
Abstract
ABT-737 is a pharmacological inhibitor of the anti-apoptotic activity of B-cell lymphoma-extra large (Bcl-xL) protein; it promotes apoptosis of cancer cells by occupying the BH3-binding pocket. We have shown previously that ABT-737 lowers cell metabolic efficiency by inhibiting ATP synthase activity. However, we also found that ABT-737 protects rodent brain from ischemic injury in vivo by inhibiting formation of the pro-apoptotic, cleaved form of Bcl-xL, ΔN-Bcl-xL. We now report that a high concentration of ABT-737 (1 μM), or a more selective Bcl-xL inhibitor WEHI-539 (5 μM) enhances glutamate-induced neurotoxicity while a low concentration of ABT-737 (10 nM) or WEHI-539 (10 nM) is neuroprotective. High ABT-737 markedly increased ΔN-Bcl-xL formation, aggravated glutamate-induced death and resulted in the loss of mitochondrial membrane potential and decline in ATP production. Although the usual cause of death by ABT-737 is thought to be related to activation of Bax at the outer mitochondrial membrane due to sequestration of Bcl-xL, we now find that low ABT-737 not only prevents Bax activation, but it also inhibits the decline in mitochondrial potential produced by glutamate toxicity or by direct application of ΔN-Bcl-xL to mitochondria. Loss of mitochondrial inner membrane potential is also prevented by cyclosporine A, implicating the mitochondrial permeability transition pore in death aggravated by ΔN-Bcl-xL. In keeping with this, we find that glutamate/ΔN-Bcl-xL-induced neuronal death is attenuated by depletion of the ATP synthase c-subunit. C-subunit depletion prevented depolarization of mitochondrial membranes in ΔN-Bcl-xL expressing cells and substantially prevented the morphological change in neurites associated with glutamate/ΔN-Bcl-xL insult. Our findings suggest that low ABT-737 or WEHI-539 promotes survival during glutamate toxicity by preventing the effect of ΔN-Bcl-xL on mitochondrial inner membrane depolarization, highlighting ΔN-Bcl-xL as an important therapeutic target in injured brain.
Similar content being viewed by others


Main
B-cell lymphoma-extra large (Bcl-xL) is a member of the Bcl2 family of proteins. Anti-apoptotic Bcl-xL1, 2 prevents homo-oligomerization of pro-apoptotic family members, such as Bax and Bak, and directly inhibits activators of Bax and Bak.3, 4, 5, 6, 7 Bcl-xL also plays a neuroprotective role by regulating neuronal outgrowth,8 enhancing cellular metabolism9, 10, 11 and promoting synaptogenesis and synaptic vesicle endocytosis.12, 13 Bcl-xL is localized both to the mitochondrial outer membrane and to the inner membrane. It binds directly to the F1 of F1FO ATP synthase, improving metabolic efficiency by preventing H+ ion leak across the inner membrane.9, 10, 14, 15
In contrast to its protective functions, Bcl-xL is also capable of playing a pro-death role.16 ΔN-Bcl-xL, the caspase-dependent N-terminal cleaved product of Bcl-xL,16, 17, 18 is produced in the brain during ischemia19 and activates large conductance mitochondrial channel activity20, 21 similar to the activity of oligomerized Bax, but implicating the activity of ΔN-Bcl-xL itself. Blocking formation of ΔN-Bcl-xL or the mitochondrial channel activity of ΔN-Bcl-xL with ABT-737 rescues neurons from ischemic death.19, 21, 22 A mouse expressing a cleavage resistant form of Bcl-xL is protected from neuronal ischemic damage.19 What is not known, however, is whether ΔN-Bcl-xL, similarly to full-length Bcl-xL, functions at the mitochondrial inner membrane.
To examine this question, we used ABT-737, a mimetic of BH3-only proteins, that binds to Bcl-xL with high affinity and antagonizes its actions.23, 24 ABT-737 facilitates apoptosis similarly to pro-death BH3-only proteins25 and activates multi-domain pro-apoptotic proteins such as Bax or Bak.26, 27, 28 ABT-737 has been tested as an anti-tumorigenic agent. Studies on the neurological actions of ABT-737 are surprisingly contradictory, however. Although its pre-synaptic injection inhibits physiological mitochondrial channel activity and slows recovery of neurotransmission after high frequency synaptic stimulation, it also prevents hypoxia-induced synaptic dysfunction.29 Treatment with ABT-737 lowers the efficiency of neuronal metabolism by increasing mitochondrial inner membrane leak.9, 10 Nevertheless, administration of ABT-737 in vivo rescues hippocampal neurons from ischemia-induced cell death.19 Thus, we hypothesize that ABT-737 is not a simple inhibitory drug, but may act through multiple pathways involved in neuronal survival or death.
In this study, we find that a low (nanomolar) concentration of ABT-737 prevents depolarization of mitochondria by ΔN-Bcl-xL and attenuates the appearance of ΔN-Bcl-xL in neurons undergoing glutamate-induced toxicity. Low ABT-737 therefore rescues neurons from death. In contrast, high (micromolar) ABT-737 inhibits the actions of full-length Bcl-xL and markedly enhances Bcl-xL proteolysis, exacerbating mitochondrial and cellular damage from glutamate-induced excitotoxicity. We find an important target of ΔN-Bcl-xL is mitochondrial permeability transition pore (mPTP) since ΔN-Bcl-xL-induced mitochondrial depolarization is equally sensitive to cyclosporine A (CsA) or to low-ABT-737. We suggest that ABT-737 either protects against or enhances mPTP-dependent cell death depending on its concentration.
Results
Bcl-xL inhibitors ABT-737 and WEHI-539 aggravate glutamate-induced neurotoxicity
To test how inhibition of Bcl-xL leads to cell dysfunction and death, we assayed the Bcl-xL inhibitor ABT-737 at two different concentrations and studied cell death in response to glutamate toxicity. During preliminary screening, we found that 5 μM glutamate did not affect neuronal viability, but 50 μM glutamate caused irreversible death in hippocampal cultures after 24 h incubation, thus we chose 20 μM to induce mild excitotoxicity (Figure 1a). Glutamate-induced neurotoxicity was blocked by application of the NMDA receptor antagonist, MK-801, therefore, glutamate-induced cell death in our in vitro system is dependent on activation of NMDA receptors (Figure 1b). Previous studies reported that 1 μM ABT-737 effectively inhibited full-length Bcl-xL function in cultured cells but did not cause cell death in the absence of death stimuli.9, 13 Primary hippocampal neurons were thus treated with 1 μM ABT-737, 20 μM glutamate or a combination of both, and cells were assayed 24 h after stimulation. There was no significant cell death in the cultures in non-treated, vehicle treated or ABT-737 (1 μM) treated cells as measured by propidium iodide (PI) uptake into neurons, a marker for late apoptotic and necrotic death. In contrast, glutamate significantly increased the number of PI-positive cells and high ABT-737 exacerbated this effect (Figures 1c and d). Calcein retention measures cell survival; the combination of high ABT-737 and glutamate caused a significant deterioration in calcein retention (Figures 1c and e). LDH release marks loss of cell membrane integrity and allows for population measurements of cytotoxicity. High ABT-737 itself did not cause LDH leakage, but it aggravated glutamate-induced LDH release (Figure 1f). ABT-737 is reported to inhibit the pro-survival function of both Bcl-xL and Bcl-2.23 To determine if our effects could be caused by specific inhibition of Bcl-xL alone, we applied a selective Bcl-xL inhibitor, WEHI-539.30 During preliminary screening, we found that 5 μM WEHI-539 served effectively as the ‘high’ concentration (Supplementary Figure 1). Similarly to ABT-737, 5 μM of WEHI-539 alone did not alter LDH leakage, but it potentiated glutamate-induced LDH leakage, confirming that specific inhibition of Bcl-xL enhances the vulnerability of neurons to excitotoxic damage (Figure 1g). To confirm the role of Bcl-xL, we also quantified the mRNA levels of Bcl-xL and Bcl2 in primary hippocampal neurons. Three-week-old mature primary hippocampal neurons contain significantly higher levels of Bcl-xL gene expression compared to those of Bcl2 (Figure 1h), therefore Bcl-xL is the main target of ABT-737/WEHI-539 in our system.

ABT-737/WEHI-539 aggravates glutamate-induced neuronal toxicity. Primary hippocampal neurons were treated with glutamate (5-50 μM) for 24 h. (a) LDH release (n=3 plates from one culture). Neurons were treated with MK801 (0.1 μM), glutamate (20 μM) or a combination of both for 24 h. (b) LDH release (n=3 independent cultures). (c-f) Neurons were treated with ABT-737 (1 μM), glutamate (20 μM) or a combination of both for 24 h. (c) PI and calcein-stained neurons. Green: calcein; red: PI. Scale bar=20 μm. (d) The number of PI-positive cells (n=3 independent cultures). (e) Calcein retention (n=3 independent cultures). (f) LDH release (n=3 independent cultures). (g) Primary hippocampal neurons were treated with WEHI-539 (5 μM), glutamate (20 μM) or a combination of both for 24 h. Bar graph shows LDH release level (n=3 independent cultures). (h) Quantification of Bcl2 or Bcl-xL mRNA level (n=3 independent cultures). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, one-way ANOVA
ABT-737 compromises mitochondrial membrane potential and Bcl-xL inhibition decreases ATP levels in neurons
Although 1 μM of ABT-737 did not influence neuronal death or survival, we previously reported that the same concentration of ABT-737 lowered ATPase activity and increased inner mitochondrial membrane leak, resulting in metabolic compromise.9 Thus, we tested if ABT-737 altered mitochondrial function with or without glutamate challenge. At 6 h incubation, neuronal mitochondrial membrane potential measured by tetramethylrhodamine (TMRM) fluorescence was decreased and it decreased further at 16 h incubation (Figure 2a). We found a significant decline in TMRM fluorescence in either the ABT-737 or glutamate-treated neurons at 6 h incubation (Figures 2b and c). The combination of ABT-737 and glutamate exacerbated the loss of TMRM (Figures 2b and c). To observe metabolic differences early in toxicity, neurons were lysed after 8 h incubation to measure ATP levels. ATP was significantly decreased in all groups compared to vehicle-treated control, and treatment with high ABT-737 in glutamate-challenged neurons demonstrated the lowest level of ATP (Figure 2d). Neurons treated with WEHI-539 consistently showed a low level of ATP production during glutamate exposure (Figure 2e), indicating that inhibition of Bcl-xL alters cellular metabolism to predispose to neuronal death during excitotoxic stimulation.

High concentration of ABT-737 compromises metabolic function. (a-d) Primary hippocampal neurons were treated with 20 μM glutamate for 1-16 h. (a) TMRM intensity (n=3 independent cultures). (b-d) Neurons were treated with ABT-737 (1 μM), glutamate (20 μM) or a combination of both. (b) TMRM (5 nM) was added to the cell culture medium at 6 h of incubation. Scale bar=10 μm. (c) TMRM intensity (n=3 independent cultures). (d) ATP level (n=3 independent cultures) at 8 h. (e) Primary hippocampal neurons were treated with WEHI-539 (5 μM), glutamate (20 μM) or a combination of both for 8 h. Bar graph shows ATP level (n=3 independent cultures). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, one-way ANOVA
Low concentration of ABT-737 or WEHI-539 rescues neurons from glutamate-induced death
Despite the inhibitory role of ABT-737 on neuronal viability, energy metabolism and mitochondrial function, we previously found that injection of ABT-737 protected rodent hippocampus against four vessel occlusion-induced global ischemia in vivo.19 This was an unexpected and contradictory result compared to our previous bioenergetic findings,9 but we hypothesized that the mechanism of ABT-737 in vivo and in vitro may not be comparable. The rat brain contains over 200 μl cerebrospinal fluid.31 Since the hippocampus is situated adjacent to the lateral ventricle, injected ABT-737 may be easily washed out after stereotaxic delivery. In vitro, ABT-737 is trapped in the dish and continuously bathes the neurons. Thus the bio-available concentration of ABT-737 may be lower in the hippocampus in vivo. Consequently, a lower concentration of ABT-737 may have a different effect on neuronal survival. After preliminary screening, we used 10 nM ABT-737, a 100-fold lower concentration than 1 μM, to protect neurons from glutamate challenge. ABT-737 (10 nM) alone did not influence PI positivity, calcein intensity or LDH leakage compared to vehicle-treated groups. However, cells exposed to 10 nM ABT-737 with glutamate, compared to glutamate alone, showed a significantly reduced PI-positive cell number (Figures 3a and b), an increased level of calcein retention (Figures 3a and c) and a lower level of released LDH (Figure 3d). Low concentrations of WEHI-539 also attenuated glutamate-induced neurotoxicity (Figure 3e and Supplementary Figure S1), consistent with specific effects of the reagents on Bcl-xL compared to Bcl-2.

Low concentration of ABT-737 protects hippocampal neurons from glutamate-induced death. (a-d) Primary hippocampal neurons were treated with ABT-737 (10 nM), glutamate (20 μM) or a combination of both for 24 h. (a) PI and calcein-stained neurons. Green: calcein; red: PI. Scale bar=20 μm. (b) Number of PI-positive cells (n=3 independent cultures). (c) Calcein retention (n=4 independent cultures). (d) LDH release (n=9 independently treated plates from three cultures). (e) Primary hippocampal neurons were treated with WEHI-539 (10 nM), glutamate (20 μM) or a combination of both for 24 h. Bar graphs show LDH release (n=3 independent cultures). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, one-way ANOVA
Low concentration of ABT-737/WEHI-539 improves cell metabolism during excitotoxic challenge
To test if the lower concentration of ABT-737 protects mitochondrial membrane potential to enhance neuronal survival, we treated TMRM-exposed cells with 10 nM ABT-737, glutamate or a combination of both. Low ABT-737 did not influence mitochondrial potential. However, 10 nM ABT-737 treatment significantly mitigated glutamate-induced loss of mitochondrial membrane potential (Figures 4a and b). Either 10 nM ABT-737 or WEHI-539 reduced the loss of ATP in glutamate-treated neurons (Figures 4c and d).

Low concentration of ABT-737 augments mitochondrial TMRM uptake. Primary hippocampal neurons were treated with ABT-737 (10 nM), glutamate (20 μM) or a combination of both. TMRM (5 nM) was added into cell the culture medium at 6 h of incubation. (a) TMRM stained neurons. Scale bar=10 μm. (b) TMRM intensity (n=10 independently treated experiments from four cultures). (c) ATP level (n=3 independent cultures) at 8 h. (d) Primary hippocampal neurons were treated with WEHI-539 (10 nM), glutamate (20 μM) or a combination of both for 8 h. Bar graph shows ATP level (n=4 independent cultures). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, one-way ANOVA
ΔN-Bcl-xL-induced mitochondrial dysfunction is prevented by low ABT-737
We previously reported that ABT-737 inhibits actions of both the anti-apoptotic full-length Bcl-xL and its pro-apoptotic fragment ΔN-Bcl-xL.19, 29 To elucidate if the neuroprotective property of ABT-737 is afforded by inhibition of the effects of ΔN-Bcl-xL, we determined the binding properties of ABT-737 to ΔN-Bcl-xL by generating a model taking into account the known crystal structure of full-length Bcl-xL (containing the BH4 domain) and ΔN-Bcl-xL (lacking the BH4 domain). We compared ABT-737 binding to the two different structures. We found that the N-terminal cleavage of Bcl-xL does not disturb complex formation of ABT-737 with its binding site within Bcl-xL (Supplementary Figure S2), suggesting that ABT-737 binds effectively within the same hydrophobic grove in either Bcl-xL or ΔN-Bcl-xL.
We next studied the direct effect of purified ΔN-Bcl-xL protein on TMRM fluorescence of isolated mitochondria from rat brains to determine if ΔN-Bcl-xL directly depolarizes brain mitochondria. We verified the quality of purified recombinant Bcl-xL and ΔN-Bcl-xL proteins with anti-Bcl-xL (Figure 5a) and anti-ΔN-Bcl-xL antibodies (Figure 5b), respectively. Since our custom-made ΔN-Bcl-xL antibody shares a common binding peptide sequence with full-length Bcl-xL, we verified the affinity of each antibody to its target protein in hippocampal lysates (Figure 5c).

ΔN-Bcl-xL decreases mitochondrial potential, prevented by CsA and ABT-737. (a) Immunoblot of purified Bcl-xL and ΔN-Bcl-xL proteins using anti-Bcl-xL antibody. (b) Immunoblot of purified Bcl-xL and ΔN-Bcl-xL proteins using anti-ΔN-Bcl-xL antibody. (c) Immunoblot of hippocampal neuron lysate using specific antibodies shown in a and b. (d) Immunoblot showing purity of isolated mitochondria. (e-g) TMRM fluorescence intensity in isolated mitochondria (see text for details; n=3-12 wells each from 12 rat brains). (h) Immunoblot showing cytochrome c release from isolated mitochondria (n=3). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, one-way ANOVA
To test the effect of ΔN-Bcl-xL on isolated brain mitochondria, we confirmed the purity of isolated subcellular fractions (Figure 5d). Brain mitochondria were then polarized by the addition of malate, glutamate and ADP. Isolated mitochondria were treated with trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP); we found a concentration-dependent decline of TMRM fluorescence indicative of mitochondrial potential depolarization (Figure 5e). Twenty minutes incubation with ΔN-Bcl-xL protein significantly lowered mitochondrial potential, a result that was effectively prevented by CsA, an inhibitor of mPTP opening (Figure 5f) or full-length Bcl-xL (Supplementary Figure 3). The loss of mitochondrial potential and the prevention of this loss by CsA suggest a role for mPTP in ΔN-Bcl-xL-induced depolarization. To understand the effect of ABT-737 on ΔN-Bcl-xL-induced mitochondrial membrane potential loss, we incubated ΔN-Bcl-xL protein with either 100 nM or 5 μM of ABT-737 (higher concentrations of ABT-737 were compared to cell culture studies due to the large mitochondrial protein amounts) and mixed with the mitochondria. After 20 min incubation, ΔN-Bcl-xL protein significantly lowered mitochondrial potential. Low ABT-737 (100 nM) rescued ΔN-Bcl-xL-induced loss of mitochondrial potential, whereas co-treatment with high ABT-737 (5 μM) did not rescue mitochondrial potential (Figure 5g) most likely because of inhibition of endogenous full-length Bcl-xL. ΔN-Bcl-xL protein also significantly enhanced cytochrome c release from isolated mitochondria, whereas co-treatment with low ABT-737 inhibited ΔN-Bcl-xL-induced cytochrome c release (Figure 5h).
Glutamate increases ΔN-Bcl-xL formation, prevented by low ABT-737
To understand if excitotoxic stimulation induces endogenous ΔN-Bcl-xL formation in our in vitro system, we treated hippocampal neurons with glutamate for varying times: 1, 6 or 16 h. ΔN-Bcl-xL started to appear at 6 h, was highly expressed at 16 h (Figure 6a) similarly to the time course of expression of activated Bax (Figure 6b). We have previously reported that the pan-specific caspase inhibitor, zVAD, blocked the appearance of ΔN-Bcl-xL.21 In our current system, we used a specific caspase 3 inhibitor, Ac-DEVD-CHO (Figures 6c and d), which effectively prevented the formation of ΔN-Bcl-xL.

ABT-737 regulates appearance of ΔN-Bcl-xL and activation of Bax. (a and b) Primary hippocampal neurons were treated with 20 μM glutamate for 1, 6 or 16 h. Immunoblot of cell lysate showing time course of appearance of ΔN-Bcl-xL (a) and active bax (b). (c) Primary hippocampal neurons were treated with Ac-DEVD-CHO (2-50 μM) and glutamate (20 μM) for 16 h. (d) Neurons were treated with Ac-DEVD-CHO (10 μM), glutamate (20 μM) or a combination of both for 16 h. Immunoblot and group data show level of ΔN-Bcl-xL (n=4, independent cultures). (e) Primary hippocampal neurons were treated with ABT-737 (1 μM), glutamate (20 μM) or a combination of both for 16 h. Immunoblot and group data show level of ΔN-Bcl-xL and BclxL (e), active bax and total bax (e′), cytochrome c and active caspase 3 (e′′) (n=3-5 independent cultures). (f) Primary hippocampal neurons were treated with low ABT-737 (10 nM), glutamate (20 μM) or combination of both for 16 h. Immunoblot and group data show level of ΔN-Bcl-xL and BclxL (f), active bax and total bax (f′) and cytochrome c and active caspase 3 (f′′) (n=3-5 independent cultures). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, one-way ANOVA
Since we found that high ABT-737 with glutamate co-treatment eliminates neurons by 24 h incubation, making it extremely challenging to collect adequate levels of protein for western blotting, we chose 16 h glutamate exposure. We observed basal levels of expression of ΔN-Bcl-xL in both vehicle- and ABT-737-treated groups without significant difference. However, high ABT-737 clearly augmented ΔN-Bcl-xL levels in glutamate-treated neurons (Figure 6e). Interestingly, treatment with high ABT-737 decreased the level of full-length Bcl-xL protein expression (Figure 6e), and glutamate did not further promote this change. The change in level of full-length Bcl-xL could be caused by decreased production or increased degradation of the protein. To differentiate between these, we repeated the experiment in the presence of the protease inhibitor MG132. MG132 prevented the decrease in Bcl-xL protein level in the presence of high ABT-737 (Supplementary Figure S4); therefore, high ABT-737 enhances proteolysis of Bcl-xL.
The effect of ABT-737 in cancer is to sequester anti-apoptotic molecules, allowing pro-apoptotic Bax to become activated and oligomerize in mitochondrial membranes.32, 33 Therefore, we examined whether high ABT-737 in neurons also activates Bax. We found that high ABT-737 does not activate Bax, but in contrast, glutamate toxicity activates Bax as previously reported,34, 35 and this activation is enhanced by high ABT-737 (Figure 6e′), suggesting that a death stimulus is needed in addition to a drop in Bcl-xL levels to activate Bax. Correlated with this, in neurons, high ABT-737 enhances cytochrome c expression and activates caspase 3 only in glutamate-exposed neurons (Figure 6e′′).
To determine if Bax activation was due to another effect of glutamate toxicity or was downstream of formation of ΔN-Bcl-xL, we performed glutamate toxicity in the presence of low ABT-737. Bcl-xL and ΔN-Bcl-xL levels were not affected by low ABT-737 (Figure 6f). No activation of Bax was measured after treatment with low ABT-737. Low ABT-737 prevented the formation of ΔN-Bcl-xL (Figure 6f′) and activation of Bax in the presence of glutamate toxicity (Figure 6f′). Thus, we conclude that Bax activation is downstream of ΔN-Bcl-xL formation in the presence of glutamate. Furthermore, addition of low ABT-737 decreases cytochrome c release and activation of caspase 3 (Figure 6f′′), consistent with a ΔN-Bcl-XL-dependent mechanism of apoptotic induction.
ΔN-BcL-xL-induced loss of mitochondrial inner membrane potential is prevented by depletion of ATP synthase c-subunit
Our hypothesis centers on the role of ΔN-Bcl-xL in activation of the inner membrane calcium ligand-gated, CsA sensitive pore known as the mPTP. We have previously reported that some full-length Bcl-xL (about 50%) localizes to the matrix of mitochondria,10 where it binds to the β-subunit of F1FO ATP synthase.14 To find the localization of ΔN-Bcl-xL, we performed immunocytochemistry. We found that glutamate increased the appearance of ΔN-Bcl-xL at mitochondria (in both soma and neurites; Figures 7a and b) because it co-localized with the β-subunit of the ATP synthase (Figures 7a and b). To determine if ΔN-Bcl-xL localized to the inner membrane, we removed the outer membrane with digitonin treatment and confirmed that, in addition to the outer membrane, both full-length Bcl-xL and ΔN-Bcl-xL are present in the inner membrane or matrix (Figure 7c).

Depletion of ATP synthase c-subunit protects from ΔN-BcL-xL-induced mitochondrial membrane potential loss. (a) Immunocytochemistry of cultured hippocampal neurons showing co-localization of the β-subunit of ATP synthase with ΔN-Bcl-xL. Green: β-subunit of ATP synthase; red: ΔN-Bcl-xL. (b) fluorescence intensity of ΔN-Bcl-xL at the β-subunit spots (n=3 independent cultures). This shows that ΔN-Bcl-xL amount is enhanced at the mitochondrial inner membrane after glutamate treatment. (c) Immunoblot of ΔN-Bcl-xL, Bcl-xL, VDAC and COX4 in intact mitochondria versus mitochondria depleted of the outer membrane. (d) Immunocytochemistry of cultured hippocampal neurons showing co-localization of HA-labeled ΔN-Bcl-xL and GFP-labeled ATP c-subunit shRNA. Red: HA; green: GFP; blue: Hoechst-stained nuclei. (e) % of co-transfected neurons/all transfected neurons. (f) Primary hippocampal neurons expressing empty vector plus scrambled GFP-labeled shRNA, empty vector plus GFP-labeled ATP c-subunit shRNA, ΔN-Bcl-xL plus GFP-labeled scrambled or ΔN-Bcl-xL plus ATP c-subunit shRNA stained with TMRM. Red: TMRM; green: GFP. (g) TMRM intensity (n=25-41 cells). Scale bar=10 μm. *P<0.05, ***P<0.001 and ****P<0.0001, one-way ANOVA
ATP synthase is an important candidate to form mPTP.14, 36, 37, 38, 39, 40 We and other have previously suggested that the c-subunit of the ATP synthase forms the mPTP.14, 36 To determine if ΔN-Bcl-xL plays a role in activation of mPTP during glutamate toxicity, we asked if depletion of the ATP synthase c-subunit prevents mitochondrial inner membrane depolarization and neuronal death caused by glutamate toxicity. Over 70% of cells co-expressed HA-tagged ΔN-Bcl-xL and GFP-tagged scrambled shRNA or c-subunit (ATP5G1) shRNA.14 (Figures 7d and e). Despite the lack of overt neuronal death in neurons expressing ΔN-Bcl-xL/scrambled shRNA, the mitochondria had a decreased intensity of TMRM signal (Figures 7f and g). In contrast, ATP c-subunit-depletion prevented the loss of mitochondrial potential in ΔN-Bcl-xL expressing neurons (Figures 7f and g).
To understand if ΔN-Bcl-xL-enhanced neuronal death works via mPTP opening during glutamate toxicity, we measured cell death in cultures depleted or not depleted of c-subunit shRNA with or without ΔN-Bcl-xL. ΔN-Bcl-xL significantly increased the vulnerability of neurons to glutamate challenge (Figures 8a and b). ATP c-subunit-depleted neurons were significantly protected from glutamate/ΔN-Bcl-xL-induced neurotoxic death (Figures 8a and b). In addition, expression of ΔN-Bcl-xL prior to glutamate toxicity caused more severe damage to neurites than glutamate treatment alone, suggesting that ΔN-Bcl-xL-expressing neurites may be on the edge of degeneration prior to glutamate challenge. This morphological change in neurites was also substantially rescued by depletion of ATP synthase c-subunit (Figure 8c).

Depletion of ATP synthase c-subunit protects neurons and neurites against ΔN-BcL-xL-aggravated glutamate-induced neuronal death. Primary hippocampal neurons expressed scrambled GFP-labeled shRNA, GFP-labeled ATP c-subunit shRNA and empty vector or ΔN-Bcl-xL with/without glutamate treatment. (a and b) Images and group data of PI staining of cultured hippocampal neurons at 24 h after glutamate treatment (n=3 independent cultures per group). (c) Number of neurites attached to soma (n=42-55 micrographs) *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001, one-way ANOVA
Discussion
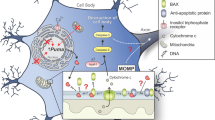
We find that ABT-737 or WEHI-539 have dual functions in neuronal death and survival. Despite lowering mitochondrial metabolic efficiency,9 high concentrations of ABT-737 (1 μM) alone do not induce neuronal death. Neurons may be able to retain energy from glycolysis in unstressed conditions. However, blocking Bcl-xL function mildly impairs mitochondrial function and increases the vulnerability of neurons to a neurotoxic stimulus. Thus, significant numbers of ABT-737-treated neurons died after mild levels of glutamate treatment. Interestingly, this was prevented with low ABT-737 (10 nM), which rescued hippocampal neurons and prevented mitochondrial membrane potential loss and loss of energy production during glutamate stimulation. Since the N terminus of Bcl-xL does not contain the ABT-737-binding site (Supplementary Figure S2), we interpret our data showing the protective effects of low ABT-737 as representing direct inhibition of ΔN-Bcl-xL leading to preserved mitochondrial membrane potential and improved survival; high ABT-737 produces the opposite effect, and additionally causes the proteolysis of full-length Bcl-xL, priming for the triggering of pro-apoptotic cascades (Figure 9).

Differential regulation of cell death by high or low ABT-737. High ABT-737 exposure does not by itself cause cell death, but it induces the loss of full-length Bcl-xL through a non-specific proteolysis-dependent mechanism and indirectly causes mitochondrial depolarization and energy loss (1). Loss of Bcl-xL also releases BID to be ready to activate Bax upon a death stimulus (primed for death status; (1)). Under conditions of glutamate toxicity, caspase activation leads to specific cleavage of full-length Bcl-xL to form ΔN-Bcl-xL (2). Both calcium influx and ΔN-Bcl-xL act together to depolarize mitochondrial inner membrane (3), leading to Bax activation (4), cytochrome c release and further activation of caspases, initiating a positive feedback loop (5) of enhanced propensity toward neuronal death. Low ABT binds to ΔN-Bcl-xL (6), preventing the depolarization of the mitochondrial inner membrane, mPT and Bax activation, thereby preventing downstream neuronal death (7)
Glutamate-induced excitotoxic stimulation causes intracellular calcium overload and ROS production, leading to early (by 1 h after in vivo ischemia) caspase activation and formation of ΔN-Bcl-xL.20 We here show that formation of ΔN-Bcl-xL is required for Bax activation in these cell death scenarios, since low ABT-737 arrests the process of Bax activation and at the level of the inner mitochondrial membrane prevents ΔN-Bcl-xL-induced, CsA-sensitive depolarization and cytochrome c release. In addition, low ABT-737 preserves levels of full-length Bcl-xL, leaving it free to inhibit cell death pathways. Impaired mitochondrial permeabilization by glutamate toxicity predicts that depletion of the c-subunit of the ATP synthase will protect neurons against glutamate/ΔN-Bcl-xL-induced membrane depolarization and cell death. We find that this is the case, further emphasizing a role for an mPTP channel in ΔN-Bcl-xL/Bcl-xL affected pathways during excitotoxicity (Figure 9).
On the other hand, high ABT-737 should bind full-length Bcl-xL and block its anti-apoptotic function. This sequestration of Bcl-xL will trigger Bax activation only upon a death stimulus as we and others have previously reported.8, 41, 42, 43 This may be equivalent to a primed-for-death state44, 45, 46 (Figure 9), since we find that neurons display no increase in cell death prior to glutamate toxicity, whereas glutamate exposure enhances caspase activation8, 34 triggers cleavage of full-length Bcl-xL to form ΔN-Bcl-xL, initiating Bax activation. Our new data lend further support to this scenario by demonstrating that low ABT-737 indirectly inhibits Bax activation by preventing ΔN-Bcl-xL-induced inner mitochondrial membrane depolarization.
We observed that treatment with ABT-737 decreased full-length Bcl-xL protein levels in a dose-dependent manner because ABT-737 binding enhances proteolysis of Bcl-xL. Czabotar et al.47 reported that the BH3-only protein, NOXA caused degradation of the anti-apoptotic Bcl-2 protein myeloid cell factor 1 (Mcl-1) during formation of the NOXA-Mcl-1 complex. It is also reported that the BH3 region of NOXA causes Mcl-1 degradation in a proteasome-dependent manner.48 Zhong et al.49 reported that Mule, an E3 ubiquitin ligase containing BH3 domain, mediates polyubiquitination of Mcl-1. In addition to caspase-dependent proteolysis, therefore, binding of ABT-737 to Bcl-xL or to ΔN-Bcl-xL protein may facilitate their degradation via ubiquitin-proteasome pathways.
A surprising finding of this study is that ΔN-Bcl-xL plays a role at the mitochondrial inner membrane, although this was predicted by our previous findings that ΔN-Bcl-xL directly enhances ATP synthase enzymatic activity.9 In the current study, we observed the loss of mitochondrial inner membrane potential in both endogenous (glutamate) and exogenous (plasmid) conditions of ΔN-Bcl-xL expression, as well as after direct application of purified protein to mitochondria. mPTP is activated during glutamate-induced death and it is reported to be an important pharmacological target in ischemic brain disease.50, 51 The retention of mitochondrial inner membrane potential is crucial to protect neurons against this form of neurotoxic insult. We previously reported that full-length Bcl-xL, by binding to the β-subunit of the ATP synthase, prevents opening of a leak channel of the inner membrane. We have described recently that a high conductance leak is formed by the c-subunit of F1FO ATP synthase9, 14 and we find here that CsA inhibits membrane depolarization directly induced by ΔN-Bcl-xL, implicating mPTP in this depolarization. In further support of this, we find here that neurons depleted of c-subunit by specific shRNA were protected from loss of mitochondrial potential associated with expression of ΔN-Bcl-xL, and were protected from death associated with glutamate alone or glutamate in combination with expression of ΔN-Bcl-xL. Depletion of the ATP synthase c-subunit also significantly protected from the increased vulnerability of neurites to degeneration, suggesting that mPTP activation is involved in triggering degenerative events.
Taken together, our findings support a multifunctional role of ABT-737 in neuronal death and survival. A strategy to apply low or high concentrations of ABT-737 may protect neurons against cytotoxic insults or potentiate apoptosis in malignant cells, respectively, in clinical applications.
Materials and methods
Primary cultures of rat hippocampal neurons
Primary rat hippocampal neurons were prepared from rat feti (Sprague-Dawley, day 18 of gestation; Harlan, Indianapolis, IN, USA) as described previously13, 52, 53 with modifications specific for this study. After isolation of hippocampi from prenatal brains, neurons were dissociated and seeded (0.2 × 106 cells/35 mm plate) onto plates containing medium with 5% FBS. After 2 h incubation, cells were maintained in neurobasal medium supplemented with B-27, glutamine and antibiotics (Invitrogen GIBCO Life Technologies, Carlsbad, CA, USA). Neurons were grown at 37 °C in 5% CO2 and 20% O2 in a humidified incubator, and assayed at DIV 20-22. Glutamate treatment: 20 μM glutamate (Sigma-Aldrich, St. Louis, MO, USA) was freshly made in sterile PBS as an aqueous solution then added to the cell culture medium as described in relevant figure legends. Bcl-xL inhibitor treatment: a stock solution of ABT-737 (Selleckbio, Houston, TX, USA), or WEHI-539 (Apex Bio, Houston, TX, USA) were prepared in dimethyl sulfoxide (DMSO). ABT-737 (1 μM or 10 nM), WEHI-539 (5 μM or 10 nM) or the same volume of DMSO was added into the culture dishes 20 min prior to glutamate treatment. Neurons were transfected at days in vitro (DIV) 7 using lipofectamin LTX with Plus Reagent (Invitrogen).
Viability assay
Lactate dehydrogenase (LDH) assay
The level of cytotoxicity in primary hippocampal neurons was assayed by measuring leakage of LDH using an in vitro toxicology assay kit (Sigma-Aldrich) as previously described.54 In brief, data were calculated by finding the activity of LDH leaked into the medium by damaged cells/total LDH activity in the culture (cells plus medium). The culture media and lysed cells were collected after treatment of neurons as described in the relevant figure legends. The LDH assay mixture was made according to the manufacturer’s protocol and added to each sample. After 20 min incubation, the reaction was terminated by adding 1 N HCl. LDH activity was spectrophotometrically measured with a VICTOR3 multilabel reader (PerkinElmer, Waltham, MA, USA) with absorbance set at 490 nm. Calcein-AM and PI: viable or dead cells were stained with calcein-AM or PI as previously described.8, 14 After treatment of neurons as described in the corresponding figure legends, 25 nM calcein-AM or 0.5 μM PI (Invitrogen, Molecular Probes, Carlsbad, CA, USA) was added into the culture medium for 30 min. Images were taken using a Zeiss Axiovert 200 microscope. The number of PI positive neurons, or calcein fluorescence densitometry per cell was analyzed using AxioVision 4.8.
Measurement of mitochondrial potential (Δψ)
Cell culture
Mitochondrial membrane potential (Δψ) was measured using the fluorescent lipophilic cationic dye tetramethylrhodamine methyl ester (TMRM, Invitrogen, Molecular Probes, Carlsbad, CA, USA), which accumulates within mitochondria in a potential-dependent manner.54, 55 After treatment with ABT-737 (Selleck Chemical, Houston, TX, USA), glutamate or a combination of both, primary hippocampal neurons were stained with 5 nM TMRM for 30 min at 37 °C in the dark. Images were taken using a Zeiss LSM 710 confocal scanning microscope and TMRM fluorescence densitometry was analyzed using ZEN software (Carl Zeiss Microscopy GmbH, Jena, Germany). Brain mitochondria: immediately after isolation of mitochondria from rat brain, mitochondria were incubated with 2 mM malate, 2 mM glutamate and 2 mM ADP (Sigma), and then treated as described in the figure legends. Mitochondria were mixed with 100 nM TMRM and loaded onto a 96-well plate (500 μg/well). Each well was treated with CsA (2 μM, Cell Signaling, Danvers, MA, USA), FCCP (100 μM, Abcam, Cambridge, UK), ABT-737 (100 nM or 5 μM), ΔN-Bcl-xL, full-length Bcl-xL or a combination and incubated for 20 min in the dark. The intensity of fluorescence was measured by a SpectraMax Gemini XS (Molecular Devices, Sunnyvale, CA, USA) with excitation 544 nm and emission 590 nm.
Measurement of ATP production
Primary hippocampal neurons were seeded on 96-well plates (0.015 × 106 neurons/well). After 3 weeks incubation, cells were treated as stated in relevant figure legends. Neuronal ATP production was measured by using ATPlite Luminescence Assay System (PerkinElmer) according to the manufacturer’s protocol. Plates were washed with sterile PBS, and cells were lysed. Cells were then incubated with substrate (luciferin) for 15 min. The reaction between ATP, luciferase and luciferin produces bioluminescence. ATP-induced-luminescence was measured with a VICTOR3 multilabel reader (PerkinElmer).
Cloning and purification of ΔN-Bcl-xL and Bcl-xL recombinant proteins
The rat ΔN-Bcl-xL cDNA (truncation lacking first 60 aa on the N terminus) was amplified by PCR using primers: (forward) 5′-GAAGGAGATACCACCATGgatagccccgcggtgaatgga-3′ and (reverse) 5′-GGGCACGTCATACGGATActtccgactgaagagtgagcc-3′. PCR product was cloned directly into pME-HA vector using Expresso CMV Cloning and Expression System (Lucigen, Middleton, WI, USA), to introduce HA-tag at the C terminus of ΔN-Bcl-xL. Next, HEK cells were transfected with pME-HA-ΔN-Bcl-xL plasmid and 24 h later cell lysates were prepared. ΔN-Bcl-xL was purified using Pierce HA tag IP/Co-IP kit (Thermo Scientific, Rockford, IL, USA) according to the instruction manual. Flag-tagged Bcl-xL construct12 was immunoprecipitated from HEK293T cell lysates using the EZview Red ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich) according to the manufacturer’s protocol. The purified protein samples were examined by western blot.
Immunoblots
After protein purification, protein concentration was determined using BCA protein reagents (Thermo Scientific). Samples were separated on a 4–12% SDS-polyacrylamide gel (Bio-Rad, Hercules, CA, USA) and probed with anti-Bcl-xL (1:1000 dilution, Cell Signaling), anti-ΔN-Bcl-xL (1:100 dilution, Aves Labs, Tigard, OR, USA), anti-active bax (1:100 dilution, Enzo Life Science, Farmingdale, NY, USA), anti-whole bax (1:1000 dilution, Cell Signaling), anti-cytochrome c (1:1000 dilution, Cell Signaling), anti-active caspase 3 (1:100 dilution, Abcam), anti-VDAC (1:1000 dilution, Cell Signaling), anti-COX IV (1:1000, Invitrogen) and anti-GAPDH (1:1000, Sigma-Aldrich). Anti-ΔN-Bcl-xL is custom-produced (peptide sequence: CZ DSP AVN GAT GHS SSL D (1:100, Aves Labs). Membranes were treated with ECL reagents (Perkin Elmer) and images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Isolation of mitochondria
Mitochondria or subfraction of mitochondria was purified from rat brain as previously described.10, 19, 56 In brief, rats (n=12) were killed and brain tissue was homogenized in isolation buffer (250 mM sucrose, 20 mM HEPES, 1 mM EDTA, 0.5% BSA). After a series of centrifugations, the nuclear material and the mitochondrial pellet containing synaptosomes were separated. Synaptosomes were disrupted by applying 1200 psi pressure for 10 min and mitochondria were separated by ultracentrifugation. Subfractionation of mitochondria was performed as described previously.10 In brief, the purified mitochondrial pellet was permeabilized and disassociated using 0.1% digitonin. Anti-VDAC (1:1000 dilution, Cell Signaling) was used as a marker for the outer membrane, and anti-COX IV (1:1000, Invitrogen) as a marker for the inner membrane to verify the quality of the fraction.
Immunocytochemistry
Primary hippocampal neurons fixed in 10% buffered formalin were blocked in 10% goat serum for 1 h, then incubated with anti-HA (1:10 dilution, Santa Cruz Biotechnology, Dallas, TX, USA), anti-GFP (1:100, 1:100, Abcam), anti-ΔN-Bcl-xL (1:10 dilution, Aves Labs), and anti-ATPB (1:100 dilution, Abcam) overnight at 4 °C. Cells were washed and incubated with Alexa-fluor 488 antibody or Alexa-568 antibody (1:200 dilution, Invitrogen, Molecular Probes, Carlsbad, CA, USA) for 1 h at room temperature and mounted on glass slides. Images were taken with a Zeiss Axiovert 200 microscope (Zeiss, Oberkochen, Germany) and processed using AxioVision 4.8.
Homology modeling and protein-ligand docking
The homology model for ΔN-Bcl-xL was built based on the crystal structure of human Bcl-xL (PDB ID: 2YXJ)57 by using the Swiss Model software (Swiss-Pdb Viewer 4.0.4, http://spdbv.vital-it.ch).58 The ABT-737 was docked into the ΔN-Bcl-xL homology model and the model was refined by using FireDock web server.59 Supplementary Figure S1 was rendered with UCSF Chimera (UCSF Chimera-visualization system for exploratory research and analysis60).
Quantitative real-time PCR
Total RNA was extracted from the primary hippocampal neurons by using the Absolutely RNA miniprep kit (Agilent, Inc., Santa Clara, CA, USA). Quantitative RT-PCR was performed with the qRT-PCR Kit (Applied Biosystems, Foster City, CA, USA) using a Step One Plus Real-Time PCR System (Applied Biosystems). TaqMan Gene Expression Assays (Life Technologies) and the following TaqMan primers were used to measure expression levels of BclXl and Bcl2: BclXl (Rn00437783_m1), Bcl2 (Rn99999125_m1). Relative mRNA expression was determined using the Step One Software v2.2.2 (Applied Biosystems). The expression values were normalized to the Actin B housekeeping gene.
Statistical analysis
Data are reported as mean±S.D. of at least three independent experiments. Difference in means was tested using one-way ANOVA with Tukey’s test. P<0.05 is considered statistically significant. P values are provided in figure legends.
References
Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA et al. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74: 597–608.
Gonzalez-Garcia M, Garcia I, Ding L, O'Shea S, Boise LH, Thompson CB et al. Bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA 1995; 92: 4304–4308.
Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 1997; 275: 983–986.
Ku B, Liang C, Jung JU, Oh BH . Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res 2011; 21: 627–641.
Soane L, Siegel ZT, Schuh RA, Fiskum G . Postnatal developmental regulation of Bcl-2 family proteins in brain mitochondria. J Neurosci Res 2008; 86: 1267–1276.
Ivanovska I, Galonek HL, Hildeman DA, Hardwick JM . Regulation of cell death in the lymphoid system by Bcl-2 family proteins. Acta Haematol 2004; 111: 42–55.
Cheng EH, Levine B, Boise LH, Thompson CB, Hardwick JM . Bax-independent inhibition of apoptosis by Bcl-XL. Nature 1996; 379: 554–556.
Park HA, Licznerski P, Alavian KN, Shanabrough M, Jonas EA . Bcl-xL is necessary for neurite outgrowth in hippocampal neurons. Antioxid Redox Signal 2015; 22: 93–108.
Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol 2011; 13: 1224–1233.
Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol 2011; 195: 263–276.
Hardwick JM, Chen YB, Jonas EA . Multipolar functions of BCL-2 proteins link energetics to apoptosis. Trends Cell Biol 2012; 22: 318–328.
Li H, Alavian KN, Lazrove E, Mehta N, Jones A, Zhang P et al. A Bcl-xL-Drp1 complex regulates synaptic vesicle membrane dynamics during endocytosis. Nat Cell Biol 2013; 15: 773–785.
Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci USA 2008; 105: 2169–2174.
Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 2014; 111: 10580–10585.
McNally MA, Soane L, Roelofs BA, Hartman AL, Hardwick JM . The N-terminal helix of Bcl-xL targets mitochondria. Mitochondrion 2013; 13: 119–124.
Clem RJ, Cheng EH, Karp CL, Kirsch DG, Ueno K, Takahashi A et al. Modulation of cell death by Bcl-XL through caspase interaction. Proc Natl Acad Sci USA 1998; 95: 554–559.
Seng NS, Megyesi J, Tarcsafalvi A, Price PM . Mimicking Cdk2 phosphorylation of Bcl-xL at Ser73 results in caspase activation and Bcl-xL cleavage. Cell Death Discov 2016; 2: 1–6.
Fujita N, Nagahashi A, Nagashima K, Rokudai S, Tsuruo T . Acceleration of apoptotic cell death after the cleavage of Bcl-XL protein by caspase-3-like proteases. Oncogene 1998; 17: 1295–1304.
Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ et al. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat Neurosci 2012; 15: 574–580.
Bonanni L, Chachar M, Jover-Mengual T, Li H, Jones A, Yokota H et al. Zinc-dependent multi-conductance channel activity in mitochondria isolated from ischemic brain. J Neurosci 2006; 26: 6851–6862.
Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y et al. Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci USA 2004; 101: 13590–13595.
Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh KM, Fujisawa S et al. Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc Natl Acad Sci USA 2008; 105: 4892–4897.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Letai A . BH3 domains as BCL-2 inhibitors: prototype cancer therapeutics. Expert Opin Biol Ther 2003; 3: 293–304.
Cristofanon S, Fulda S . ABT-737 promotes tBid mitochondrial accumulation to enhance TRAIL-induced apoptosis in glioblastoma cells. Cell Death Dis 2012; 3: e432.
Premkumar DR, Jane EP, DiDomenico JD, Vukmer NA, Agostino NR, Pollack IF . ABT-737 synergizes with bortezomib to induce apoptosis, mediated by Bid cleavage, Bax activation, and mitochondrial dysfunction in an Akt-dependent context in malignant human glioma cell lines. J Pharmacol Exp Ther 2012; 341: 859–872.
Sutton VR, Sedelies K, Dewson G, Christensen ME, Bird PI, Johnstone RW et al. Granzyme B triggers a prolonged pressure to die in Bcl-2 overexpressing cells, defining a window of opportunity for effective treatment with ABT-737. Cell Death Dis 2012; 3: e344.
Wang C, Youle RJ . Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1's inhibitory effect on Bak. Oncogene 2012; 31: 3177–3189.
Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA . Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. J Neurophysiol 2008; 99: 1515–1522.
Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol 2013; 9: 390–397.
Chiu C, Miller MC, Caralopoulos IN, Worden MS, Brinker T, Gordon ZN et al. Temporal course of cerebrospinal fluid dynamics and amyloid accumulation in the aging rat brain from three to thirty months. Fluids Barriers CNS 2012; 9: 3.
Buron N, Porceddu M, Brabant M, Desgue D, Racoeur C, Lassalle M et al. Use of human cancer cell lines mitochondria to explore the mechanisms of BH3 peptides and ABT-737-induced mitochondrial membrane permeabilization. PLoS ONE 2010; 5: e9924.
Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ 2008; 15: 820–830.
D'Orsi B, Bonner H, Tuffy LP, Dussmann H, Woods I, Courtney MJ et al. Calpains are downstream effectors of bax-dependent excitotoxic apoptosis. J Neurosci 2012; 32: 1847–1858.
Simonishvili S, Jain MR, Li H, Levison SW, Wood TL . Identification of Bax-interacting proteins in oligodendrocyte progenitors during glutamate excitotoxicity and perinatal hypoxia-ischemia. ASN Neuro 2013; 5: e00131.
Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013; 12: 674–683.
Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 2013; 110: 5887–5892.
Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynela J et al. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014; 55: 69–77.
Azarashvili TS, Tyynela J, Odinokova IV, Grigorjev PA, Baumann M, Evtodienko YV et al. Phosphorylation of a peptide related to subunit c of the F0F1-ATPase/ATP synthase and relationship to permeability transition pore opening in mitochondria. J Bioenerg Biomembr 2002; 34: 279–284.
Couoh-Cardel S, Hsueh YC, Wilkens S, Movileanu L . Yeast V-ATPase proteolipid ring acts as a large-conductance transmembrane protein pore. Sci Rep 2016; 6: 24774.
Pavlov EV, Priault M, Pietkiewicz D, Cheng EH, Antonsson B, Manon S et al. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol 2001; 155: 725–731.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Liu Q, Leber B, Andrews DW . Interactions of pro-apoptotic BH3 proteins with anti-apoptotic Bcl-2 family proteins measured in live MCF-7 cells using FLIM FRET. Cell Cycle 2012; 11: 3536–3542.
Davids MS, Letai A . Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol 2012; 30: 3127–3135.
Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011; 334: 1129–1133.
Clerc P, Carey GB, Mehrabian Z, Wei M, Hwang H, Girnun GD et al. Rapid detection of an ABT-737-sensitive primed for death state in cells using microplate-based respirometry. PLoS ONE 2012; 7: e42487.
Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci USA 2007; 104: 6217–6222.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 2005; 19: 1294–1305.
Zhong Q, Gao W, Du F, Wang X . Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121: 1085–1095.
Luetjens CM, Bui NT, Sengpiel B, Munstermann G, Poppe M, Krohn AJ et al. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci 2000; 20: 5715–5723.
Concannon CG, Tuffy LP, Weisova P, Bonner HP, Davila D, Bonner C et al. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol 2010; 189: 83–94.
Kaech S, Banker G . Culturing hippocampal neurons. Nat Protoc 2006; 1: 2406–2415.
Beaudoin GM 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 2012; 7: 1741–1754.
Park HA, Khanna S, Rink C, Gnyawali S, Roy S, Sen CK . Glutathione disulfide induces neural cell death via a 12-lipoxygenase pathway. Cell Death Differ 2009; 16: 1167–1179.
Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA . Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther 2005; 312: 509–516.
Sacchetti S, Alavian KN, Lazrove E, Jonas EA . F1FO ATPase vesicle preparation and technique for performing patch clamp recordings of submitochondrial vesicle membranes. J Vis Exp 2013: e4394.
Lee EF, Czabotar PE, Smith BJ, Deshayes K, Zobel K, Colman PM et al. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ 2007; 14: 1711–1713.
Guex N, Peitsch MC . SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997; 18: 2714–2723.
Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ . FireDock: a web server for fast interaction refinement in molecular docking. Nucleic Acids Res 2008; 36 (Web Server issue): W229–W232.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004; 25: 1605–1612.
Acknowledgements
We thank Dr. J Marie Hardwick for the original ΔN Bcl-xL construct that served as a template for our new ΔN Bcl-xL construct, and for her exceptional advice to improve the manuscript. R37NS045876 funded to EAJ.
Author contributions
H-AP and EAJ designed research. H-AP, PL, NM, YN, SS, JW, BMP and KNA performed research. H-AP and EAJ wrote this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by L Greene
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Park, HA., Licznerski, P., Mnatsakanyan, N. et al. Inhibition of Bcl-xL prevents pro-death actions of ΔN-Bcl-xL at the mitochondrial inner membrane during glutamate excitotoxicity. Cell Death Differ 24, 1963–1974 (2017). https://doi.org/10.1038/cdd.2017.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.123
This article is cited by
-
Interaction Between HCN and Slack Channels Regulates mPFC Pyramidal Cell Excitability in Working Memory Circuits
Molecular Neurobiology (2024)
-
Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions
Cell Death & Differentiation (2023)
-
Neuroprotective effects of ATPase inhibitory factor 1 preventing mitochondrial dysfunction in Parkinson's disease
Scientific Reports (2022)
-
Molecular mechanisms and consequences of mitochondrial permeability transition
Nature Reviews Molecular Cell Biology (2022)
-
Cell death in cancer in the era of precision medicine
Genes & Immunity (2019)

{kind=link}
{kind=link}
{kind=link}
{kind=link}