
Abstract
miR-148a has been shown to regulate inflammation, immunity and the growth of certain tumors, but its roles in colitis and colorectal tumorigenesis remain largely undetermined. Here we found miR-148a-deficient mice to be more susceptible to colitis and colitis-associated tumorigenesis. Both were associated with increased nuclear factor κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3) signaling. Bone marrow- and non-bone marrow-derived miR-148a contributed to colitis and colitis-associated tumorigenesis. miR-148a loss of heterozygosity exacerbated Apcmin/+ colon and small intestinal spontaneous tumor development. Restoring miR-148a expression prevented both spontaneous and carcinogen-induced colon tumor development. miR-148a was downregulated in human inflammatory bowel disease (IBD) and colorectal cancer patient tissues. This correlated with a high degree of miR-148a promoter methylation mediated by a complex comprised of P65 and DNA methyltransferase 3 alpha (DNMT3A). miR-148a directly targets several well-accepted upstream regulators of NF-κB and STAT3 signaling, including GP130, IKKα, IKKβ, IL1R1 and TNFR2, which leads to decreased NF-κB and STAT3 activation in macrophages and colon tissues. Our findings reveal that miR-148a is an indirect tumor suppressor that modulates colitis and colitis-associated tumorigenesis by suppressing the expression of signaling by NF-κB and STAT3 and their pro-inflammatory consequences.
Similar content being viewed by others


Main
Colorectal cancer (CRC) is a leading cause of cancer-related deaths. Inflammation, as observed in inflammatory bowel disease (IBD), has important roles in CRC development. Strong links have been found between colorectal inflammation and increased colitis-associated tumorigenesis as indicated by numerous epidemiological and clinical studies.1, 2 In colitis-associated tumorigenesis, cancer susceptibility is enhanced by the activation of inflammation-associated factors such as signal transducer and activator of transcription 3 (STAT3) and nuclear factor (NF)-κB, which contribute to CRC development. Their activation not only promotes CRC growth and survival but also stimulates the production of numerous growth factors and cytokines.1, 2, 3, 4, 5, 6, 7 Colitis also alters colonic epithelial cellular DNA genetically or epigenetically thereby further contributing to transformation.8, 9
miRNAs are critical mediators and regulators of inflammatory signaling pathways, contributing to the pathogenesis of different human inflammatory diseases, including IBD. miRNA expression is also abnormal in IBD patients, indicating that this family of molecules may be involved in IBD pathogenesis.10, 11 However, specific roles for miRNAs in IBD remain to be elucidated. One such miRNA, miR-148a, was previously demonstrated to impair the innate response and antigen presentation of TLR-triggered dendritic cells,12 to repress NF-κB signaling and inflammatory gene expression in human aortic valve cells,13 to regulate B-cell tolerance and autoimmunity14 and to inhibit tumor growth.15 However, the intrinsic physiological roles of miR-148a in colitis and colitis-associated tumorigenesis have not been reported. Here we show that miR-148a-deficienct mice are highly susceptible to intestinal inflammation and colitis-associated tumorigenesis. miR-148a protects against colon tumorigenesis by inhibiting several upstream regulators of NF-κB and STAT3 signaling. These findings suggest that miR-148a has a critical role in colitis-associated tumorigenesis by negatively regulating the pro-inflammatory environment.
Results
Ablation of miR-148a enhances DSS-induced colitis
To identify potential tumor-suppressor miRNAs in colon cancer, 1255 individual human miRNA expression vectors were screened for their ability to inhibit the viability of colon cancer cells and miR-148a was identified as a candidate (Supplementary Figure S1A). To study the physiological roles of miR-148a in the colon, a knockout (KO) mouse model was generated (Supplementary Figure S1B) and the loss of miR-148a was confirmed (Supplementary Figure S1C). As shown in Supplementary Figure S1D, the expression of miR-148a-3p/5p was undetectable in a variety of tissues from miR-148a KO mice; however, this did not affect the expression of several other miRNAs such as miR-124-3p.
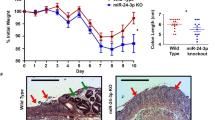
The consequences of miR-148a’s absence were first examined in DSS-induced colitis after providing mice 3.5% DSS in their drinking water for 6 days. The severity of colitis was evaluated by assessing survival, body weight, stool consistency, rectal bleeding and colon length. Drastically reduced miR-148a-3p/5p expression was observed after DSS-treated in wild-type (WT) colons (Figure 1a). Compared with WT animals, miR-148a KO mice had much more severe colitis as indicated by greater weight loss, worse survival and increased colon shortening and rectal bleeding (Figures 1b–f).

miR-148a KO mice are more susceptible to dextran sodium sulfate (DSS)-induced colitis. (a) Changes in miR-148a-3p/5p expression in colon tissues from WT mice at the indicated times after DSS treatment (n=3). (a–j) WT and miR-148a KO mice were treated with 3.5% DSS in drinking water for 6 days and followed by regular drinking water. (b) Body weight changes. (c) Survival curves. (d) Rectal bleeding (Left) and stool consistency (Right) were scored daily. (e) Colon length was measured at the indicated times after killing. (f) Typical colon images on day 9 after DSS treatment. (g) Activation of NF-κB and STAT3 signaling was determined in colon tissues at the indicated times. Each lane represents an individual mouse. (h) The level of each indicated cytokines was determined by ELISA in colon tissues. (i and j) Hematoxylin and eosin (H&E) images of colon tissues (i) and immuno-histochemical stains for the macrophage marker F4/80 (j). Scale bars, 100 μm. Data indicate the mean±S.D. in panels (a, b, d, e and h). *P<0.05, **P<0.01, ***P<0.001
To evaluate the impact of miR-148a deficiency on NF-κB and STAT3 activity, colons were isolated and analyzed by western blotting. The colons of miR-148a KO mice had higher levels of phosphorylated P65, IκBα, AKT1, STAT3 and ERK1/2 compared with colons from WT animals (Figure 1g). KO colons also showed elevated levels of mRNAs (Supplementary Figure S1E) and proteins (Figure 1h) for pro-inflammatory cytokines, such as G-CSF, IL-1β, IL6, IL8, KC and tumor necrosis factor-α (TNFα). Elevated serum levels of the above cytokines were confirmed in miR-148a KO mice (Supplementary Figure S1F). Significantly worse colon inflammation was observed by H&E staining and F4/80 immuno-staining in miR-148a KO mice (Figures 1i and j). Consistent with these findings, more apoptotic cells were observed on days 9 and 12 after DSS treatment in the colons of miR-148a KO mice (Supplementary Figure S1G). These data show that miR-148a deficiency significantly enhances the severity of DSS-induced colitis.
miR-148a deficiency exacerbates AOM-DSS-induced CRC
To investigate the effects of miR-148a deficiency in colorectal tumorigenesis, we used an established colitis-associated colorectal tumorigenesis model in which mice were treated with the DNA-methylation agent azoxymethane (AOM) followed by three rounds of DSS treatment (Supplementary Figure S2A).3, 6 Notably, the miR-148a-3p/5p expression was drastically reduced in the colons of WT mice 30 or 120 days after AOM/DSS treatment (Figure 2a). miR-148a KO mice showed significantly more weight loss (Figure 2b) and shorter survival (Figure 2c). Although colon weights were increased in these mice, the overall length was less than that of WT mice (Figures 2d and e). This was likely due to the fact that miR-148a KO mice developed significantly more and larger-sized CRCs (Figures 2f and g) compared with WT mice that tended to be associated with more pronounced inflammation and hyperplasia (Figure 2h). The colonic tissue from miR-148a KO mice also showed enhanced activation of NF-κB and STAT3 signaling (Figure 2i) and increased production of pro-inflammatory cytokines (Figure 2j and Supplementary Figures S2B and C). These results suggested that miR-148a normally suppresses colorectal tumorigenesis.

miR-148a KO mice are more susceptible to AOM/dextran sodium sulfate (DSS)-induced colorectal tumorigenesis. (a) Changes of miR-148a-3p/5p expression in colon tissues collected from WT mice at days 0, 30 and 120 after AOM/DSS treatment. (a–j) WT and miR-148a KO mice were injected with AOM on day 0 and were treated with three rounds of DSS for 7 days. (b) Body weight changes of WT (n=26) and miR-148a KO (n=37) mice. (c) Survival analysis. (d) Typical images of colon tumors from WT and miR-148a KO mice 120 days after AOM/DSS treatment. (e) Colon length (Top) and weight (Bottom) were determined from WT (n=13) and miR-148a KO (n=16) mice on day 120 after AOM/DSS treatment. (f and g) Colon tumor number (f, Left), average volume (f, right) and size (g) in WT (n=13) and miR-148a KO (n=16) mice from panel (d). (h) Colon tissues collected from panel (d) were fixed, stained with hematoxylin and eosin (H&E; Top) and immuno-stained for Ki-67 (Bottom). Scale bars, 100 μm. (i) Activation of NF-κB and STAT3 signaling were determined in the colons collected from WT and miR-148a KO mice from panel (d). Each lane represents an individual mouse. (j) The levels of the indicated cytokines were determined by ELISA in colon tissues collected from WT and miR-148a KO mice from panel (d). Data present mean±S.D. in panels (a, b and g). *P<0.05, **P<0.01, ***P<0.001. Significance was determined using Wilcoxon signed-rank test in panels (e, f and j). The horizontal lines in the box plots represent the median, the boxes represent the interquartile range and the whiskers represent the minimal and maximal values
miR-148a expression in both bone marrow and non-bone marrow compartments suppresses colitis and colitis-associated tumorigenesis
In additional to intestinal tissues, miR-148a-3p/5p was also widely expressed in macrophages, dendritic cells and mesenteric lymph nodes (Supplementary Figure S3A). Four groups of miR-148a bone marrow chimeric animals were therefore generated in order to study which cell populations were the most critical for colitis and colitis-associated tumorigenesis.3, 6, 16 Six weeks after bone marrow transplantation, these mice were exposed to DSS and AOM/DSS as described above. As predicted, miR-148a KO mice transplanted with bone marrow from miR-148a KO mice were more susceptible to colitis and colitis-associated tumor than WT mice receiving bone marrow from WT mice (Figures 3a–k and Supplementary Figures S3B–G). However, WT mice transplanted with miR-148a KO bone marrow also showed increased susceptibility to colitis and colitis-associated CRC compared with WT mice that received WT bone marrow. miR-148a KO mice transplanted with WT bone marrow were more susceptible to colitis and colitis-associated CRC as well (Figures 3a–k and Supplementary Figures S3B–G). These data indicate that miR-148a expression both in the bone marrow and intestine is critical for colitis and colitis-associated tumorigenesis and that both tissue compartments participate in these processes.

miR-148a expression in both bone marrow and non-bone marrow contributes to attenuation of colitis and colorectal tumorigenesis. (a) Body weight changes of the indicated miR-148a bone marrow chimera mice after dextran sodium sulfate (DSS) treatment (n=10–12). (a–g) miR-148a bone marrow chimera mice were treated with 3.5% DSS in drinking water for 6 days. (b) Rectal bleeding (Left) and stool consistency (Right) were scored daily. (c) Survival of the indicated miR-148a bone marrow chimera mice after DSS treatment. (d) Colon length from indicated miR-148a bone marrow chimera mice on day 9 after DSS treatment. (e) Activation of NF-κB and STAT3 signaling in colon tissues was determined by immuno-blotting on day 9. Each lane represents an individual mouse. (f) mRNA (Top) and protein (Bottom) levels of the indicated cytokines were determined by reverse transcription quantitative-PCR and ELISA, respectively, in colon tissues on day 9. (g) Colon tissues on day 9 after DSS treatment were fixed, stained with hematoxylin and eosin (H&E; Left) and immuno-stained for the macrophage marker F4/80 (Right). Scale bars, 100 μm. (h) Typical images of colon tumors from the indicated miR-148a bone marrow chimera mice 120 days after AOM/DSS treatment. (h–k) miR-148a bone marrow chimera mice were injected with AOM on day 0 and treated with three rounds of 2.5% DSS in drinking water for 7 days. (i and j) Colon tumor number (i, Left), averge tumor volume (i, right) and size (j) in the indicated miR-148a bone marrow chimera mice 120 days after AOM/DSS treatment. (k) Colon weight from mice in panel (i). Data indicate the mean±S.D. in panels (a, b, f and j). *P<0.05, **P<0.01, ***P<0.001
miR-148a deficiency enhances activation of NF-κB and STAT3 signaling in macrophages
Given that miR-148a expression in bone marrow is important for colitis and colitis-associated tumorigenesis, bone marrow-derived macrophages from WT and miR-148a KO mice were stimulated with lipopolysaccharide (LPS) and examined for the expression of NF-κB and STAT3.6 miR-148a-3p/5p expression was drastically reduced in the macrophages of WT mice 1, 2 or 4 h after LPS treatment (Supplementary Figure S3H). As shown in Supplementary Figure S3I, increased expression of p-P65, p-IκBα, p-AKT1, p-STAT3 and p-ERK1/2 was observed in miR-148a KO macrophages. Significantly increased expression of multiple cytokine mRNAs and proteins were also detected in LPS-stimulated miR-148a KO macrophages (Supplementary Figures S3J and S3K).
miR-148a KO mice were also more susceptible to LPS-triggered death than their WT counterparts (Supplementary Figure S3L). Moreover, they demonstrated greater weight loss (Supplementary Figure S3M), lower body temperature (Supplementary Figure S3N) and increased serum cytokine production (Supplementary Figure S3O). These results showed that the more severe colitis and colitis-associated tumorigenesis seen in miR-148a KO mice is associated with enhanced pro-inflammatory cytokine production and activation of NF-κB and STAT3 signaling in macrophages.
miR-148a attenuates spontaneous tumor development in Apcmin/+ mice
Adenomatous polyposis coli (APC) is the most frequently mutated gene in sporadic CRC cancer in humans and is nearly always mutated in familial adenomatous polyposis coli (FAP).1, 2 Apcmin/+ mice contain a germ-line mutation in the Apc gene, which predisposes to spontaneous colonic and intestinal polyposis and serves as a model for human FAP.17, 18 Reverse transcription quantitative-PCR analysis showed significantly decreased miR-148a-3p/5p expression in Apcmin/+ mice compared with WT animals (Figure 4a). To investigate how miR-148a deficiency affects spontaneous tumor development, miR-148a KO mice were crossed with Apcmin/+ mice and the offspring were observed for tumor development. Interestingly, these crosses yielded only miR-148a+/−Apcmin/+ mice. Nevertheless, miR-148a haplo-insufficiency was sufficient to lead to an increase in both the number and size of both colon and small intestinal tumors (Figures 4b–f,Supplementary Figures S4A–D) compared with WT littermates in the Apcmin/+ mice background. These tumors also showed increased activation of NF-κB and STAT3 signaling (Figure 4g), production of cytokines (Figure 4h) and proliferation rates (Figures 4i–j and Supplementary Figure S4E). Collectively, these data demonstrate that endogenous levels of miR-148a also attenuate spontaneous colon and small intestinal tumorigenesis in Apcmin/+ mice.

miR-148a attenuates spontaneous colon tumor development in Apcmin/+ mice. (a) miR-148a-3p/5p expression in colon tissues collected from 14-week-old WT and Apcmin/+ mice (n=3). (b) Survival curve of WT, miR-148a+/−, Apcmin/+ and miR-148a+/−Apcmin/+ mice (n=12–16). (c) Rectal bleeding (Left) and stool consistency (Right) were scored from 14-week-old WT and Apcmin/+ mice (n=12). (d) Body weights of 14-week-old WT, miR-148a+/−, Apcmin/+ and miR-148a+/−Apcmin/+ mice (n=12). (e) Typical images of colon tumors from 14-week-old WT, miR-148a+/−, Apcmin/+ and miR-148a+/−Apcmin/+ mice. (f) Colon tumor number (Top) and average tumor volume (Bottom) of mice in panel (e). (g) Activation of NF-κB and STAT3 was determined by immune-blotting of colon lysates prepared from mice in panel (e). (h) G-CSF, IL-1β, IL6, IL8, KC and TNFα transcript levels were determined by reverse transcription quantitative-PCR in colon tissues collected from mice in panel (e). (i and j) Colon tissues collected from panel (e) were stained with hematoxylin and eosin (H&E) (i) and immuno-stained for Ki-67 (j). Scale bars, 100 μm. Data present mean±S.D. in panels (a and h). *P<0.05, **P<0.01, ***P<0.001
Restoring miR-148a expression shows therapeutic effects on both spontaneous and carcinogen-induced tumor mice models
To test the therapeutic potential of manipulating miR-148a in vivo, we asked whether restoring its expression would inhibit tumor growth in CRC mouse models. Concentrated preparations of lentivirus encoding Pri-miR-148a were therefore administrated to AOM/DSS-treated WT mice for 3 weeks and their tumor burdens were assessed on day 120 (Supplementary Figure S5A). Compared with mice treated with a control lentiviral vector, these animals showed less pronounced colitis and colitis-associated tumorigenesis (Figures 5a–d and Supplementary Figures S5B–D), reduced expression of NF-κB and STAT3 and lower levels of pro-inflammatory cytokine production (Figures 5e–f and Supplementary Figures S5E and F). Restoration of miR-148a expression in miR-148a KO mice also suppressed colitis-associated tumorigenesis (Figures 5a–f and Supplementary Figures S5A–F). Similarly, Pri-miR-148a lentiviral administration to Apcmin/+ mice also suppressed spontaneous tumorigenesis, reduced NF-κB and STAT3 signaling and attenuated pro-inflammatory cytokine production (Figures 5g–i,Supplementary Figures S5G–J).

Restoring miR-148a expression attenuates spontaneous and AOM/dextran sodium sulfate (DSS)-induced tumorigenesis. (a) Body weight changes of WT and miR-148a KO mice treated with lentivirus containing Pri-miR-148a or control expression vector during AOM/DSS treatment (n=9–12). (b) Typical images of colon tumors of mice from (a) 120 days after AOM/DSS treatment. (c and d) Colon tumor number (c, Left), averge volume (c, right) and size (d) in mice from panel (b). (e) Inhibition of NF-κB and STAT3 signaling was determined in the colons collected in mice from panel (b). Each lane represents an individual mouse. (f) Cytokine levels were determined by ELISA in colon tissues collected in mice from panel (b). (g and h) Colon tumor number (g, Top), average volume (g, Bottom) and size (h) in Apcmin/+ mice treated with lentivirus containing Pri-miR-148a or a control expression vector. (i) Small intestine tumor number (Top) and average volume (Bottom) in Apcmin/+ mice from panel (g) (n=9–10). Data present mean±S.D. in panels (a, d and h). *P<0.05, **P<0.01, ***P<0.001
Attenuated expression of miR-148a in human IBD and CRC patient tissues
To clarify the clinical relevance of miR-148a expression in human IBD and CRC, we analyzed the miR-148a expression in colonic tissues of human IBD and CRC patients from Zhongnan Hospital and Wuhan Union Hospital (WUH). miR-148a-3p/5p expression was significantly lower in these samples compared with normal colon tissues (Figures 6a and b and Supplementary Figure S6). To confirm the above results, the public data set of CRC cases from the GSE7828 collection was analyzed. In agreement with WUH cases, miR-148a expression was also significantly lower in these CRC samples compared with normal colon tissues (Figure 6c). Thus attenuated miR-148a expression is a common and shared feature of primary human IBD and CRC.

miR-148a is downregulated in human IBD and CRC. (a) Expression of miR-148a-3p/5p in human IBD samples and normal colon tissues. miR-148a-3p/5p RNA levels normalized for U6 were measured by stem-loop reverse transcription quantitative-PCR in RNAs purified from the above tissues. (b) Expression of miR-148a-3p/5p in 31 CRC samples and their adjacent non-cancerous tissues. (c) miR-148a-3p expression in normal colon and CRC tissues from the GSE7828 data set. (a and c) Significance was performed using Wilcoxon signed-rank test
P65 directly regulates miR-148a expression
To investigate why miR-148a expression is downregulated in human IBD and CRC samples, we analyzed the Pri-miR-148a promoter for transcription factor-binding sites and identified three highly conserved NF-κB-binding sites, located within 5 kb upstream of the transcription start site (Figure 7a). The cytokines TNFα and interleukin-1β (IL1β) can activate NF-κB signaling, which is highly activated in and important for CRC progression.5, 19, 20, 21 Thus we first determined whether TNFα or IL1β treatment or P65 affects the Pri-miR-148a expression. As shown in Figures 7b–d and Supplementary Figures S7A and B, TNFα and IL1β treatment and ectopic P65 expression all decreased Pri-miR-148a and miR-148a-3p/5p expression while inhibiting P65 led to the opposite results in HCT116 and RKO colon cancer cells.

P65 and DNMT3A regulate miR-148a expression. (a) Left: Schematic diagram showing the location of NF-κB-binding sites (B1-B3) in the miR-148a promoter. 1–5 are amplicons of ChIP-PCR. Right: quantitative PCR ChIP analyses of P65 and DNMT3A binding to different miR-148a promoter regions in HCT116 and RKO CRC cells transfected with shP65 or shCtrl vectors. (b) Effects of TNFα and IL1β treatment on the expression of endogenous miR-148a and miR-148a-3p/5p in HCT116 and RKO CRC cells. (c) Effects of ectopic P65 and DNMT3A expression on the expression of endogenous Pri-miR-148a and miR-148a-3p/5p in HCT116 and RKO CRC cells. (d) Effects of P65 and DNMT3A inhibition on the expression of endogenous Pri-miR-148a and miR-148a-3p/5p in HCT116 and RKO CRC cells. (e) Alterations in DNA methylation around the miR-148a promoter region were determined by bisulfite genomic sequencing in HCT116 and RKO CRC cells with or without P65 inhibition. PCR products were cloned into a TA cloning vector and 12 randomly selected clones were sequenced. Each row represents a single cloned allele. Filled circles, methylated CpG; open circles, unmethylated CpG. The percentage of methylated CpG sites is shown for each analysis. (f) P65 is necessary for DNMT3A-mediated Pri-miR-148a suppression. Pri-miR-148a and miR-148a-3p/5p RNA levels were normalized for U6 and were measured by stem-loop reverse transcription quantitative-PCR in RNA purified from HCT116 and RKO CRC cells stably expressing exogenous DNMT3A or inhibition of endogenous DNMT3A with or without P65 inhibition. (g) Co-IP of endogenous P65 and DNMT3A from HCT116 CRC cells. (h) The relation among miR-148a promoter methylation, miRNA level of miR-148a-3p/5p, mRNA level of P65 and DNMT3A expression in 310 CRC samples from TCGA data set is shown in a clustered heatmap. A vertical branch shows the expression or promoter methylation pattern of the selected miRNAs or genes in each CRC sample from TCGA data set. (i) P65 and DNMT3A mRNA expression in human IBD and normal colon tissues. (j) P65 and DNMT3A mRNA expression in normal colon and CRC tissues from TCGA data sets. (i and j) Significance was performed using Wilcoxon signed-rank test. Data present mean±S.D. in panels (a, b, c, d and f). *P<0.05, **P<0.01, ***P<0.001
A chromatin IP (ChIP) assay was next conducted and showed that, in HCT116 and RKO cells, P65 binds to the miR-148a promoter (Figure 7a). miR-148a has been previously proposed to be a putative tumor suppressor that is epigenetically silenced by CpG island promoter hypermethylation.15 Therefore, to further study whether P65 affects miR-148a promoter methylation, a promoter CpG island methylation assay using bisulfate sequencing was conducted after P65 inhibition. As shown in Figure 7e, miR-148a promoter CpG islands were found to be hypermethylated in HCT116 and RKO cells but decreased significantly in response to P65 inhibition. These results show that P65 affects the extent of CpG island methylation of the miR-148a promoter and directly participates in inhibiting its expression.
DNMT3A methylates miR-148a promoter CpG islands
In mammals, there are three major DNA methyltransferases (DNMTs): DNMT1, DNMT3A and DNMT3B.22 We performed ChIP experiments to investigate which of these binds the miR-148a promoter to methylate its CpG islands. The results showed that the antibody against DNMT3A efficiently immuno-precipitated the miR-148a promoter both in HCT116 and RKO cells stably transfected with a control short hairpin RNA (shRNA) vector but not with a vector expressing an shRNA against P65. Of note, the binding sites for DNMT3A corresponded to P65-binding sites (Figure 7a and Supplementary Figure S7C). Next we performed Co-IP experiments to determine which DNMT forms a co-repressor complex with P65 to inhibit the miR-148a expression. We tested the exogenous interaction between DNMT and P65 in HEK293 cells. As shown in Supplementary Figure S7D, exogenous DNMT3A interacted strongly with P65. Their endogenous interaction was also confirmed by Co-IP experiments (Figure 7g and Supplementary Figure S7E). Furthermore, we found that residues 189–337 of P65 were required to interact with DNMT3A (Supplementary Figure S7F) while the 1–615 region of DNMT3A was required to interact with P65 (Supplementary Figure S7G). Collectively, these results show that P65 binds the miR-148a promoter in association with DNMT3A, which then mediates specific hypermethylation of nearby CpG islands.
Next, we evaluated the potential role of DNMT3A in regulating miR-148a expression. DNMT3A inhibition significantly increased the Pri-miR-148a and miR-148a-3p/5p expression in HCT116 and RKO cells stably transfected with a shCtrl vector but not with a shP65 vector (Figures 7d and f) whereas enforced DNMT3A expression had the opposite effects (Figure 7c). DNMT3A-mediated miR-148a inhibition was also blocked by P65 inhibition (Figure 7f).
We then tested the effects of P65 and DNMT3A in a miR-148a promoter-driven luciferase reporter assay. Notably, P65 and DNMT3A both repressed the activity of a promoter containing the putative P65-binding sites but had no effect on a promoter with a mutated binding site. In the former case, repression was blocked by P65 inhibition in HCT116 and RKO cells (Supplementary Figures S7H and I).
Finally, we analyzed the relationship among miR-148a promoter methylation, miR-148a-3p/5p, P65 and DNMT3A RNA levels in CRC cases from TCGA data sets and IBD patients. As shown in Figure 7h and Supplementary Figures S7J–M, a negative correlation was determined between the RNA levels of miR-148a-3p/5p and the mRNA levels of P65, DNMT3A or the β-value of miR-148a promoter methylation. These results indicate that P65 and DNMT3A repress miR-148a expression through promoter CpG island methylation.
In contrast to miR-148a expression, P65 and DNMT3A expression were significantly upregulated in IBD and CRC tissues compared with normal colon tissues (Figures 7i and j); indeed, CRC patients with stage III/IV tumors showed significantly higher levels of P65 and DNMT3A compared with patients with stage I/II disease (Supplementary Figure S7N).
miR-148a directly inhibits several upstream regulators of NF-κB and STAT3 signaling
Identifying miR-148a targets is critical for understanding its putative biological functions. Therefore, we used several database prediction tools to identify putative miR-148a-3p and 5p targets in the 3′-UTRs of several upstream regulators of NF-κB and STAT3 signaling, including GP130, IKKα, IKKβ, TNFR2 and IL1R1 (Figure 8a).4, 20, 23, 24, 25, 26, 27 We then evaluated the effect of miR-148a on the expression of the genes encoding these proteins. As expected, miR-148a deficiency increased the expression of endogenous mRNA and protein levels in murine colon (Figures 8b and c), and opposing results were obtained following administration of lentivirus encoding Pri-miR-148a (Supplementary Figures S8A and B). Restoring miR-148a expression in miR-148a KO mice prevented the increase (Supplementary Figures S8C and D).

miR-148a-3p/5p directly targets several upstream regulators of NF-κB and STAT3. (a) Schematic of human GP130, IKKα, IKKβ, TNFR2 and ILR1 3′-untranslated regions (UTRs). The predicted miR-148a-3p/5p-binding sites, which are conserved between human and mouse, are indicated. (b) miR-148a deletion upregulates mRNA level of miR-148a-3p/5p targets. The above targets mRNA levels, normalized for glyceraldehyde 3-phosphate dehydrogenase, were measured by reverse transcription quantitative-PCR (RT-qPCR) in RNA purified from colon tissues of WT and miR-148a KO mice (n=3). (c) miR-148a deletion enhances the protein levels of miR-148a-3p/5p targets. Immuno-blotting for the above targets proteins in colon tissues from WT and miR-148a KO mice. Each lane represents an individual mouse. (d) Luciferase activity of the reporter vector containing the WT or miR-148a-3p and 5p binding mutant 3′-UTR of miR-148a-3p/5p targets was determined after co-transfection with control, Pri-miR-148a or miR-148a-3p sponge expressing vectors. (e) RT-qPCR analysis was performed to detect miR-148a-3p/5p targets RNA level incorporated into RISC derived from colon tissues of WT and miR-148a KO mice. (f) miR-148a-3p/5p targets expression in human IBD and normal colon tissues. (g) The expression of GP130, IKKα, IKKβ, TNFR2 and ILR1 is negatively correlated with miR-148-3p and 5p, respectively. Each point is an individual IBD or normal colon sample. r, Spearman correlation coefficient. (h) The expression of miR-148a-3p/5p and their targets in 323 CRC samples from TCGA data set is shown in a clustered heatmap. A vertical branch shows the expression pattern of the selected miRNAs or genes in each CRC sample from the TCGA data set. (i) Schematic summary. In normal epithelium, miR-148a directly targets GP130, IL1R1, IKKα, IKKβ and TNFR2 transcripts and thus inhibits NF-κB and STAT3 signaling pathways. miR-148a suppression activates NF-κB and STAT3 to promote colitis and colitis-associated tumorigenesis. In a negative feedback loop, NF-KB inhibits miR-148a expression by recruiting DNMT3A to methylate the miR-148a promoter. Data present mean±S.D. in panels (b, d and e). *P<0.05, **P<0.01, ***P<0.001
To further study whether these targets are direct or indirect, miR-148a-3p/5p and luciferase reporter assays were conducted to determine whether the putative miR-148a-3p/5p-binding sites in the 3′-UTRs of target genes are critical for miR-148a-3p/5p-mediated inhibition. Indeed, ectopic miR-148a expression inhibited the activity of human 3′-UTR reporter vector in dual luciferase reporter assays, while mutations in miR-148a-3p/5p-binding sites abrogated this inhibition. Repression of miR-148a-3p and 5p using its sponge and inhibitor, respectively, increased the activity of 3′-UTR reporter vectors in the same assays, while mutation in miR-148a-3p/5p-binding site abrogated this upregulation (Figure 8d and Supplementary Figure S8E). Thus the mRNAs of miR-148a-3p/5p targets are directly regulated by miR-148a-3p/5p via seed-matching sequences. We then used Co-IP with an antibody to AGO-2 to precipitate RNAs associated with the RNA-induced Silencing Complex (RISC). As shown in Figure 8e, the above target RNAs were significantly enriched in WT colon extracts versus those from miR-148a KO mice. Taken together, these data indicate that miR-148a-3p/5p directly inhibit these targets by binding to the predicted binding sites in their 3′-UTRs.
Finally, we analyzed mRNA levels of these targets in human IBD and normal tissues. As shown in Figure 8f, all transcripts were significantly overexpressed in IBD samples compared with normal colon tissues. Furthermore, a negative correlation was seen between the mRNA level of these targets and the expression of miR-148a-3p/5p, respectively (Figures 8g and h). Collectively, our results indicate that miR-148a negatively regulates the development and severity of colitis and colitis-associated tumorigenesis in both mouse models and human patients.
Discussion
An important function for miRNAs in the intestinal epithelium has been previously reported in mice lacking Dicer1, the enzyme responsible for miRNA processing. Intestinal barrier function was found to be impaired in these mice, resulting in intestinal inflammation with lymphocyte and neutrophil infiltration.28
miRNAs other than miR-148a have been reported to modulate inflammation, colitis-associated tumorigenesis and the pathogenesis of IBDs.10, 11, 29, 30, 31, 32, 33 For example, miR-192, which inhibits chemokine production, is decreased in tissue samples from IBD patients.10 IL6 upregulates STAT3-mediated transcription of miR-214 in colon tissues, which reduces the levels of PDLIM2 and PTEN, increases AKT phosphorylation and activates NF-κB. The activity of this circuit correlates with disease activity in patients with IBD and progression to colorectal cancer.10 miR-223 was also reported to be upregulated in IBD and its activity was regulated through the IL23 pathway. Antagomir inhibition of miR-223 reactivated CLDN8 and improved a number of signs associated with TNBS-induced colitis in mice. This suggested that miR-223 is a novel mediator of the crosstalk between the IL23 signaling pathway and CLDN8 in the development of IBD.31 miR-301a expression is increased in IBD patients and miR-301a-deficient mice are resistant to DSS-induced colitis.32 miR-665 is also upregulated in mucosal colonic biopsies of IBD. miR-665 negatively regulated XBP1 and ORMDL3 by directly targeting their 3′-UTRs and indirectly upregulating JNK. Upregulation of miR-665 promoted apoptosis and DSS-induced colitis, while inhibition of miR-665 had the opposite effects.33
We have demonstrated here that miR-148a downregulation during CRC development is a consequence of CpG island hypermethylation. The resultant loss of miR-148a stabilizes its downstream target transcripts and this, in turn, upregulates NF-κB and STAT3 signaling.4, 20, 23, 24, 25, 26, 27 The resultant pro-inflammatory environment activates P65, which together with DNMT3A, is recruited to the miR-148a promoter, thereby promoting CpG island hypermethylation. Together, these factors constitute an important feedback loop for NF-κB activation, inflammation and cytokine production (Figure 8i). Based on the findings reported here, similar mechanisms likely operate in human IBD and CRC as well. However, it will be necessary in future work to examine larger numbers of patient-derived samples in order to establish the universality of these findings. Other questions that remain unanswered include whether these findings apply to all forms of IBD, whether miR-148a has additional non-cytokine-related targets that may influence IBD and CRC pathogenesis and how miR148a and other of the miRNAs discussed above interact to influence disease pathogenesis.
Mice lacking negative regulators of NF-κB such as A20 and NLRP12 are also more susceptible to colitis and colitis-associated cancer.3, 6, 34, 35, 36 Together with our current work, these findings indicate that NF-κB signaling is critical for driving colitis and colitis-associated cancer and that its inflammation- and cancer-modulating activities are regulated by what are likely to be multiple tightly controlled and interdependent transcriptional and posttranscriptional processes.
In some cases, the therapeutic delivery of miRNAs or their inhibitors in vivo can effectively suppress colitis-associated colorectal cancer.11, 37, 38 Our findings show that restoring miR-148a expression reduced the severity of inflammation, decreased NF-κB and STAT3 activation and repressed both spontaneous and carcinogen-induced colon cancer development in mice while restoring miR-148a expression. Further studies using miRNA mimetics or other chemically modified miRNAs that allow for prolonged miR-148-like expression may help to determine the potentially utility of such agents for therapeutic trials.
In summary, our study has demonstrated a critical role for miR-148a in murine models of colitis and colitis-associated tumorigenesis. Importantly, our observations in clinical IBD and CRC samples strongly suggest that a similar role for miR-148a exists in humans. Therapeutic regulation of miR-148a expression could reduce the risk of colitis-associated cancer development in susceptible individuals.
Materials and methods
CRC and IBD patient specimens
CRC specimens were obtained from patients undergoing tumor resection and samples from human IBD patients were taken at the time of endoscopy. Informed consent was obtained at Zhongnan Hospital and the WUH (Wuhan, China). The diagnosis of samples was confirmed in each case by histological review.
Mice
All mouse studies were approved by the Animal Care Committee of Wuhan University. Apcmin/+ mice were ordered from Model Animal Research Center (Nanjing, China). To generate miR-148a KO mice, briefly, a targeting vector with 953 base pairs (bp) of genomic sequence containing mouse miR-148a was replaced with a neomycin expression cassette and was electroporated into mouse ES cells on the C57BL/6 background at The Model Animal Research Center, Nanjing University. To generate miR-148a+/−Apcmin/+ mice, miR-148a KO mice were crossed with Apcmin/+ mice.
DSS-induced colitis and AOM/DSS-induced CAC mouse models
The DSS and AOM/DSS-induced colitis and CAC mouse models followed previously described protocols.3, 6 Briefly, for colitis, WT or miR-148a KO mice were provided with 3.5% DSS (216011080, MW=36-50KD; MP Biomedicals, Santa Ana, CA, USA) in drinking water for 6 days, followed by regular drinking water. For CAC, 2-month-old WT or miR-148a KO mice were intraperitoneally injected once with 10 mg/kg AOM (A5486, Sigma, St. Louis, USA), followed by three rounds of 2.5% DSS in drinking water for 7 days.
Bone marrow chimeras
Bone marrow chimeric mice were established as described.3 Briefly, 2-month-old WT (CD45.1) and miR-148a KO (CD45.2) mice were lethally irradiated and intravenously injected with 107 bone marrow cells isolated from either CD45.1+ WT or miR-148a KO mice 24 h after body irradiation. Recipient mice were housed for 2 months to fully reconstitute bone marrow before being treated with DSS or AOM/DSS.
Administration of lentivirus-expressing miR-148a in AOM/DSS-induced mouse CRC and Apcmin/+ mice
Lentiviral injections were performed as described previously.39 For administration of lentivirus in the AOM/DSS-induced CRC mice model, female C57BL/6 mice were first treated with AOM/DSS and then randomly distributed into 2 groups of 10 animals each, which were then treated with lentivirus-expressing pHAGE-GFP miR-148a. Concentrated virus in a volume of 100 μl was administered intraperitoneally twice weekly for 3 weeks. At day 120, tumor burdens were evaluated. For administration of lentivirus in the Apcmin/+ model, 3-week-old female Apcmin/+ mice were randomly distributed into 2 groups of 10 animals each, administered the above viruses on weeks 3–4 and continued twice weekly for 3 weeks. Tumor burdens were evaluated on week 14 by enumerating all neoplasms >1 mm in size. Tumor and adjacent normal colon tissues were harvested for RNA and protein assays as well as for pathological studies.
Cytokine assays from serum and colon tissues
The levels of serum and colon tissue cytokines were determined with commercially available ELISA kits using the conditions recommended by the supplier (Shanghai Jijin Chemistry Technology, Shanghai, China).
Bioinformatics and statistics
Online softwares miRanda (http://www.miRanda-im.org/), miRDB (http://www.miRdb.org/), PICTAR5 (http://pictar.mdc-berlin.de/), miRwalk (http://www.ma.uni-heidelberg.de/apps/zmf/miRwalk/) and Targetscan (http://www.targetscan.org/) were used to predict miR-148a targets.40 CRC data sets were downloaded from The Cancer Genome Atlas (TCGA) data portal (http://tcga-data.nci.nih.gov). Gene expression was assessed in human CRC tissues from the TCGA RNA-seq data set. Experimental data were analyzed using unpaired, two-tailed Student’s t-test, Wilcoxon signed-rank test or Mann–Whitney U-test, and the correlation was analyzed using a Spearman rank correlation test. Kaplan–Meier curves for survival were analyzed with the GraphPad software (Version5.01, Graphpad software, San Diego, CA, USA) using log-rank test. Results are represented as the mean±S.D. or S.E.M. and P<0.05 was considered statistically significant.35
References
Haggar FA, Boushey RP . Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg 2009; 22: 191–197.
Terzic J, Grivennikov S, Karin E, Karin M . Inflammation and colon cancer. Gastroenterology 2010; 138: 2101–2114.
Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 2012; 36: 742–754.
Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009; 15: 91–102.
Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013; 23: 634–646.
Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011; 20: 649–660.
Boulard O, Kirchberger S, Royston DJ, Maloy KJ, Powrie FM . Identification of a genetic locus controlling bacteria-driven colitis and associated cancer through effects on innate inflammation. J Exp Med 2012; 209: 1309–1324.
Chiba T, Marusawa H, Ushijima T . Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 2012; 143: 550–563.
Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE et al. Normalizing microbiota-induced retinoic acid deficiency stimulates protective CD8(+) T cell-mediated immunity in colorectal cancer. Immunity 2016; 45: 641–655.
Kalla R, Ventham NT, Kennedy NA, Quintana JF, Nimmo ER, Buck AH et al. MicroRNAs: new players in IBD. Gut 2015; 64: 504–517.
Koukos G, Polytarchou C, Kaplan JL, Morley-Fletcher A, Gras-Miralles B, Kokkotou E et al. MicroRNA-124 regulates STAT3 expression and is down-regulated in colon tissues of pediatric patients with ulcerative colitis. Gastroenterology 2013; 145: 842–852.
Liu X, Zhan Z, Xu L, Ma F, Li D, Guo Z et al. MicroRNA-148/152 impair innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMKIIα. J Immunol 2010; 185: 7244–7251.
Patel V, Carrion K, Hollands A, Hinton A, Gallegos T, Dyo J et al. The stretch responsive microRNA miR-148a-3p is a novel repressor of IKBKB, NF-κB signaling, and inflammatory gene expression in human aortic valve cells. FASEB J 2015; 29: 1859–1868.
Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM et al. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol 2016; 17: 433–440.
Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 2008; 105: 13556–13561.
Kesselring R, Glaesner J, Hiergeist A, Naschberger E, Neumann H, Brunner SM et al. IRAK-M expression in tumor cells supports colorectal cancer progression through reduction of antimicrobial defense and stabilization of STAT3. Cancer Cell 2016; 29: 684–696.
Koblansky AA, Truax AD, Liu R, Montgomery SA, Ding S, Wilson JE et al. The innate immune receptor NLRX1 functions as a tumor suppressor by reducing colon tumorigenesis and key tumor-promoting signals. Cell Rep 2016; 14: 2562–2575.
Hu S, Peng L, Kwak YT, Tekippe EM, Pasare C, Malter JS et al. The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep 2015; 13: 1922–1936.
Chen R, Li M, Zhang Y, Zhou Q, Shu HB . The E3 ubiquitin ligase MARCH8 negatively regulates IL-1β-induced NF-κB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proc Natl Acad Sci USA 2012; 109: 14128–14133.
Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ et al. NF-kappaB-independent role of IKKα/IKKβ in preventing ripk1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell 2015; 60: 63–76.
Li Q, Yan J, Mao AP, Li C, Ran Y, Shu HB et al. Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β-triggered NF-κB activation by targeting TAK1 for K63-linked polyubiquitination. Proc Natl Acad Sci USA 2011; 108: 19341–19346.
Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet 2015; 47: 469–478.
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ et al. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004; 118: 285–296.
Hamilton KE, Simmons JG, Ding S, Van Landeghem L, Lund PK . Cytokine induction of tumor necrosis factor receptor 2 is mediated by STAT3 in colon cancer cells. Mol Cancer Res 2011; 9: 1718–1731.
Koliaraki V, Pasparakis M, Kollias G . IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J Exp Med 2015; 212: 2235–2251.
Stoffels B, Hupa KJ, Snoek SA, van Bree S, Stein K, Schwandt T et al. Postoperative ileus involves interleukin-1 receptor signaling in enteric glia. Gastroenterology 2014; 146: 176–187.
Xiao H, Yin W, Khan MA, Gulen MF, Zhou H, Sham HP et al. Loss of single immunoglobulin interlukin-1 receptor-related molecule leads to enhanced colonic polyposis in Apc(min) mice. Gastroenterology 2010; 139: 574–585.
McKenna LB, Schug J, Vourekas A, McKenna JB, Bramswig NC, Friedman JR et al. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology 2010; 139: 1654–1664 1664.e1651.
Shi C, Yang Y, Xia Y, Okugawa Y, Yang J, Liang Y et al. Novel evidence for an oncogenic role of microRNA-21 in colitis-associated colorectal cancer. Gut 2016; 65: 1470–1481.
Neudecker V, Haneklaus M, Jensen O, Khailova L, Masterson JC, Tye H et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J Exp Med 2017; 214: 1737–1752.
Wang H, Chao K, Ng SC, Bai AH, Yu Q, Yu J et al. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol 2016; 17: 58.
He C, Yu T, Shi Y, Ma C, Yang W, Fang L et al. MicroRNA 301A promotes intestinal inflammation and colitis-associated cancer development by inhibiting BTG1. Gastroenterology 2017; 152: 1434–1448.
Li M, Zhang S, Qiu Y, He Y, Chen B, Mao R et al. Upregulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell Death Dis 2017; 8: e2699.
Vereecke L, Sze M, Mc Guire C, Rogiers B, Chu Y, Schmidt-Supprian M et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med 2010; 207: 1513–1523.
Fang L, Lu W, Choi HH, Yeung SC, Tung JY, Hsiao CD et al. ERK2-dependent phosphorylation of CSN6 is critical in colorectal cancer development. Cancer Cell 2015; 28: 183–197.
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011; 145: 745–757.
Chivukula RR, Shi G, Acharya A, Mills EW, Zeitels LR, Anandam JL et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell 2014; 157: 1104–1116.
Valeri N, Braconi C, Gasparini P, Murgia C, Lampis A, Paulus-Hock V et al. MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell 2014; 25: 469–483.
Lamas B, Richard ML, Leducq V, Pham HP . CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 2016; 22: 598–605.
Han H, Li W, Shen H, Zhang J, Zhu Y, Li Y . microRNA-129-5p, a c-Myc negative target, affects hepatocellular carcinoma progression by blocking the Warburg effect. J Mol Cell Biol 2016; 8: 600–610.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81772609, 81472549, 31521091) and National Key R&D Program of China (2016YFC1302300, 2014CB910600). We thank Professor Hongbing Shu (Wuhan University, Wuhan, China) for providing reagents and the Model Animal Research Center of Nanjing University for miR-148a KO mice.
Author contributions
YZ, LG, YL and YL designed the study, analyzed and interpreted the data and wrote the manuscript. YZ, LG and YL performed most of the experiments. BZ and EP provided the reagents. JZ, FZ and QZ provided the human colon samples. YL and EP wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by R De Mari
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Zhu, Y., Gu, L., Li, Y. et al. miR-148a inhibits colitis and colitis-associated tumorigenesis in mice. Cell Death Differ 24, 2199–2209 (2017). https://doi.org/10.1038/cdd.2017.151
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.151
This article is cited by
-
Expression profiles of hsa-miR-148a-3p and hsa-miR-125b-5p in human breast milk and infant formulae
International Breastfeeding Journal (2022)
-
Systematic characterization of seed overlap microRNA cotargeting associated with lupus pathogenesis
BMC Biology (2022)
-
Updated immunomodulatory roles of gut flora and microRNAs in inflammatory bowel diseases
Clinical and Experimental Medicine (2022)
-
Transcriptome analysis of potential candidate genes and molecular pathways in colitis-associated colorectal cancer of Mkp-1-deficient mice
BMC Cancer (2021)
-
MicroRNA: Could It Play a Role in Bovine Endometritis?
Inflammation (2021)
