
Abstract
Formation of secretary endometrial glands in the uterus known as adenogenesis is a typical process of branching morphogenesis involving dynamic epithelial growth and differentiation. Unsuccessful adenogenesis often leads to female infertility. However, it remains largely unexplored so far regarding the epigenetic machinery governing normal endometrial gland formation. Here, we demonstrated that PR-Set7, an epigenetic regulator for H4K20me1 modification, was extensively expressed in the postnatal uteri, and its conditional deletion resulted in a complete lack of endometrial glands and infertility in mice. Subsequent analysis revealed that uterine PR-Set7 deficiency abolishes the dynamic endometrial epithelial population growth during the short span of gland formation from postnatal days 3 to 9. This markedly reduced epithelial population growth in PR-Set7-null mutant uteri is well associated with DNA damage accumulation and massive apoptotic death in the epithelium, due to blockade of 53BP1 recruitment to DNA damage sites upon reduced levels of H4K20me1/2. Using PgrCre/+/Rosa26DTA/+ mouse line and postnatal progesterone injection mouse model, we further confirmed that an impaired epithelial cell population growth either by inducing epithelial death in the diphtheria toxin-A (DTA)-mouse model or attenuating epithelial growth upon postnatal progesterone treatment similarly hampers uterine adenogenesis. Collectively, we establish here a novel ‘epithelial population growth threshold’ model for successful gland development. Besides further shedding light on the regulatory machinery governing uterine gland formation, our findings raise a safety concern on progesterone supplementation to prevent preterm birth in women bearing a female fetus, as exogenous progesterone may hamper uterine adenogenesis via attenuating epithelial population growth.
Similar content being viewed by others


Main
Uterine endometrium consists of a layer of luminal epithelium (LE) supported by stromal cells that contain coiled endometrial glands. As an intricate and highly organized network throughout the stroma, glandular epithelial (GE) cells synthesize and secrete proteins as well as related substances to nourish the developing embryos.1, 2, 3 Disruption of uterine gland development often leads to permanent infertility in the adulthood.4, 5, 6
In mice, uterine adenogenesis begins postnatally. The uterus derives from the Müllerian duct, being quite rudimentary at birth (postnatal day (PND) 0) with a simple layer of LE and the surrounding mesenchyme.7 At PNDs 5 and 6, through active cell proliferation, adenogenesis begins with the invagination of LE (bud formation). LE-GE differentiation happens at the tips of LE buds at around PND 9. Then, these buds gradually invade and migrate into the underlying uterine stroma. Through coiling and branching, uterine glands already can be observed in the uterus by PND12. Meanwhile, the mesenchyme undergoes extensive morphological and functional differentiation and forms three layers: stroma, circular myometrium and longitudinal myometrium.1 Thus, epithelial growth and LE-GE differentiation are key events during uterine morphogenesis and gland development.
The complexity of uterine adenogenesis has been presenting a challenge to understand the orchestration of signaling events that contribute to the formation of this delicately patterned ductal network. In recent years, a variety of genetically engineered mouse models have helped us identify many crucial genes contributing to uterine adenogenesis, including Foxa2, Wnt7a, Lgr4 and Dlx5/6.4, 6, 8, 9, 10, 11, 12, 13 However, potential contribution of epigenetic regulators during uterine adenogenesis remains mysterious and barely explored yet.
PR-Set7, encoded by Kmt5a, is the sole enzyme known to catalyze the monomethylation of histone H4 Lys 20 (H4K20me1) that serves as the primary substrate for H4K20me2.14, 15, 16 Extensive studies have demonstrated its central role during early embryonic development and organ development.15, 17, 18, 19 However, it remained elusive as to whether and how PR-Set7-mediated signaling regulates uterine adenogenesis.
In the current study, we used a variety of mouse models and demonstrated that PR-Set7-mediated signaling guarantees the endometrial epithelial population growth to a threshold level, thus ensuring successful uterine gland formation.
Results
PR-Set7 is essential for normal endometrial gland development and female fertility
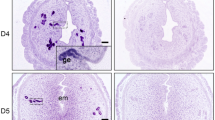
To explore the potential physiological roles of PR-Set7 during postnatal uterine development, we examined PR-Set7 expression in mouse uterus at both mRNA and protein levels on PNDs 6–12, the critical period of uterine adenogenesis. We found that PR-Set7 was persistently expressed among the stages examined (Figures 1a and b). Moreover, PR-Set7 was strongly expressed in uterine epithelial cells revealed by immunohistochemistry (Figure 1c), suggesting that PR-Set7 might be an important player during uterine gland development.

PR-Set7 is critical for normal uterine adenogenesis. (a) RT-PCR analysis of PR-Set7 mRNA levels in wild-type uteri during adenogenesis at indicated PNDs. (b) Western blot analysis of PR-Set7 protein levels in wild-type uteri at PNDs 6, 9 and 12. (c) Immunohistochemistry staining of PR-Set7 during adenogenesis. LE, luminal epithelium; S, stroma; G, gland. Scale bars: 200 μm and 50 μm. (d) Generation strategy for uterine PR-Set7 conditional knockout mice (PR-Set7d/d, d/d). (e and f) Knockout efficiency was analyzed by RT-PCR (e) and Western blot (f). In (e), the expression of exon7 of PR-Set7 mRNA was assayed. Wild type, 299 bp; excised, 108 bp. (g–j) PR-Set7 deficiency led to failure of uterine gland formation. Immunostaining analysis of cytokeratin (CK, red), smooth muscle action (SMA, green) and vimentin (pink) in PR-Set7f/f (f/f) and PR-Set7d/d (d/d) uteri at indicated PNDs (g). Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Arrows indicated uterine glands. Scale bar: 100 μm. Failed adenogenesis in PR-Set7d/d uteri was evidenced by immunostaining of Foxa2 (h). Nuclei were stained with DAPI. Scale bars: 100 μm. Quantitative analysis of Foxa2-positive glands (i) and real time RT-PCR analysis of Foxa2 mRNA (j) on indicated PNDs. Data shown represent the mean±S.E.M., *P<0.05
To identify the function of PR-Set7 during uterine development, we established the mouse model with conditional ablation of PR-Set7 in the uterus by crossing PR-Set7flox/flox (PR-Set7f/f) mice with progesterone receptor Cre knock-in (PgrCre/+) mice (Figure 1d). The Cre-mediated recombination results in the deletion of SET domain of PR-Set7. By PCR analysis, we found that the excised bands were observed in PR-Set7d/d uteri as early as PND 3, whereas only wild-type bands were observed in PR-Set7f/f uteri (Figure 1e), demonstrating an excision of PR-Set7 in PR-Set7d/d uteri during adenogenesis, which was further confirmed by the data from western blot (Figure 1f). To further examine the spatiotemporal efficiency of PR-Cre, we analyzed the β-gal activity in PgrCre/+ mice mated with Rosa26 lacZ reporter line. As shown in Supplementary Figure S1, consistent with postnatal uterine PR expression, Cre recombinase activity was already initiated in both epithelial and stromal compartments by PND 6 before the onset of uterine adenogenesis, which was confirmed by the immunostaining analysis of PR-Set7 expression (Supplementary Figure S2). To investigate the phenotype of PR-Set7 conditional knockout mouse, we first carried out fertility analysis and found that PR-Set7d/d females were completely infertile (Supplementary Figure S3A), with thinner tubular uterus compared with that of PR-Set7f/f mice (Supplementary Figure S3B). These observations made us to suppose that uterine PR-Set7 deficiency might lead to severe uterine developmental defects. Indeed, immunostaining analysis of endometrial sub-cell type markers further revealed that PR-Set7d/d uteri were devoid of endometrial glands (Figures 1g–i). For example, immunopositive cells for Foxa2, a well-recognized uterine GE marker, were not detected in PR-Set7d/d uteri even when analyzed at PND 12 (Figures 1h and i). Moreover, the real-time PCR analysis showed that no Foxa2 mRNA was detected in PR-Set7d/d uteri (Figure 1j), verifying the loss of gland formation. These results indicate that PR-Set7 is a critical driver for normal uterine gland development.
Uterine PR-Set7 deficiency induces massive endometrial epithelial cell death
Since the formation of most mammalian branching systems, including uterine adenogenesis, is accompanied by massive cell proliferation and thus the enlargement of organic size, we speculated that the failed adenogenesis induced by PR-Set7 deficiency may be due to defective proliferation of uterine epithelial cells. To test this hypothesis, we first statistically analyzed the sum of epithelial cells from PNDs 3 to 9, the critical duration of mouse uterine development. As shown in Figure 2a, epithelial cell population growth exhibited as a generalized S-shaped growth curve in PR-Set7f/f mice (Figure 2a). For example, the amount of epithelial cells increased slowly in a positive acceleration phase from PNDs 3 to 5. Then, it increased rapidly to approach an exponential growth rate exhibiting as a J-shaped curve from PNDs 5 to 7, during which luminal epithelial invagination happens. From PND 8 to 9, the epithelial population gradually stabilized when the differentiation of the GE cells initiates. By contrast, the epithelial population in PR-Set7d/d uteri was significantly decreased, especially on PND 6, the critical stage for uterine adenogenesis (Figure 2b). These observations suggest that initiation and progression of uterine adenogenesis requires epithelial population growth overriding certain threshold level, while PR-Set7 deficiency inhibits the epithelial population from reaching this critical threshold.

Uterine PR-Set7 deficiency induces massive endometrial epithelial cell death. (a) Growth curve of epithelial population per cross-section from PNDs 3 to 9. (b) The total number of epithelial cells per cross-section was quantitatively analysed in PR-Set7f/f (f/f) and PR-Set7d/d (d/d) uteri at PNDs 3, 6 and 9. (c) Immunostaining analysis of the proliferation marker Ki67. White line indicates the uterine epithelium. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Scale bar: 50 μm. (d) Quantitative analysis of the ratio of Ki67-positive cells in the epithelium. (e) Immunohistochemistry analysis of the apoptotic marker cleaved Caspase-3 in PR-Set7f/f and PR-Set7d/d uteri. e, epithelium. Scale bars: 50 μm. (f) Quantitative analysis of the ratio of cleaved caspase-3-positive cells in the epithelium. Data shown represent the mean±S.E.M., *P<0.05, **P<0.01 and ***P<0.001
Since it is well known that PR-Set7 acts as a multifunctional switch of cell cycle as well as cell survival,16 we surmised that the decreased epithelial population growth upon PR-Set7 deletion might be due to impeded proliferation of luminal epithelial cells and/or epithelial cell death. To test this hypothesis, we firstly detected the proliferation state of the luminal epithelial cells by immunostaining analysis of the proliferation marker Ki67. Unexpectedly, epithelial cell proliferation was instead increased in the PR-Set7d/d uteri (Figures 2c and d). This contradictory finding of decreased epithelial population coupled with increased proliferation activity made us to speculate that adenogenesis failure in PR-Set7d/d mice was accused of reduced cell survival ability. To test this hypothesis, we analyzed the expression of cleaved caspase-3, an apoptosis marker. Indeed, while few cleaved caspase-3-positive cells were detected in PR-Set7f/f epithelia, a great portion of cells were positive for cleaved caspase-3 in PR-Set7d/d epithelia at PNDs 6 and 9 (Figures 2e and f). These findings indicate that PR-Set7 deficiency robustly enhances the death of uterine epithelial cells, thus limiting the growth of epithelial population, eventually hampering the process of uterine adenogenesis.
PR-Set7 deficiency derails DNA damage repair pathway resulting in increased uterine epithelial cell death during adenogenesis
To further explore the mechanism underlying the increased apoptosis of uterine epithelial cells upon PR-Set7 deletion, we sequenced the transcriptome of PR-Set7f/f and PR-Set7d/d uteri on PNDs 3, 6 and 9, and integrated the sequencing data into a comprehensive view of the developing transcriptomes. The hierarchical clustering analysis of gene-expression profiles and data from the volcano plot separated PR-Set7f/f and PR-Set7d/d uteri at different developmental stages, and defined a bunch of differentially expressed genes (DEGs) with respect to their mean expression levels (Figure 3a and Supplementary Figures S4A and B). Consistent with the above findings, the uterine adenogenesis-related genes were significantly downregulated, suggesting that the signaling network ensuring normal gland development was disturbed in the absence of PR-Set7 (Figures 3b and c). To identify the processes dysregulated upon PR-Set7 deletion and classify these DEGs between PR-Set7f/f and PR-Set7d/d uteri, we performed gene ontology (GO) enrichment analysis of these DEGs on PNDs 6 and 9. Consistent with the functions of PR-Set7 as a survival factor, DEGs were enriched in the GO term of cell death regulation (GO: 0010942, positive regulation of cell death; GO: 0060548, negative regulation of cell death) (Supplementary Figures S4C and D), supporting the notion that increased cell death resulted from PR-Set7 deletion. More importantly, DEGs upon PR-Set7 deficiency were largely enriched in GO categories related to several aspects of DNA damage response (GO: 0006974, response to DNA damage stimulus; GO: 0000077, DNA damage checkpoint; GO: 0006281, DNA repair) (Figures 3d and e and Supplementary Figure S4E), which can be further confirmed by real-time RT-PCR analysis (Figure 3f). In addition, we noted a significantly increased number of PR-Set7d/d uterine epithelial cells expressing γH2AX (Figures 3g and h), a well-known marker for DNA damage. These data indicate that PR-Set7 deficiency derails DNA damage repair pathway, inducing massive apoptotic cell death during postnatal uterine development.

PR-Set7 deficiency derails DNA damage repair pathway resulting in increased uterine epithelial cell death during adenogenesis. (a) Heatmap illustrating DEGs (P<0.05, 1.5-fold change) in PR-Set7f/f (f/f) and PR-Set7d/d (d/d) uteri at indicated PNDs. (b) Heatmap illustrates the expression levels of genes involved in uterine gland development. (c) Quantitative RT-PCR analysis of representative genes regulating adenogenesis. Data shown represent the mean±S.E.M., *P<0.05 and ***P<0.001. (d and e) Expression levels of representative DEGs enriched in the GO terms relating to DNA damage. (f) Quantitative RT-PCR analysis of representative DEGs in the selected GO categories at PND 9. Data shown represent the mean±S.E.M., *P<0.05, **P<0.01 and ***P<0.001. (g) Immunostaining analysis of γH2AX indicates massive DNA damage in PR-Set7d/d uterine epithelial cells. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Scale bar: 50 μm. (h) Quantitative analysis of the ratio of γH2AX in PR-Set7f/f and PR-Set7d/d epithelial cells. Data shown represent the mean±S.E.M., ***P<0.001
PR-Set7 is required for 53BP1 recruitment to DNA damage sites against apoptotic death in endometrial epithelia
PR-Set7-mediated H4K20me1 has been shown to facilitate the generation of H4K20me2 necessary for 53BP1 binding, which identifies PR-Set7 as an essential component for DNA double-strand break repairing.20, 21, 22 To address the mechanisms underlying the accumulated DNA damage upon PR-Set7 deficiency, we first detected the levels of H4K20me1 in PND 6 uteri. As shown in Figure 4a, we found that the nuclear staining of H4K20me1 was decreased significantly in PR-Set7d/d epithelia in comparison with PR-Set7f/f uteri, which was confirmed by subsequent western blot analysis of H4K20me1 and H4K20me2 (Figure 4b). Moreover, we further observed a significant decline of 53BP1, an essential DNA repair factor, in the chromatin fraction of PR-Set7d/d uteri (Figure 4c), in line with early evidence that 53BP1 is recruited into the nuclei by H4K20me2 upon the detection of DNA damage.23

PR-Set7 is required for 53BP1 recruitment to DNA damage sites against apoptotic death in endometrial epithelia. (a) Epithelial H4K20me1 was decreased by PR-Set7 deletion. Immunostaining of H4K20me1 was carried out at PND 6. White dashed line indicates the epithelial compartment. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Scale bar: 25 μm. (b) Western blot analysis of H4K20me1 and H4K20me2 in PR-Set7f/f (f/f) and PR-Set7d/d (d/d) uteri at PNDs 6 and 9. (c) The protein levels of 53BP1 and γH2AX were analyzed by western blot in the chromatin fraction of PR-Set7f/f and PR-Set7d/d uteri. (d) The decreased levels of PR-Set7, H4K20me1, H4K20me2 and increased level of γH2AX upon PR-Set7 knockdown were revealed by western blot analysis in Ishikawa cells. The control siRNA (si-Ctrl) was served as a negative control. (e) MTS assay showed an impaired increase of cell number upon PR-Set7 knockdown. (f) Fluorescence-activated cell sorting (FACS) analysis revealed G2-phase arrest at 24 h in siR-PR-Set7-transfected cells. *P<0.05, **P<0.01. (g) Immunostaining of Ki67 in Ishikawa cells at 24 h post transfection. Nuclei were stained with DAPI. Scale bar: 50 μm. (h) TUNEL assay was carried out in Ishikawa cells at 24 h post transfection. Nuclei were stained with DAPI. Scale bar: 50 μm. (i) Immunostaining of γH2AX in Ishikawa cells at 24 h post transfection. Nuclei were stained with DAPI. Scale bar: 50 μm. (j) Quantitative analysis of the ratio of γH2AX-positive cells in Ishikawa cells at 24 h post transfection. Data shown represent the mean±S.E.M., ***P<0.001. (k) Colocalization of 53BP1 (red) and γH2AX (green) in nuclear foci by immunostaining. Nuclei were stained with DAPI. Scale bar: 10 μm
Using PR-Set7 siRNA knockdown approach in human endometrial Ishikawa cells, we confirmed the content of adverse correlation of decreased H4K20me1 and H4K20me2 levels with increased γH2AX levels and DNA damage upon PR-Set7 silencing (Figure 4d). MTS assay further revealed a significantly impeded cell population growth curve in line with an impaired cell cycle progression upon PR-Set7 siRNA transfection (Figures 4e and f). Moreover, PR-Set7 knockdown significantly increased the apoptotic cell death occurrence rate as well as the ratio of γH2AX-positive cells, with no apparent influence on the cell proliferation (Figures 4g–j). To further understand the interplay between PR-Set7 knockdown and 53BP1 nucleation, we analyzed the cellular expression status of 53BP1 together with the nuclear DNA damage marker γH2AX. As shown in Figure 4k, while γH2AX and 53BP1 were colocalized in si-Ctrl cells, 53BP1 was barely detectable in si-PR-Set7 cells. The results collectively indicate that an inefficient recruitment of 53BP1 into the DNA damage sites is one of the causes for extensively accumulated DNA damage and aberrantly elevated γH2AX levels in endometrial epithelia in the absence of PR-Set7.
Epithelial population growth threshold is a prerequisite for uterine adenogenesis: evidences from other models
On the basis of the findings from PR-Set7-deficient mouse model, in particular the association of epithelial population growth with the duration and consequence of endometrial gland formation, we proposed that there is an endometrial epithelial population threshold during uterine adenogenesis. To further confirm this concept, we established two other mouse models with restricted epithelial population growth. We first generated PgrCre//Rosa26DTA/+ mice by breeding Rosa26DTA/+ with PgrCre/+ mice to conditionally induce cell death upon PR-Cre-mediated expression of DTA (Figures 5a and b). Meanwhile, we established the other mouse model using postnatal injection of progesterone to inhibit the epithelial cell proliferation as reported previously24, 25, 26 (Figures 5c and d). These two mouse models provided us with ideal tools to test the existence of 'epithelial population growth threshold' for normal adenogenesis. Cell population analysis showed that the total epithelial cell number was decreased notably in both the DTA-mouse model and progesterone-treated mice (Figure 5e). More importantly, no Foxa2-positive cells were detected in the uterine epithelia in both conditions, indicating the failure of adenogenesis in these uterine epithelial growth-curbed models (Figure 5f). These findings further confirm the existence of epithelial population growth threshold for normal uterine gland formation (Figure 6).

Epithelial population growth beyond the threshold level is critical for uterine adenogenesis. (a) Generation strategy for DTA (PgrCre/+/Rosa26DTA/+) mouse model. (b) Immunohistochemistry staining of cleaved caspase-3 reveals massive cell death in the epithelium of DTA model at PND 9. The right pictures show the magnified images of the left ones, respectively. Scale bars: 50 μm. (c) Postnatal progesterone-injection mouse model. (d) Epithelial proliferation was inhibited upon progesterone injection evidenced by immunostaining of Ki67 at PND 9. The right pictures show the magnified images of the left ones, respectively. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Scale bars: 50 μm. (e) Quantitative analysis of the total number of epithelial cells per cross-section in wild-type, DTA and progesterone-injection uteri at PND 9. Data shown represent the mean±S.E.M., ***P<0.001. (f) Failed adenogenesis in the DTA and progesterone-injection uteri was evidenced by immunostaining of Foxa2. Nuclei were stained with DAPI. Scale bars: 50 μm

Proposed 'epithelial population threshold' model for uterine adenogenesis. (a) Epithelial population growth to threshold level is critical for uterine gland formation. (b) Aberrantly enhanced death of epithelial cells in PR-Set7d/d uteri limits cell population growth, hampering gland formation
Discussion
Previous studies on uterine adenogenesis have been focusing on the classic pathway settings, such as developmental genes and transcription factors.27 However, little information was available regarding whether and how epigenetic regulators have a role during uterine gland development. Combining multiple approaches, we demonstrated that null mutation of histone methyltransferase PR-Set7 diminishes H4K20me1/2 levels and consequently abolishes the recruitment of 53BP1 to the DNA damage sites, leading to massive DNA damage accumulation and cell death in the uterine epithelia, thus failure of gland formation.
As a process of branching morphogenesis, uterine adenogenesis contains the restructure of the epithelium to the complex yet delicate tubular networks with crucial transporting or secretory functions. It has been shown that the formation of most mammalian branching systems is accompanied by massive cell proliferation, and the organ itself expands significantly in size as it is being built.28 In this respect, we also observed that uterine epithelial cell number markedly increases through active cell proliferation, gradually approaching a stable level followed with the emergence of Foxa2-positive GE cells toward the endometrial bed. Logically, we raised for the first time the hypothesis that there is certain epithelial population threshold for the initiation of adenogenesis. In line with this hypothesis, we indeed proved that PR-Set7 deficiency inhibits uterine adenogenesis by curbing the rise of epithelial cell population. Using DTA-mouse model as well as postnatal progesterone injection mouse model, we further confirmed that an impaired epithelial cell population growth either by induction of epithelial death in DTA-mouse model or attenuation of epithelial growth upon postnatal progesterone treatment will similarly hamper the process of uterine adenogenesis. We thus establish here a model of 'epithelial population threshold' that turns on the LE-GE differentiation during postnatal uterine development in mice.
The importance of well-developed maternal uterine glands and their secretions on conceptus survival and implantation provide strong evidence that pregnancy loss and complications in women may have their origin in uterine gland dysfunction.5, 13, 25 Different from being a postnatal event in domestic and laboratory animals, human adenogenesis occurs in utero, continues postnatally and is completed during puberty.1 Since progesterone was suggested to be important for maintaining uterine quiescence,29 its administration in the latter half of gestation has been used to prevent preterm birth for over decades.30, 31, 32 However, it has never been questioned regarding the potential hazards of exogenous progesterone supplementation on fetal endometrial gland development in utero. Even previous trials of daily vaginal progesterone during late gestation showed no harm on outcomes in children until 2 years of age,33 further information on adulthood outcomes following progesterone treatment is required to determine the risk–benefit ratio of this therapy.
Collectively, in the present study, we demonstrated for the first time that epithelial population growth threshold is a prerequisite for successful uterine gland formation. We provided here direct genetic evidence for a unique contribution of histone methyltransferase PR-Set7 to uterine adenogenesis, proving that PR-Set7-dependent genomic stability contributes to the success of postnatal uterine adenogenesis. Besides further shedding light on the regulatory machinery governing uterine gland development, our findings raise a safety concern on progesterone supplementation for prevention of preterm birth in women bearing female fetus, as exogenous progesterone may hamper the process of uterine adenogenesis via attenuating epithelial population growth.
Materials and methods
Animals and treatments
PR-Set7f/f mice were generated as described previously.17 Rosa26DTA/+ mice34 were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Uterine-specific mutant mice were generated by crossing PR-Set7f/f mice with PgrCre/+ mice.35 DTA (PgrCre/+/Rosa26DTA/+) mice were generated by crossing Rosa26DTA/+ mice with PgrCre/+ mice. Postnatal progesterone injection mouse model was applied as described previously.26 All mice were housed in the animal care facility of the Institute of Zoology, Chinese Academy of Sciences, according to the institutional guidelines for the care and use of laboratory animals.
Immunostaining and X-gal staining
Immunostaining and X-gal staining analysis was performed as described previously.36 Antibodies used were listed in Supplementary Table S1. TUNEL assay was used to detect apoptotic cells according to the manufacturer’s instructions (DeadEnd Fluorometric TUNEL System; Promega, Madison, WI, USA).
Western blot analysis
Protein extraction and western blot analysis were performed as described previously.37 Antibodies used were listed in Supplementary Table S1.
Cell counting
Paraffin-embedded sections of uterine samples at distinct developmental time points were stained using the antibody against cytokeratin (CK) to mark the epithelial cells, of which the nuclei was stained with DAPI. Then, Imaris8.0.2 was used to count CK and DAPI double-positive cells per cross-section. Six individual mice were used for quantification at each developmental time point and three discontinuous cross-sections of each mouse were counted.
Real-Time RT-PCR
A total of 3 μg RNA was used to synthesize cDNA. The expression levels of different genes were validated by real-time RT-PCR analysis using the ABI 7500 sequence detector system (Applied Biosystems, Carlsbad, CA, USA). All the real-time RT-PCR experiments were repeated at least three times. All primers for real-time PCR were listed in Supplementary Table S2.
Cell culture and siRNA knockdown
The human endometrial Ishikawa cells were maintained at 37 °C in an atmosphere of 5% CO2/95% air in DMEM medium supplemented with 10% (v/v) FBS. PR-Set7 and control siRNAs were purchased from RIBO Biology (Guangzhou, China). Ishikawa cells were transfected with 50 nM PR-Set7 siRNA or control siRNA and collected at indicated time points for further analysis. Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used for transfection experiments in Ishikawa cells according to the manufacturer’s instructions.
Cell viability (MTS assay)
Ishikawa cells were seeded in 96-well plates, and were analyzed using the MTS viability assay according to the manufacturer’s protocol (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega), and absorption value was measured at 490 nm.
RNA-Seq and data analysis
Total RNA of the whole uteri was extracted from 12 samples using ARCTURUS PicoPure RNA Isolation Kit (Life Technologies, Invitrogen). Sequencing libraries were generated using NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) following the manufacturer’s recommendations, and sequenced on an Illumina Hiseq 2500 platform at RiboBio Co. Ltd (Guangzhou, China), and finally 125 bp paired-end reads were obtained. TopHat2 was selected as the mapping tool to map these reads to mouse reference genome GRCm38 (ENSEMBL release 69). Then RPKM (Reads Per Kilobase of exon model per Million mapped reads) of each gene was calculated, and differential expression analysis was performed using DEGseq R package. Functional annotation of DEGs was performed with DAVID (the Database for Annotation, Visualization and Integrated Discovery, https://david.ncifcrf.gov). GO terms with corrected P-value <0.05 were considered significantly enriched by differential expressed genes. All heatmaps were generated based on the Z-score with R script.
Statistical analysis
All data are presented as mean±S.E.M. Each experiment included at least three independent samples. Comparison between two groups was made by unpaired Student’s two‐tailed t‐test. P<0.05 was considered to indicate a significant result.
References
Gray CA, Bartol FF, Tarleton BJ, Wiley AA, Johnson GA, Bazer FW et al. Developmental biology of uterine glands. Biol Reprod 2001; 65: 1311–1323.
Spencer TE, Hayashi K, Hu J, Carpenter KD . Comparative developmental biology of the mammalian uterus. Curr Top Dev Biol 2005; 68: 85–122.
Brody JR, Cunha GR . Histologic, morphometric, and immunocytochemical analysis of myometrial development in rats and mice: II. Effects of DES on development. Am J Anat 1989; 186: 21–42.
Jeong JW, Kwak I, Lee KY, Kim TH, Large MJ, Stewart CL et al. Foxa2 is essential for mouse endometrial gland development and fertility. Biol Reprod 2010; 83: 396–403.
Filant J, Spencer TE . Endometrial glands are essential for blastocyst implantation and decidualization in the mouse uterus. Biol Reprod 2013; 88: 93.
Spencer TE . Biological roles of uterine glands in pregnancy. Semin Reprod Med 2014; 32: 346–357.
Kobayashi A, Behringer RR . Developmental genetics of the female reproductive tract in mammals. Nat Rev Genet 2003; 4: 969–980.
Bellessort B, Le Cardinal M, Bachelot A, Narboux-Neme N, Garagnani P, Pirazzini C et al. Dlx5 and Dlx6 control uterine adenogenesis during post-natal maturation: possible consequences for endometriosis. Hum Mol Genet 2016; 25: 97–108.
Kim YS, Kim HR, Kim H, Yang SC, Park M, Yoon JA et al. Deficiency in DGCR8-dependent canonical microRNAs causes infertility due to multiple abnormalities during uterine development in mice. Sci Rep 2016; 6: 20242.
Dunlap KA, Filant J, Hayashi K, Rucker EB 3rd, Song G, Deng JM et al. Postnatal deletion of Wnt7a inhibits uterine gland morphogenesis and compromises adult fertility in mice. Biol Reprod 2011; 85: 386–396.
Cooke PS, Spencer TE, Bartol FF, Hayashi K . Uterine glands: development, function and experimental model systems. Mol Hum Reprod 2013; 19: 547–558.
Farah O, Biechele S, Rossant J, Dufort D . Porcupine-dependent Wnt signaling controls stromal proliferation and endometrial gland maintenance through the action of distinct WNTs. Dev Biol 2016; 422: 58–69.
Kelleher AM, Peng W, Pru JK, Pru CA, DeMayo FJ, Spencer TE . Forkhead box a2 (FOXA2) is essential for uterine function and fertility. Proc Natl Acad Sci USA 2017; 114: E1018–E1026.
Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA et al. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev 2005; 19: 1444–1454.
Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell 2002; 9: 1201–1213.
Beck DB, Oda H, Shen SS, Reinberg D . PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev 2012; 26: 325–337.
Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM et al. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol 2009; 29: 2278–2295.
Driskell I, Oda H, Blanco S, Nascimento E, Humphreys P, Frye M . The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J 2012; 31: 616–629.
Nikolaou KC, Moulos P, Chalepakis G, Hatzis P, Oda H, Reinberg D et al. Spontaneous development of hepatocellular carcinoma with cancer stem cell properties in PR-SET7-deficient livers. EMBO J 2015; 34: 430–447.
Dulev S, Tkach J, Lin S, Batada NN . SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep 2014; 15: 1163–1174.
Tuzon CT, Spektor T, Kong X, Congdon LM, Wu S, Schotta G et al. Concerted activities of distinct H4K20 methyltransferases at DNA double-strand breaks regulate 53BP1 nucleation and NHEJ-directed repair. Cell Rep 2014; 8: 430–438.
Dantuma NP, van Attikum H . Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J 2016; 35: 6–23.
Baldock RA, Day M, Wilkinson OJ, Cloney R, Jeggo PA, Oliver AW et al. ATM localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with gammaH2AX. Cell Rep 2015; 13: 2081–2089.
Allison GC, Bartol FF, Taylor KM, Wiley AA, Ramsey WS, Ott TL et al. Ovine uterine gland knock-out model: effects of gland ablation on the estrous cycle. Biol Reprod 2000; 62: 448–456.
Gray CA, Burghardt RC, Johnson GA, Bazer FW, Spencer TE . Evidence that absence of endometrial gland secretions in uterine gland knockout ewes compromises conceptus survival and elongation. Reproduction 2002; 124: 289–300.
Cooke PS, Ekman GC, Kaur J, Davila J, Bagchi IC, Clark SG et al. Brief exposure to progesterone during a critical neonatal window prevents uterine gland formation in mice. Biol Reprod 2012; 86: 63.
Zhang S, Lin H, Kong S, Wang S, Wang H, Wang H et al. Physiological and molecular determinants of embryo implantation. Mol Aspects Med 2013; 34: 939–980.
Affolter M, Bellusci S, Itoh N, Shilo B, Thiery JP, Werb Z . Tube or not tube: remodeling epithelial tissues by branching morphogenesis. Dev Cell 2003; 4: 11–18.
Norwitz ER, Caughey AB . Progesterone supplementation and the prevention of preterm birth. Rev Obstet Gynecol 2011; 4: 60–72.
Navathe R, Berghella V . Progesterone as a tocolytic agent for preterm labor: a systematic review. Curr Opin Obstet Gynecol 2016; 28: 464–469.
Fonseca EB, Celik E, Parra M, Singh M, Nicolaides KH . Progesterone and the risk of preterm birth among women with a short cervix. N Engl J Med 2007; 357: 462–469.
Iams JD, Romero R, Culhane JF, Goldenberg RL . Primary, secondary, and tertiary interventions to reduce the morbidity and mortality of preterm birth. Lancet 2008; 371: 164–175.
Norman JE, Marlow N, Messow C-M, Shennan A, Bennett PR, Thornton S et al. Vaginal progesterone prophylaxis for preterm birth (the OPPTIMUM study): a multicentre, randomised, double-blind trial. Lancet 2016; 387: 2106–2116.
Ivanova A, Signore M, Caro N, Greene ND, Copp AJ, Martinez-Barbera JP . In vivo genetic ablation by Cre-mediated expression of diphtheria toxin fragment A. Genesis 2005; 43: 129–135.
Soyal SM, Mukherjee A, Lee KY, Li J, Li H, DeMayo FJ et al. Cre-mediated recombination in cell lineages that express the progesterone receptor. Genesis 2005; 41: 58–66.
Lu J, Zhang S, Nakano H, Simmons DG, Wang S, Kong S et al. A positive feedback loop involving Gcm1 and Fzd5 directs chorionic branching morphogenesis in the placenta. PLoS Biol 2013; 11: e1001536.
Wang Q, Lu J, Zhang S, Wang S, Wang W, Wang B et al. Wnt6 is essential for stromal cell proliferation during decidualization in mice. Biol Reprod 2013; 88: 5.
Acknowledgements
We are grateful to Dr. Francesco DeMayo (National Institute of Environmental Health Sciences, USA) for his generosity in providing us with the PgrCre/+ mice. This work was supported by National Key R&D Program of China (2017YFC1001402 to HW, 2017YF0104603 to SK and JL) and the National Natural Science Foundation (81330017 and 81490744 to HW, 81601285 to SK and 31600945 to JL).
Author contributions
WH and LJ designed research; CT, HB, KS, ZC, NZ and BH performed research; WH, LJ, CT, ZH, QJ and XQ analyzed the data; DR and JPL provided mouse models; CT, LJ and WH wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Cui, T., He, B., Kong, S. et al. PR-Set7 deficiency limits uterine epithelial population growth hampering postnatal gland formation in mice. Cell Death Differ 24, 2013–2021 (2017). https://doi.org/10.1038/cdd.2017.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.120
This article is cited by
-
De novo reconstruction of a functional in vivo-like equine endometrium using collagen-based tissue engineering
Scientific Reports (2024)
-
Epigenetic control of embryo–uterine crosstalk at peri-implantation
Cellular and Molecular Life Sciences (2019)
