and increasing concentration of abexinostat. MK displaying PPT were counted as previously described.48
Western blot analysis
CD41+ cell population was harvested at different times of culture, washed once in PBS, lysed in 2 × Laemmli buffer (100 mM Tris pH 6.8, 20% glycerol, 4% SDS, 0.05% bromophenol blue and 10 mM DTT). Lysates were gently sonicated on ice. Samples were boiled 5 min in loading buffer and subjected to 10 or 12% SDS-PAGE gel. After transfer, nitrocellulose membranes were blotted with the following antibodies: anti-pSTAT3, STAT3, pSTAT5, STAT5, pJAK2, JAK2, pERK, ERK, pH2AX, pP53, P53, pATM, ATM (all from Cell Signaling, Ozyme, Saint Quentin Yvelines, France), RAD51 polyclonal (Millipore, Molsheim, France), BAX monoclonal (Oncogene Research, Millipore) and a rat monoclonal anti-HSC70 (Stressgen, British Columbia, VIC, Canada). Primary antibodies were revealed with appropriate secondary antibodies conjugated with horseradish peroxidase and the filters were developed with an enhanced chemiluminescence system (ECL detection kit, Amersham, Orsay, France).
Cell cycle analysis
CD34+ cells were grown in liquid serum-free medium in the presence of TPO (10 ng/ml). Cultures were treated continuously for 48 h or for only 24 h with abexinostat. Non-treated cultures served as control. Cells were stained for 30 min at 4 °C with propidium iodide (50 μg/ml; Sigma, Saint-Quentin Fallavier, France) in hypotonic sodium citrate solution containing 50 μg/ml RNAse (Sigma-Aldrich, Lyon, France) followed by flow cytometry analysis.
Assessment of apoptosis by Annexin-V staining
To assess apoptosis in the CD34+ cell population, untreated and abexinostat-treated cells were stained with annexin-allophycocyanin and propidium iodide. At the level of mature MKs, untreated and abexinostat-treated cells were stained with a monoclonal anti-CD41+ allophycocyanin-conjugated antibody for 30 min at 4 °C, washed and stained with Annexin-V PE (BD Bioscience, Pharmingen, Le Pont de Claix, France) and 7AAD. The percentage of apoptotic cells was determined by flow cytometry.
Assessment of percentage of viable cells
CD34+ cells were seeded in triplicate in the presence or absence of increasing doses of abexinostat (1 to 100 nM). After treatment, cells were stained with trypan blue. The numbers of viable cells were determined with a hemocytometer and results are expressed as the total alive number cells/ml.
Gene expression arrays
Two replicates S1 and S2 of CD34- CD41+-sorted cells at day 7 were grown in serum-free medium with TPO (10 ng/ml) and treated with abexinostat for 24 h. Total RNA was extracted from non-treated and treated cells using a RNA extraction kit according to manufacturer protocol (Norgen Biotek Corp, Thorold, ON, Canada). Briefly, probes were synthesized from 500 ng of total RNA in two steps, according to the manufacturer’s instructions. In each comparison, the two samples were labeled separately with different fluorescent dyes, cyanine-3 (Cy3) and cyanine-5 (Cy5). For each sample, 1 mg of purified cy5-labeled cRNA was mixed with the same amount of cy3-labeled cRNA. Label incorporation was checked on a NanoDrop spectrophotometer. Hybridizations were performed with a dye-swap strategy on whole-human-genome dual color 8 × 60 K oligonucleotide microarrays (design 028004; Agilent Technologies, Massy, France). Feature extraction software provided by Agilent (version 10.7.3.1) was used to quantify the intensity of fluorescent images and to apply a linear normalization to correct for artifacts caused by nonlinear rates of dye incorporation and inconsistent relative fluorescence intensities between some green and red dyes. All data were imported into Resolver software (Rosetta Biosoftware, Boston, MA, USA) for database management, quality control, computational recombination of dye-swaps and statistical analysis. Gene set enrichment analysis was performed between the two groups of samples (treated and non-treated by abexinostat).
Data availability
The microarray data related to this paper have been submitted to the Array Express data repository at the European Bioinformatics Institute (http://www.ebi.ac.uk/arrayexpress/) under the accession number EMTAB-1180.
shRNA cloning and cell transduction
Blunt vector of pRRL-PGK-GFP after digestion by XhoI and blunt inserts of pH1-shRNAp53 after XhoI/EcoRI digestion of SuperRetro-pH1-shp53-PGK-Puro were ligated as previously described.49, 50
CD34+ cells cultured 5 days in serum-free medium with TPO (10 ng/ml) and SCF (50 ng/ml) were transduced with lentiviral particles for 6 h followed by a second transduction. Forty-eight hours after transduction, cells were sorted at day on the expression of GFP and CD41 by flow cytometry, and then cultured in the presence of TPO without or with abexinostat.
Assessment of polyploidization
Cells were incubated for 2 h at 37 °C in 0.01 mM Hoechst 33342 (Sigma-Aldrich, St Quentin Fallavier, France) and then stained for 30 min at 4 °C with a PE-conjugated anti-CD41a MoAb. Ploidy was acquired on an InfIux cytometer (Becton Dickinson) and analysis was performed using FlowJo software.
RT PCR and qRT-PCR
Total RNA was isolated using RNA/DNA/Protein Purification Kit (Norgen Biotek Corp). Reverse transcription was performed using SuperScript VILO cDNA Synthesis Kit (Life technologies) before PCR amplification. qRT-PCRs were performed using an Applied Biosystems 7500 Real-Time PCR System with Power SYBR Green Master Mix (Life technologies) using the manufacturer’s recommendations. Expression levels of genes (BAX, CDKN1A (P21)) were measured relatively to HPRT. Primer sequences are listed in Supplementary Table 1.
Accession codes
Abbreviations
- DSB:
-
double-strand break
- HDACi:
-
histone deacetylase inhibitor
- HDAC:
-
HDACi
- Megakaryocyte:
-
MK
- Myeloproliferative neoplasms:
-
MPN
- Thrombopoietin:
-
TPO
- PE:
-
phycoerythrin
- PPT:
-
proplatelet
References
Boyes J, Byfield P, Nakatani Y, Ogryzko V . Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 1998; 396: 594–598.
Gu W, Roeder RG . Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997; 90: 595–606.
Janke C, Bulinski JC . Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 2011; 12: 773–786.
Minucci S, Pelicci PG . Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6: 38–51.
Yang XJ, Seto E . HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007; 26: 5310–5318.
Yang XJ, Seto E . Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 2008; 31: 449–461.
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325: 834–840.
Yang XJ, Seto E . The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 2008; 9: 206–218.
Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002; 419: 411–415.
Peterson CL, Cote J . Cellular machineries for chromosomal DNA repair. Genes Dev 2004; 18: 602–616.
Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature 1998; 391: 815–818.
Guidez F, Howell L, Isalan M, Cebrat M, Alani RM, Ivins S et al. Histone acetyltransferase activity of p300 is required for transcriptional repression by the promyelocytic leukemia zinc finger protein. Mol Cell Biol 2005; 25: 5552–5566.
Marks PA, Xu WS . Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem 2009; 107: 600–608.
Lane AA, Chabner BA . Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009; 27: 5459–5468.
Duvic M, Vu J . Vorinostat in cutaneous T-cell lymphoma. Drugs Today (Barc) 2007; 43: 585–599.
Martinez-Iglesias O, Ruiz-Llorente L, Sánchez-Martínez R, García L, Zambrano A, Aranda A . Histone deacetylase inhibitors: mechanism of action and therapeutic use in cancer. Clin Transl Oncol 2008; 10: 395–398.
Marson CM . Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med Chem 2009; 9: 661–692.
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109: 31–39.
Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol 2009; 27: 2052–2058.
Galli M, Salmoiraghi S, Golay J, Gozzini A, Crippa C, Pescosta N et al. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol 2010; 89: 185–190.
Kavanaugh SM, White LA, Kolesar JM . Vorinostat: a novel therapy for the treatment of cutaneous T-cell lymphoma. Am J Health Syst Pharm 2010; 67: 793–797.
Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 2009; 27: 5410–5417.
Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J 2010; 29: 2586–2597.
Bishton MJ, Harrison SJ, Martin BP, McLaughlin N, James C, Josefsson EC et al. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 2011; 117: 3658–3668.
Iancu-Rubin C, Gajzer D, Mosoyan G, Feller F, Mascarenhas J, Hoffman R . Panobinostat (LBH589)-induced acetylation of tubulin impairs megakaryocyte maturation and platelet formation. Exp Hematol 2012; 40: 564–574.
Matsuoka H, Unami A, Fujimura T, Noto T, Takata Y, Yoshizawa K et al. Mechanisms of HDAC inhibitor-induced thrombocytopenia. Eur J Pharmacol 2007; 571: 88–96.
Evans A, Ai W, Balasubramanian S, Sirisawad M, Mani C, Guerra M et al. Phase 1 analysis of the safety and pharmacodynamics of the novel broad spectrum HDACi PCI-24781 in relapsed and refractory lymphoma. Blood 2009; 114 [abstract: 2726].
Besancenot R, Chaligné R, Tonetti C, Pasquier F, Marty C, Lécluse Y et al. A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol 2010; 8 pii: e1000476.
Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009; 114: 5024–5033.
Jeanpierre S, Nicolini FE, Kaniewski B, Dumontet C, Rimokh R, Puisieux A et al. BMP4 regulation of human megakaryocytic differentiation is involved in thrombopoietin signaling. Blood 2008; 112: 3154–3163.
Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ . HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci USA 2007; 104: 19482–19487.
Siegel D, Hussein M, Belani C, Robert F, Galanis E, Richon VM. et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol 2009; 2: 31.
Guerini V, Barbui V, Spinelli O, Salvi A, Dellacasa C, Carobbio A et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia 2008; 22: 740–747.
Boyault C, Sadoul K, Pabion M, Khochbin S . HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007; 26: 5468–5476.
Kapuria V, Levitzki A, Bornmann WG, Maxwell D, Priebe W, Sorenson RJ. et al. A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination. Cell Signal 2011; 23: 2076–2085.
Lee JH, Choy ML, Ngo L, Foster SS, Marks PA . Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci USA 2010; 107: 14639–14644.
Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol 2010; 17: 1144–1151.
Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R. et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471: 74–79.
Vyas P, Ault K, Jackson CW, Orkin SH, Shivdasani RA . Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood 1999; 93: 2867–2875.
Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, Jackson CW, Hunt P, Saris CJ et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell 1995; 81: 695–704.
Lamonica JM, Vakoc CR, Blobel GA . Acetylation of GATA-1 is required for chromatin occupancy. Blood 2006; 108: 3736–3738.
Hong W, Nakazawa M, Chen YY, Kori R, Vakoc CR, Rakowski C. et al. FOG-1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J 2005; 24: 2367–2378.
Miccio A, Wang Y, Hong W, Gregory GD, Wang H, Yu X et al. NuRD mediates activating and repressive functions of GATA-1 and FOG-1 during blood development. EMBO J 2010; 29: 442–456.
Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F . HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol 2008; 18: 291–297.
Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol 2008; 28: 1688–1701.
Gilles L, Bluteau D, Boukour S, Chang Y, Zhang Y, Robert T et al. MAL/SRF complex is involved in platelet formation and megakaryocyte migration by regulating MYL9 (MLC2) and MMP9. Blood 2009; 114: 4221–4232.
Debili N, Louache F, Vainchenker W . Isolation and culture of megakaryocyte precursors. Methods Mol Biol 2004; 272: 293–308.
Norol F, Vitrat N, Cramer E, Guichard J, Burstein SA, Vainchenker W et al. Effects of cytokines on platelet production from blood and marrow CD34+ cells. Blood 1998; 91: 830–843.
Brummelkamp TR, Bernards R, Agami R . A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296: 550–553.
Koprinarova M, Botev P, Russev G . Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair (Amst) 2011; 10: 970–977.
Acknowledgements
We are grateful to F Wendling for helpful suggestions on the manuscript. Labex GR-Ex is funded by the program ‘Investissements d’avenir’, an ANR Blanc (I Plo) and a research grant from Servier (N Debili). This work has also been supported by a grant from the Ligue Nationale contre le Cancer (Equipe labellisée 2013). O Bluteau and S Boukour were supported by a postdoctoral fellowship from ANR and AP by a PhD fellowship from the GEHT. N Debili, H Raslova and W Vainchenker are recipients of a research fellowship from CHU Bordeaux-INSERM, AP-HP-INSERM.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
N Debili received research support from the Institut de Recherches Internationales Servier. L Kraus-Berthier, A Jacquet-Bescond, H Lelièvre and S Depil are employed by the Institut de Recherches Internationales Servier. The remaining authors declare no conflict of interest.
Additional information
Edited by D Aberdam
Author Contributions
AA designed and performed most of the experiments and analyzed data, OB performed QRT-PCR analysis and analyzed data; KM, AP, SB performed experiments, LL did the lentiviral shRNA p53, YL and PR performed cell sorting; LKB, AJB, HL and SD provided the abexinostat, PD performed transcriptome experiments. ES, HR, WV, IP and ND wrote the manuscript. ND designed and analyzed data and was responsible for coordination.
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ali, A., Bluteau, O., Messaoudi, K. et al. Thrombocytopenia induced by the histone deacetylase inhibitor abexinostat involves p53-dependent and -independent mechanisms. Cell Death Dis 4, e738 (2013). https://doi.org/10.1038/cddis.2013.260
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.260
Keywords
This article is cited by
-
Critical role of the HDAC6–cortactin axis in human megakaryocyte maturation leading to a proplatelet-formation defect
Nature Communications (2017)
-
First-in-human study of the toxicity, pharmacokinetics, and pharmacodynamics of CG200745, a pan-HDAC inhibitor, in patients with refractory solid malignancies
Investigational New Drugs (2015)
-
Pharmacokinetic/Pharmacodynamic modeling of abexinostat-induced thrombocytopenia across different patient populations: application for the determination of the maximum tolerated doses in both lymphoma and solid tumour patients
Investigational New Drugs (2014)
