
Abstract
High Mobility Group A1 (HMGA1) is an architectural chromatin protein whose overexpression is a feature of malignant neoplasias with a causal role in cancer initiation and progression. HMGA1 promotes tumor growth by several mechanisms, including increase of cell proliferation and survival, impairment of DNA repair and induction of chromosome instability. Autophagy is a self-degradative process that, by providing energy sources and removing damaged organelles and misfolded proteins, allows cell survival under stress conditions. On the other hand, hyper-activated autophagy can lead to non-apoptotic programmed cell death. Autophagy deregulation is a common feature of cancer cells in which has a complex role, showing either an oncogenic or tumor suppressor activity, depending on cellular context and tumor stage. Here, we report that depletion of HMGA1 perturbs autophagy by different mechanisms. HMGA1-knockdown increases autophagosome formation by constraining the activity of the mTOR pathway, a major regulator of autophagy, and transcriptionally upregulating the autophagy-initiating kinase Unc-51-like kinase 1 (ULK1). Consistently, functional experiments demonstrate that HMGA1 binds ULK1 promoter region and negatively regulates its transcription. On the other hand, the increase in autophagosomes is not associated to a proportionate increase in their maturation. Overall, the effects of HMGA1 depletion on autophagy are associated to a decrease in cell proliferation and ultimately impact on cancer cells viability. Importantly, silencing of ULK1 prevents the effects of HMGA1-knockdown on cellular proliferation, viability and autophagic activity, highlighting how these effects are, at least in part, mediated by ULK1. Interestingly, this phenomenon is not restricted to skin cancer cells, as similar results have been observed also in HeLa cells silenced for HMGA1. Taken together, these results clearly indicate HMGA1 as a key regulator of the autophagic pathway in cancer cells, thus suggesting a novel mechanism through which HMGA1 can contribute to cancer progression.
Similar content being viewed by others


Main
The High Mobility Group A (HMGA) family includes three proteins: HMGA1a, HMGA1b, (encoded by the same gene through alternative splicing) and HMGA2.1 These proteins bind the minor groove of AT-rich DNA sequences through three short basic repeats, named ‘AT-hooks’, thereby altering chromatin structure and facilitating the assembly of multiprotein complexes of transcriptional factors, thus regulating the transcription of several genes involved in a wide spectrum of biological processes, such as embryogenesis, cell differentiation, cell cycle, cell migration, apoptosis and cell transformation.2 The HMGA protein levels are low or absent in normal cells and adult tissues, whereas are abundant during development and in malignant cancers.1 Indeed, overexpression of HMGA genes represents a constant feature of malignant neoplasias, and their expression levels correlate with metastatic potential, resistance to anti-cancer therapies3, 4 and reduced survival of cancer patients.5 The causal role of HMGA proteins in the process of carcinogenesis has been clearly demonstrated as the blockage of their expression prevents neoplastic transformation6 and leads neoplastic cells to death.7 Consistently, transgenic mice overexpressing the HMGA genes develop several neoplasias.8, 9, 10 Although several mechanisms by which HMGA proteins contribute to cancer development have been described so far,1 other ones, such as autophagy regulation, may be envisaged.
In recent years, the study of the intertwined connections between cancer, apoptosis and autophagy has gained increasing interest, also in consideration of the potential translational applications of such discoveries. Autophagy is an intracellular self-digestive process that has a critical role in maintaining energy homeostasis, and its regulation seems to be crucial for cancer initiation and progression. Autophagy involves the formation of autophagosomes, which assemble around and encapsulate damaged organelles, misfolded proteins or cellular debris, and then fuse with lysosomes to degrade their content.11 Autophagy is essential for long-term survival of mammalian cells, by allowing supply of nutrients under starvation and other stress conditions. Interestingly, disruption of autophagic pathways is associated with multiple diseases, including cancer, in which it can exert either an oncogenic or tumor suppressor activity depending on cellular context and tumor stage.12 Indeed, many human neoplasias show aberrant autophagy, and its inhibition may contribute to cancer progression. In particular, autophagy inhibition may favor necrotic cell death, exacerbating local inflammation, promoting tumor growth11 or leading to genomic instability.13 Moreover, defective autophagy protects cancer cells from autophagic cell death (type II programmed cell death),14 and resistance to both apoptotic and autophagic cell death can be acquired during cancer progression.15
Here, we report that depletion of HMGA1 expression dysregulates autophagy in human epidermal squamous carcinoma SCC-13 cells as well as in HeLa cells. This effect appears mediated by autophagy initiation ULK genes,16 whose expression is transcriptionally increased upon HMGA1-knockdown (KD). Taken together, these data indicate a critical role of HMGA1 in the regulation of autophagy, thus suggesting a novel mechanism by which HMGA1 may contribute to cancer progression.
Results
HMGA1 depletion induces autophagy in skin cancer cells
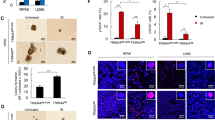
To investigate the role of HMGA1 proteins in skin cancer cells, HMGA1 expression was silenced by siRNA-transfection in human squamous carcinoma SCC-13 cells (Figures 1a and b). We evaluated the effect of HMGA1-KD on cell proliferation by performing a growth curve assay, observing a strong reduction in the number of HMGA1-interfered cells (HMGA1i) with respect to the scrambled oligonucleotide-transfected cells (Ctli) 48, 72 and 96 h after transfection (Figure 1c). Consistently, HMGA1i cells expressed higher levels of the CDK inhibitors p27Kip1 and p21Cip1 and lower levels of cyclin D1 and cyclin E (Figure 1d). However, we did not find β-galactosidase-positive cells (Supplementary Figure S1), concluding that HMGA1-KD does not induce permanent cell cycle arrest (i.e. senescence) in SCC-13 cells. However, we found that the viability of the HMGA1i cells was significantly lower (~40%) as compared with control cells (Figure 1e). Furthermore, HMGA1-KD does not significantly induce apoptosis in SCC-13 cells, as assessed by measuring the cleavage of two marker proteins of the apoptotic cascade, namely poly (ADP-ribose) polymerase (PARP) and caspase-7 (Figure 1f). Therefore, we investigated whether the reduction of cell viability induced by HMGA1 silencing was due to autophagy activation, which may lead to another type of programmed cell death.14 A connection between HMGA1 and autophagy regulation has already been suggested by the ability of HMGA1 overexpression to activate the PI3K/AKT signaling pathway,17 which in turn control the mTORC1, a negative regulator of autophagy.18 Hence, we analyzed the phosphorylation of AKT and S6 ribosomal protein, two proteins downstream of the mTORC1 pathway, which was reduced in HMGA1i cells (Figure 2a), suggesting that a reduction of HMGA1 expression may trigger the mTOR-dependent activation of the autophagic pathway in skin cancer cells. Subsequently, we evaluated the LC3-I to LC3-II conversion, which occurs during autophagosomes formation.19 The levels of LC3-II were increased in HMGA1-depleted cells (Figure 2b). We also performed the experiments in the presence of the lysosomal pump inhibitor Bafilomycin A1, which allows one to infer about the rate of LC3-II neosynthesis.19, 20, 21 Indeed, the levels of LC3-II were higher in HMGA1i cells also in the presence of Bafilomycin A1, suggesting that there was an induction of autophagosomes synthesis. In addition, we analyzed the expression levels of p62/SQSTM1, which is recruited into autophagosomes during their assembly and then degraded inside autolysosomes.21 Surprisingly, we did observe a modest accumulation of p62/SQSTM1 in HMGA1-depleted cells. Albeit in apparent contradiction with the LC3-II flux analysis, such a phenomenon could be explained by a concomitant reduction of the later stages of autophagic pathway, as previously reported.19, 22

HMGA1 depletion impairs proliferation and viability of SCC-13 cells. Control (Ctli) and HMGA1-interfered (HMGA1i) SCC-13 cells were tested for the expression of HMGA1 by qRT-PCR (a) and western blotting (b). Actin was used as loading control. (c) Growth curves of Ctli and HMGA1i SCC-13 cells. Cells were plated as described in ‘Materials and Methods’ and counted at 48, 72 and 96 h. (d) Proteins extracted from Ctli and HMGA1i cells were analyzed by western blotting for Cyclin E, Cyclin D1, p21 and p27 protein levels. Actin was used as loading control. (e) Cell viability assay of Ctli and HMGA1i cells was evaluated as described in ‘Materials and Methods’ section. Bars represent the mean absorbance±S.D. of a representative experiment performed in triplicate. (f) Proteins extracted from Ctli and HMGA1i cells were analyzed by western blotting for PARP and Caspase-7 protein levels. Actin was used as loading control

HMGA1 depletion perturbs autophagic activity. (a) Immunoblot detection of p-AKT (Ser473), total AKT, p-rpS6 and total rpS6 expression levels in Ctli and HMGA1i cells. Actin was used as loading control. (b) Proteins extracted from Ctli and HMGA1i cells untreated or treated with 100 nM Bafilomycin A1 (Bafi) for 6 h were tested for LC3-I>II conversion level and p62/SQSTM1 expression levels by western blotting. Actin was used as loading control. The graphs represent the relative optical density of LC3-II and p62/SQSTM1 normalized to Actin levels (mean±S.D. of three independent experiments). (c–d) Ctli and HMGA1i cells were stained with p62/SQSTM1 and LC3-specific antibodies detected, respectively, with Alexa-488 and Alexa-546 conjugated secondary antibodies (c) or with fluorescent dyes, monodansylcadaverin (MDC) and lysotracker (d), as described in ‘Materials and Methods’. Serial confocal sections were collected. Bars, 10 μm. For each condition, pictures at higher magnification are shown. Bars, 5 μm. Number of puncta per cells in Ctli and HMGA1i cells is shown for p62/SQSTM1. Mean fluorescence intensity (arbitrary unit, a.u.) in Ctli and HMGA1i cells is shown for other markers. In (d) the area occupied by MDC and lysotracker was measured in Ctli and HMGA1i cells. Experiments were performed at least three independent times (n≥50 cells). The statistical analysis has been performed including experiments conducted with both HMGA1-interfering approaches. Error bars, means±S.D.; *P<0.0001
To clarify and further corroborate the above described biochemical data, we assessed the distribution of the autophagy markers p62/SQSTM1 and LC3 by confocal microscopy. We observed a strong increase of p62/SQSTM1- and LC3-positive dots (or puncta) in HMGA1-depleted cells, indicating their recruitment in autophagosomes and autophagolysosomes (Figure 2c). Furthermore, we assessed the effect of HMGA1 silencing on the properties of autophagic compartments. We stained Ctli and HMGA1i cells with monodansylcadaverine (MDC), a fluorescent dye that labels in vivo late-stage autophagosomes, whose staining rises under conditions that increase autophagy.21, 23, 24 HMGA1 depletion caused an increase in both the number and size of MDC-positive structures with respect to Ctli cells, as well as of lysosomes, labeled with the Lysotracker dye (Figure 2d). Interestingly, a whole drastic expansion (in particular, a three/fourfold increase in the area positive for both markers) of the autophagosomal/lysosomal compartment was observed upon HMGA1 depletion (Figure 2d).
Moreover, to test whether HMGA1 depletion closely mimics mTOR inhibition-induced autophagy induction, we performed amino-acid-starvation experiments. As expected, HBSS treatment reduced the activity of mTORC1 pathway (Supplementary Figure S2). However, in terms of autophagic flux we noticed how the overall effects observed in response to HBSS (namely, the increased turnover of p62/SQSTM1 and LC3-II) were remarkably different from the ones observed upon HMGA1 depletion (Figure 2). Finally, to rule out any potential ‘off-target’ effects generated by the siRNA pools employed, we performed the main experiments with a mix of three different siRNA oligonucleotides in SCC-13 cells (HMGA1i§) (Supplementary Figure S3), confirming the aforementioned results. Altogether, these data suggest that HMGA1 regulates autophagy in SCC-13 cells.
Induction of autophagy by the impairment of HMGA1 expression is not restricted to skin cancer cells
As HMGA1 is an architectural transcription factor that activates, recruits to or displaces from specific promoters other transcriptional regulators, its activity strongly depends on the set of proteins concomitantly co-expressed by a particular cell type, therefore its effects may vary on the basis of the cellular context. To verify whether the autophagy increase induced by the HMGA1-KD is not restricted to skin cancer cells, we analyzed the effect of HMGA1 silencing also in human cervix cancer-derived HeLa cells. Indeed, depletion of HMGA1 induces autophagy in HeLa cells, as demonstrated by both western blot and confocal imaging analysis (Figure 3), and the effects of HBSS treatment on autophagic markers are less consistent than those induced by HMGA1-KD (Supplementary Figure S4).

HMGA1 depletion affects autophagy also in HeLa cells. (a) Immunoblot detection of HMGA1, (b) p-rpS6 and total rpS6 expression levels in Ctli and HMGA1i cells. Actin was used as normalization. (c) Proteins extracted from Ctli and HMGA1i cells, untreated or treated with 100 nM Bafilomycin A1 (Bafi) for 6 h, were tested for LC3-I>II conversion and p62/SQSTM1 expression level by western blotting. Actin was used as loading control. (d) Ctli and HMGA1i cells were stained with p62/SQSTM1 and LC3-specific antibodies or with fluorescent dyes, MDC and lysotracker. Serial confocal sections were collected. Bars, 10 μm. Number of puncta per cells in Ctli and HMGA1i cells is shown for p62/SQSTM1. Mean fluorescence intensity (arbitrary unit, a.u.) in Ctli and HMGA1i cells is shown for other markers. Experiments were performed at least three independent times (n≥50 cells). The statistical analysis has been performed including experiments conducted with both HMGA1-interfering approaches. Error bars, means±S.D.; *P<0.00001
To better evaluate the autophagic flux, we used a GFP-mRFP-tandem tagged LC3, which allows to discriminate between early autophagic organelles (GFP and mRFP positive) from acidified autophagolysosomes, which result positive only for mRFP because of higher sensitivity of GFP fluorescence to the acidic lumen of the lysosomal compartment.23 To this aim, we performed HMGA1-KD in HeLa cells stably transfected with tandem GFP-mRFP-LC3.25 In agreement with the results obtained by immunofluorescence experiments, we observed that the number of GFP-positive puncta was increased in HMGA1i cells with respect to control cells, or upon HBSS treatment (Figure 4a and Supplementary Figure S5). Strikingly, the puncta extended almost throughout cytosol of HMGA1i cells (occupied area was twofold higher in HMGA1i versus Ctli cells). Furthermore, by quantifying the number of red-only LC3 dots, we found that it is higher in Ctli than in HMGA1i cells, as also evidenced by the higher overlap coefficient (>0.5, graph at right) for GFP and mRFP signals (Figure 4a). Moreover, the fluorescence intensity of red puncta is significantly higher in HMGA1i cells, possibly resulting from homotypic and/or heterotypic fusion events between autophagosomes and lysosomes, respectively. Hence, the number of autophagosomes is increased in HMGA1i cells and such an increase is not mirrored by an equal increase in their maturation. Consistently, in the large majority of HMGA1i cells, p62/SQSTM1-positive structures do accumulate and co-localize with either the lysosomal marker LAMP-1 or LC3 (Figure 4b), confirming that the autophagic flux may be partially delayed in HMGA1-depleted cells. Importantly, the results obtained in HeLa cells strongly suggest that the interplay between HMGA1 and autophagy is not restricted to skin cancer cells.

Effects of HMGA1 depletion on autophagic flux through GFP-mRFP-tandem LC3 analysis. (a) HeLa stably expressing GFP-mRFP-LC3 cells were transfected with siRNAs targeting a scrambled sequence (Ctli) or HMGA1 (HMGA1i), fixed and analyzed by confocal microscopy. Bars, 10 μm. For each condition, pictures at higher magnification are shown. Bars, 5 μm. Quantification of the number of fluorescent puncta exhibiting green (GFP) or red-only fluorescence (obtained by subtracting green dots to the total red dots) is shown (right graphs). The mean fluorescence intensity (arbitrary unit, a.u.) of red puncta, and the percentage of cells exhibiting overlap coefficient lower or higher 0.5 upon the different conditions are shown in the lower graphs. Experiments were performed three independent times (n≥250 cells). Error bars, means±S.D.; *P<e-7. The same results were obtained in living cells as shown in Supplementary Figure S5a. (b) SCC-13 and HeLa HMGA1i cells were subjected to double immunofluorescence assays. Specifically, cells were stained with p62/SQSTM1 and Lamp-1 or LC3-specific antibodies detected with Alexa-488 and Alexa-546 conjugated secondary antibodies, respectively. Serial confocal sections were collected. Bars, 10 μm. Arrows indicate co-localization areas. The percentage of cells exhibiting overlap coefficient lower or higher 0.5 in randomly chosen Region Of Interest (ROI) is shown. Experiments were performed three independent times (n≥250 cells). The statistical analysis has been performed including experiments conducted with both HMGA1-interfering approaches. Error bars, means±S.D.; *P<e-7
HMGA1-negative regulation of ULK1 and ULK2 expression likely accounts for the involvement of HMGA1 in autophagy
The results described above suggest that HMGA1-KD might alter autophagy at different steps. Starting from the observation that Ulk2 was upregulated in Hmga1 knock-out (KO) with respect to wild type (WT) MEFs by microarray analysis (data not shown) and qRT-PCR (fold-change ≈3), we hypothesized that HMGA1 could regulate the expression of the Unc-51-like kinase (ULK) genes, coding for the Ser/Thr kinases belonging to the autophagy-initiating complex.26 Moreover, also the Ulk2 closely related paralogue Ulk1 was moderately overexpressed in KO versus WT MEFs (fold-change ≈1.9) (Figure 5a). Consistent with this, the mRNA levels of both ULK genes were substantially upregulated upon HMGA1-KD in HeLa and SCC-13 cell lines, as assessed by qRT-PCR analysis (Figure 5a). Interestingly, the fold-change of ULK1 (≈3-4) was higher than that of ULK2 (≈2.5) expression in both cell lines. We also confirmed the upregulation of ULK1 by western blotting analysis (Figure 5b). Moreover, we analyzed the effect of HMGA1 depletion on the phosphorylation of the ULK1 selective substrate ATG13, a critical event for the early step of the phagophore formation.21 Interestingly, both HMGA1-KD and HBSS treatment (used as positive control) increased the amount of Ser318 phosphorylated-ATG13, despite a concomitant decrease in the total amount of ATG13 (Figure 5c). To verify whether HMGA1 proteins directly bind to ULK1 and ULK2 promoters, we performed chromatin immunoprecipitation (ChIP) assays followed by qRT-PCR. The occupancy of AT-rich sequences of ULK1 and ULK2 promoters by HMGA1 has been detected in the anti-HMGA1-precipitated chromatin from HeLa cells, whereas no amplification was observed in samples immunoprecipitated with rabbit IgG (Figure 5d). Finally, to evaluate the ability of HMGA1 protein to regulate ULK1 and ULK2 promoter activity, we performed luciferase activity assays and demonstrated how HMGA1 depletion increased the transcriptional activity of both ULK1 and ULK2 promoters (Figure 5e). To further verify the ability of HMGA1 to regulate the transcription of ULK1 promoter, we used HEK293 cells, which represent a useful system to study the effects of either depletion or overexpression of HMGA1 protein. As expected, HMGA1 depletion increased ULK1 promoter activity in HEK293 cells, whereas its overexpression significantly reduced it (Figure 5f). Altogether, these data strongly support a critical role of HMGA1 in the negative regulation of ULK genes.

HMGA1 protein regulates ULK1 and ULK2 transcription. (a) RNA extracted from control (Ctli) or HMGA1-interfered (HMGA1i) SCC-13, HeLa cells or from WT and KO MEFs were analyzed by qRT-PCR for ULK1 and ULK2 expression. The actin expression levels have been used for data normalization. Data are mean±S.D. of a representative experiment performed in triplicate. (b) Proteins extracted from Ctli and HMGA1i SCC-13 and HeLa cells were tested for ULK1 expression by western blotting. Tubulin and Vinculin were used as loading control. (c) Proteins extracted from HeLa cells in control conditions (Ctl) or treated with HBSS for 4 h and Ctli and HMGA1i HeLa cells were tested for phosho-Ser318-ATG13 and total ATG13 expression by western blotting. Vinculin was used as loading control. (d) ChIP was performed in HeLa cells. Soluble chromatin was immunoprecipitated with anti-HMGA1 antibodies. The DNAs were amplified by qPCR using primers covering specific regions of human ULK1 and ULK2 promoters (−878/−713 and +129/+352, respectively). IgG were used as negative control of immunoprecipitation. Amplification of the immunoprecipitated DNA using primers for the GAPDH gene promoter was used as control of specificity. Data are mean±S.D. of a representative experiment performed in triplicate. (e) Analysis of ULK1 and ULK2 luciferase-reporter activity in Ctli and HMGA1i HeLa cells. All transfections were performed in triplicate. Data are mean±S.D. of three independent experiments. The same analysis was carried out for ULK1 promoter activity in Ctli and HMGA1i HEK293 cells (f, left panel). Analysis of ULK1 promoter activity was also evaluated in HEK293 cells transiently transfected with empty vector (CV) or 0.8 μg of pcDNA3.1-Hmga1b expression vector (f, right panel). All transfections were performed in triplicate. Data are mean±S.D. of three independent experiments
The effects of HMGA1-KD are prevented by silencing of ULK1
As ULK1 and ULK2 are master regulators of autophagy initiation, we hypothesized that the regulation of their expression may account for the effects on autophagy caused by HMGA1-KD. In particular, we focused on ULK1, the most upregulated in response to HMGA1 depletion in both SCC-13 and HeLa cells. Therefore, HeLa cells were interfered for ULK1 expression using a pool of three specific siRNAs (Figure 6a). We found that ULK1 silencing strongly prevented LC3-I>LC3-II conversion induced by HMGA1 depletion (Figure 6b). Moreover, similarly to the pattern observed in ULK1 silenced cells, in the double KD cells (HMGA1i+ULK1i), the number of p62/SQSTM1-positive puncta was consistently reduced (Figure 6c). Furthermore, in the GFP-mRFP-LC3 stable cell line, ULK1 silencing significantly antagonized the increase in autophagosomes number mediated by HMGA1-KD (Figure 6d). Altogether, these data suggest that HMGA1 regulates autophagy, at least in part, through ULK1.

Effects of HMGA1 silencing in autophagy are mediated by ULK1. Proteins extracted from Ctli, HMGA1i, ULK1i and HMGA1i-ULK1i HeLa cells were tested for the expression of ULK1 and HMGA1 (a) or LC3 (b) by western blotting. Actin was used as loading control. (c) Ctli, HMGA1i, ULK1i and HMGA1i-ULK1i cells were stained with p62/SQSTM1 specific antibody (labeled with FITC-conjugated secondary antibody). Bars, 11 μm. Number of puncta per cells in Ctli and HMGA1i cells is shown. Experiments were performed three independent times (n≥50 cells). (d) HeLa stably expressing GFP-mRFP-LC3 cells were transfected with siRNAs targeting ULK1 (ULK1i) or both HMGA1 and ULK1 (HMGA1i+ULK1i), fixed and analyzed by confocal microscopy. Bars, 10 μm. For each condition, pictures at higher magnification are shown. Bars, 5 μm. Quantification of the number of fluorescent puncta exhibiting green (GFP) (left graph) or red-only fluorescence (obtained by subtracting green dots to the total red dots, middle graph) is reported. The percentage of cells exhibiting overlap coefficient lower or higher 0.5 upon the different conditions is shown (right graph). Experiments were performed three independent times (n≥250 cells). Error bars, means±S.D.; *P<e-7
Subsequently, considering that ULK1-dependent cell death has been described in cancer cells in several conditions,27 we examined whether ULK1 is involved in the suppression of SCC-13 cell number and viability observed in HMGA1-depleted cells (Figure 1). Strikingly, the reduction of viable cells was almost completely prevented by ULK1-KD, therefore suggesting that HMGA1 depletion leads to ULK1-dependent cell death in these cells (Figure 7a). To further investigate the relationship between ULK1 and the cell cycle arrest induced by HMGA1-KD, we performed a colony formation assay. HMGA1i formed less and smaller clones than Ctli cells, and ULK1-KD was able to rescue their clonogenic potential (Figure 7b). To rule out the possibility that the observed effects were mere consequences related to cell cycle arrest, we performed a propidium iodine (PI) exclusion assay, and we found that the percentage of HMGA1i PI-positive cells was consistently higher in comparison with control cells. Interestingly, the ULK1 depletion reduced the effects mediated by the HMGA1-KD, without affecting per se the percentage of PI-positive cells (Figure 7c). Furthermore, we reproduced similar results by using deconvoluted siRNA oligonucleotides against ULK1, ruling out any off-target effect (Supplementary Figure S6). Finally, to test whether the effects of HMGA1-KD on cellular proliferation and viability induced were due to autophagy induction and not to other functions of ULK1-dependent functions, we tested the KD of ATG5, a key component of the autophagic machinery and found that the number of metabolically viable cells was only moderately affected by HMGA1-KD in cells where ATG5 was concomitantly depleted (ATG5i) (Figure 8), therefore reinforcing the concept that the effect of HMGA1 depletion on cell viability is indeed autophagy-dependent. Altogether, these data indicate that depletion of HMGA1 induces a reduction of cell viability and proliferation that require autophagy to be fully enacted.

ULK1 silencing rescues the decrease in cell viability induced by HMGA1 depletion in SCC-13 cells. (a) Cell viability assays of Ctli, HMGA1i, ULK1i and HMGA1i-ULK1i cells were evaluated as described in ‘Materials and Methods’ section. Bars represent the mean absorbance±S.D. of a representative experiment performed in triplicate. (b) Equal numbers of Ctli, HMGA1i, ULK1i and HMGA1i-ULK1i cells were plated 24 h post transfection and then stained with Crystal violet after 10 days of culture. The graph shows the mean±S.D. of the number of colonies formed in three independent experiments. (c) The propidium iodide (PI) exclusion assay using flow cytometry was used to discriminate between live and dead Ctli, HMGA1i, ULK1i and HMGA1i+ULK1i SCC-13 cells 96 h post transfection. A representative experiment is shown

ATG5 silencing reduces the decrease in cell viability induced by HMGA1 depletion in SCC-13 cells. (a) The efficacy of siRNAs directed against ATG5 was tested by western blotting. Actin was used as loading control. The siRNA(s) used to KD ATG5 in ATG5i#1, ATG5i#2, ATG5i#3, ATG5i#4 and ATG5i cells are indicated in ‘Materials and Methods’ section. (b) Proteins extracted from Ctli, HMGA1i, ATG5i and HMGA1i-ATG5i SCC-13 cells were tested for HMGA1 and ATG5 expression by western blotting. Actin was used as loading control. (c) Cell viability assay of Ctli, HMGA1i, ATG5i, HMGA1i-ATG5i, ATG5i#1, HMGA1i-ATG5i#1, ATG5i#2 and HMGA1i-ATG5i#2 cells were evaluated as described in ‘Materials and Methods’ section. Bars represent the mean absorbance±S.D. of a representative experiment performed in triplicate
Discussion
HMGA overexpression is a frequent event in human malignancies, and correlates with cancer progression, high aggressiveness and poor prognosis.1, 2, 3, 4, 5, 6, 7 HMGA proteins contribute to carcinogenesis by several mechanisms: (a) inactivation of p53-induced apoptosis28 and regulation of cancer stem cell division;29 (b) impairment of DNA repair;3 (c) transcriptional regulation of miRNAs and genes involved in the control of the cell cycle30, 31 and induction of epithelial-mesenchymal transition;32, 33 (d) induction of AP-1 activity;34 (e) induction of chromosome instability.35, 36 Nevertheless, it is general opinion that HMGA1 exerts its oncogenic activity also by other unidentified mechanisms.
Here, we demonstrate that HMGA1 expression has a key role in autophagy regulation. Indeed, HMGA1 silencing induces a ‘dual effect’ on the autophagic pathway associated to a decrease in cellular proliferation and viability in human epidermal squamous SCC-13 cells and uterine cervix HeLa cancer cells. HMGA1-depleted cells show typical features of increased autophagy, including reduced phosphorylation of AKT and ribosomal S6 proteins, increased rate of LC3-II conjugation, and re-distribution of LC3 and p62/SQSTM1 to both autophagosomes and autophagolysosomes (Figures 2 and 3). Nevertheless, by using a range of assays, we have shown how such an increase in autophagosomes formation is not mirrored by an equal increase in their maturation, with a consequent reduction of the autophagic flux. The observed increase in LC3-II levels is consistent with this dual effect of HMGA1 depletion on the autophagic pathway. In fact, an increase in levels of LC3-II can have different possible causes: increased autophagosome formation, a block or delay in autophagosome maturation, or both. Such a dual effect is very similar to that induced by the depletion of Sly1, whose effects on both LC3-II levels and GFP-mRFP-LC3 distribution are superimposable to those caused by depletion of HMGA1.22 A similar detrimental effect on the autophagic flux has been reported also for some dual PI3K/mTOR inhibitors (DKI), which are canonically considered to be autophagy activators and currently under study for the potential treatment of several types of cancers. Indeed, DKIs such as PI-103 and NVP-BEZ-235 strongly compromise autophagic flux, by concomitantly inhibiting the class-III PI3K Vps34, which in turn causes the blockade to autophagosome–lysosome fusion.37 In such a scenario, the autophagy perturbation exerted by these molecules may ultimately contribute to cell death in a way similar to the one we propose here for the HMGA1 depletion.
Furthermore, the evaluation of MDC/Lysotracker staining indicates that the loss of HMGA1 appears to impair the structure (i.e. numbers and/or size) of the lysosomal compartment, which is suggestive of an altered autophagosome–lysosome fusion. As such, this could also explain the negative effect on the activity of the mTOR pathway38 observed in HMGA1-depleted cells. Moreover, the increase in autophagosomes formation induced by HMGA1 depletion, augmenting the ratio between autophagosomes and lysosomes, may favor the fusion of several autophagosomes with one lysosome, as suggested by the increase in the fluorescence intensity of red dots found in HMGA1-depleted cells using the GFP-mRFP-LC3 reporter assay. This may partially compensate the decrease in red-only dots and contribute to explain the autolysosomal degradative capacity shown by HMGA1i cells. The observed delay of the autophagic flux is not so surprising. In fact, HMGA1 may not be involved in the regulation of the late stages of autophagy, differently from other transcription factors, such as TFEB and ZKSCAN3.39 In fact, our data does not necessarily imply that HMGA1 depletion impairs or decreases lysosomal capacity per se, rather that the increase in autophagosomes formation induced by HMGA1 depletion is not associated to a proportionate increase in autophagosomes maturation and lysosomal capacity sufficient to guarantee an appropriate turnover of autophagosomes. Consistently with this hypothesis, we performed a fluorogenic assay to measure Cathepsin L, which infers about the activity of the lysosomal acidic hydrolases, and we found no significative difference between Ctli and HMGA1i HeLa cells (data not shown), indicating that HMGA1 depletion does not alter the activity of the lysosomal compartment.
Nevertheless, the effect of HMGA1-KD on autophagy eventually results in a decrease in cellular proliferation and viability, as strongly indicated by the autophagy inhibition experiments (namely, ULK1- and ATG5-KD). Interestingly, autophagic cell death is usually associated to a delay/block of the autophagic flux owing to saturation of the degradative capacity of lysosomes, rather than death mediated by autophagy.40 We can hypothesize that also in our conditions the overall effect in terms of cell viability might be due, at least in part, to the impaired turnover of autophagosomes.
The differences we have observed in basal levels of autophagy (higher in SCC-13 than in HeLa cells) might explain the minor differences and only apparent discrepancies (in particular, the effect on p62/SQSTM1 levels and autophagosomes accumulation) between the two cell lines. In addition, as p62/SQSTM1 is an oxidative stress-inducible protein,41 the observed changes could be also consequent to other mechanisms, rather than to its autophagy-dependent turnover. Noteworthy, we have confirmed how a prolonged serum and amino-acid starvation (HBSS for 4 h) can paradoxically reduce LC3-II protein levels in SCC-13 cells, while it increases the number of LC3- and p62-positive puncta (Supplementary Figure S2). Hence, an increased number of autolysosomes, which correlates with the enhanced starvation-induced flux of LC3 to lysosomes, can be indeed reflected by reduced LC3-II levels as a direct consequence of the fractional amount of LC3-II (or p62/SQSTM1) degraded upon delivery to the lysosomal compartment.42, 43
Other studies have suggested a possible interplay between HMGA proteins and autophagy. Indeed, HMGA1 overexpression activates the (PI3K)/AKT and mTORC1 signaling pathways17, 18 and positively regulates the transcription of the glucose transporter SLC2A3/GLUT3 gene, thus increasing glucose uptake and ATP levels leading to AMPK inactivation with the consequent inhibition of autophagy.44 Furthermore, also HMGB1 protein, a member of other HMG subfamily, has been demonstrated to act as a pro-autophagic protein.45
Here, we provide further evidence that HMGA1 proteins are involved in autophagy by regulating ULK1 transcriptional activity. Indeed, the ULK1 gene is upregulated upon HMGA1 depletion in both HeLa and SCC-13 cells (Figure 5). In consideration of the role exerted by ULK1 in the early steps of autophagosome biogenesis, its upregulation is coherent with the increased rate of autophagosomes synthesis observed upon HMGA1 depletion. In addition, the fact that ULK1 silencing significantly prevents the effects of HMGA1-KD on autophagic pathway (Figures 6 and 7) strongly support that the HMGA1 effects on this pathway are, at least in part, directly mediated by ULK1 protein.
Considering that several HMGA1 interactors, such as p53 and NF-κB, are involved in transcriptional and post-transcriptional regulation of autophagy,39 the role of HMGA1 in the modulation of ULK genes expression may depend on these interactions and on the cellular context. In particular, as HMGA proteins impairs p53-mediated regulation of apoptotic genes46 and p53 positively regulates ULK1 expression,47 it is conceivable to speculate that HMGA1 and p53 could regulate the expression of the same autophagy-related genes in an opposite way, as already described for apoptosis-related genes.28 Moreover, resistance to autophagic cell death has also been correlated to other cellular processes in which HMGA1 proteins are involved, such as chromosomal instability and impairment of DNA repair.3, 13, 48 Therefore, the possibility that HMGA1 would enhance the survival of cancer cell and, thereby, cancer progression by inhibiting, at the same time, autophagy and apoptosis, can be envisaged. Furthermore, the ULK1-KD seems to be more effective than ATG5-KD in preventing the effects on cell viability induced by depletion of HMGA1. Hence, some additional ULK1-downstream effectors may be involved in the regulation of the entire phenomenon in an autophagy-independent manner. Indeed, ULK1 is able to exerts an autophagy-independent pro-death role in response to ROS-induced damage through PARP49 and to contribute to cellular homeostasis by regulating ER-to-Golgi trafficking.50
The role of autophagy in cancer is not completely understood, as it strongly depends on cellular context and tumor stage. Autophagy may be oncogenic allowing the survival of cancer cells by providing new energy sources under stress conditions, but it may also represent a mechanism of tumor suppression, reducing oxidative stress, activating the DNA damaging response, preventing inflammation, genomic instability and ultimately inducing cell death.13, 51 Notably, both autophagy impairment and resistance to autophagic cell death are features frequently acquired during cancer progression accounting, along with resistance to apoptosis, for enhanced cancer cell survival and chemoresistance.52 Although further investigation is needed to define the influence of HMGA1-mediated autophagy on the biology of cancer cells, one can speculate that autophagy modulation could be another way by which HMGA1 regulates cell survival and genomic stability, consistent with its pro-survival activity and its role in the development of chemoresistance.
In conclusion, the data reported here imply that HMGA1 overexpression inhibits autophagy by, at least in part, negatively regulating the expression of ULK1 gene. The roles of ULK1 in disease pathogenesis are still poorly understood and often controversial. Nevertheless, alterations in ULK/ATG1 expression and autophagy-related functions have been implicated in the prognosis of some cancers.53 The one described here represents a novel mechanism by which HMGA overexpression would enhance survival of cancer cells and contribute to cancer progression, further supporting the need of innovative antineoplastic therapies based on the selective inactivation of HMGA1/ULK1 functions, rather than on the more general autophagy inhibitors and activators.
Materials and methods
Cell cultures
HeLa cells were grown in RPMI supplemented with 10% FBS, L-glutamine, and antibiotics (Invitrogen, Carlsbad, CA, USA). Human epidermal squamous carcinoma SCC-13 cell line was kindly provided by Rheinwald JG.54 Cells were cultured in keratinocyte serum-free medium (KSFM, Invitrogen) with 25 mg/ml bovine pituitary extract, penicillin, streptomycin, 0.2 ng/ml epidermal growth factor, and CaCl2 to a final Ca2+ concentration of 0.4 mmol/l. To maintain healthy confluent cultures, after cultures reached 40% confluence, they were re-fed daily with 1:1 medium (1:1 vol/vol Ca2+-free DMEM/KSFM, supplemented as above described). HEK293 cells were cultured in DMEM with 10% FBS, l-glutamine, and antibiotics (Invitrogen). Bafilomycin A1 (B1793) has been purchased from Sigma-Aldrich (Saint Louis, MO, USA). HBSS buffer (#14025092) has been purchased from Gibco (Waltham, MA, USA).
Transfections and plasmids
Cells were transfected with plasmids by Lipofectamine plus reagent (Invitrogen) according to the manufacturer’s instructions. Cells were transiently transfected with pcDNA3.1-Hmga1b.55 Luciferase activity was analysed by LightSwitch Luciferase Assay kit (Switch Gear Genomics, Carlsbad, CA, USA), according to manifacturer’s instructions. ULK1-luc (cod. SKU:S707592) and ULK2-luc (cod. SKU:S709931) plasmids were from Switch Gear Genomics. HeLa cells stably expressing GFP-mRFP-LC3 were a kind gift of D. Rubinzstein (Cambridge Institute for Medical Research).25
RNA interference, RNA extraction and quantitative real-time PCR (qRT-PCR)
RNA interference was obtained using Lipofectamine RNAiMAX (Invitrogen) as previously described37 with the following specific siRNAs:
-
HMGA1i: Hs_HMGA1_5 (SI02662023) (Sense strand GGACAAGGCUAACAUCCCATT and Antisense strand UGGGAUGUUAGCCUUGUCCAG) (Qiagen, Hilden, Germany)
-
HMGA1i§: Stealth siRNAs (Set of 3) HSS142459, HSS142460, HSS142461, Cat. #1299001, (Life Technologies-Invitrogen)
-
ULK1i: Stealth siRNAs (Set of 3) HSS140824, HSS140825, HSS141051, Cat. #1299001, (Life Technologies-Invitrogen)
-
ULK1i#1: single Stealth siRNA HSS140824 (Life Technologies-Invitrogen)
-
ULK1i#2: single Stealth siRNA HSS140825 (Life Technologies-Invitrogen)
-
ULK1i#3: single Stealth siRNA HSS141051 (Life Technologies-Invitrogen)
-
ATG5i: Set of 4: siGENOME ATG5 siRNA, M-004374-04 (Dharmacon, LA, USA)
-
ATG5i#1: deconvoluted siRNA J-004374-07 (Dharmacon, LA, USA)
-
ATG5i#2: deconvoluted siRNA J-004374-08 (Dharmacon, LA, USA)
-
ATG5i#3: deconvoluted siRNA J-004374-09 (Dharmacon, LA, USA)
-
ATG5i#4: deconvoluted siRNA J-004374-10 (Dharmacon, LA, USA)
As negative control, Qiagen AllStars control (SI03650318) was used. Cells were transduced with 25 nM of siRNA using RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions and analyzed after 48, 72 and 96 h. Total RNA was isolated as previously described.56 qRT-PCR analysis was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) with following primer sequences:
humanHMGA1-Fw 5′-CAACTCCAGGAAGGAAACCA-3′
humanHMGA1-Re 5′-AGGACTCCTGCGAGATGC-3′
humanULK1-Fw 5′-CAGACAGCCTGATGTGCAGT-3′
humanULK1-Re 5′-CAGGGTGGGGATGGAGAT-3′
humanULK2-Fw 5′-TTTAAATACAGAACGACCAATGGA-3′
humanULK2-Re 5′-GGAGGTGCCAGAACACCA-3′
humanACTIN-Fw 5′-CCAACCGCGAGAAGATGA-3′
humanACTIN-Re 5′-CCAGAGGCGTACAGGGATAG-3′
mouseUlk1-Fw 5′-GGATCCATGGTGTCACTGC-3′
mouseUlk1-Re 5′-CAAGGGCAGCTGATTGTACC-3′
mouseUlk2-Fw 5′-CACCATCTTGTCGCTTTGC-3′
mouseUlk2-Re 5′-GGATAAGTTTTCTTCCTGAATATGCT-3′
mouseActin-Fw 5′-CTAAGGCCAACCGTGAAAAG-3′
mouseActin-Re 5′-ACCAGAGGCATACAGGGACA-3′
Growth curve and cell viability assay
SCC-13 cells were seeded in six-well plates, interfered and transfected as above described. Ctli and HMGA1i cells were counted after 48, 72 and 96 h as described in.57
Cell viability of cells was quantified by MTS (3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium) assay (Promega's CellTiter 96 AQueous One Solution, Promega Fitchburg, WI, USA). Cells were seeded in 96-well plates at 5 × 103 cells per well, then interfered and transfected as above described. After 72 h, absorbance was measured at 490 nm.
β-Galactosidase staining
β-Galactosidase staining was performed using Senescence Cells Histochemical Staining Kit (CS0030, Sigma-Aldrich) kit (Sigma-Aldrich) according to the manufacturer’s instructions 6 days after transfection. Over/night treatment with 400 μM H2O2 was used as a positive control. Cells have been observed with Axiovert 25 and photographed with Canon GC5 (final magnification × 40).
Colony assay
To perform the colony assays, the cells were transfected with siRNAs as described before. After 24 h of transfection, the cells were trypsinized, counted and re-plated in equal numbers. After 10 days of culture, the cells were fixed in methanol for 10 min, stained with Crystal Violet (C3886, Sigma-Aldrich) for 25 min, washed several times with water, and then photographed.
PI exclusion assay
For the determination of cell viability, cells were analyzed 96 h after trasfection with a FACS Calibur cytofluorimeter using CellQuest software (BD Biosciences, Mississauga, ON, Canada), following addition of a Propidium Iodide solution at a final concentration of 1 μg/ml.
Western blotting and antibodies
Protein extraction and western blotting were performed as described elsewhere.58 Antibodies directed against HMGA1 proteins were already described.59 Commercial antibodies were: anti-pSer473-AKT (#9271), anti-AKT (#9272), anti-Caspase-7 (#9492), anti-phospho S6 ribosomal protein (Ser240/244) (D68F8, #5364), anti-S6 ribosomal protein (54D2, #2317S), anti-LC3A/B (#4108), anti-PARP (46D11, #9532), anti-ATG5 (D5F5U, #12994) and anti-ULK1 (R600, #4773) antibodies from Cell Signaling Technology (MA, USA); anti-β-Actin (I-19, sc-1616), anti-β-Tubulin (D-10, sc-5274), anti-p21 (C-19, sc-397), anti-Cyclin D1 (A12, sc-8396), anti-Cyclin E (E20, sc-481), and anti-Vinculin (N-19, sc-7649) were from Santa Cruz Biotechnology (Dallas, TX, USA); anti-p27 (610241) and anti-p62/SQSTM1 (610833) were from BD Biosciences (Franklin Lakes, NJ USA); anti-phospho ATG13 (Ser318) (PAB19948) was from Abnova (Taipei City,Taiwan); anti-ATG13 (SAB4200100) was from Sigma-Aldrich. ECL System was purchased from Amersham Pharmacia (Buckinghamshire, UK).
ChIP assay
After transfection, cells have been treated with formaldehyde 1%, washed and then lysed isolating the nuclei. Then the nuclei have been in turn lysed and chromatin has been sonicated. Then, samples have been processed and immunoprecipitated using anti-HMGA1 antibody28 or normal rabbit IgG as negative control. For PCR analysis, 2 μl out of 150 μl immunoprecipitated DNA was used with primers described below. GAPDH promoter amplicon was used as negative control in all the experiments. Input DNA was used as positive control.
Primers used were:
ULK1-prom-Fw 5′-TGCCCTGTTCCATATTTTGC-3′
ULK1-prom-Re 5′-ACCCAAACCAACGACATAGC-3′
ULK2-prom-Fw 5′-AGCTGGGGATGGAGAGTACC-3′
ULK2-prom-Re 5′-AGAGACCGGAGCGGAAACT-3′
GAPDH-prom-Fw 5′-CCCAAAGTCCTCCTGTTTCA-3′
GAPDH-prom-Re 5′-GTCTTGAGGCCTGAGCTACG-3′
Fluorescence microscopy
Lysotracker Probe (Molecular Probes, Eugene, OR, USA) and MDC (50 μM, Sigma) were used to label lysosomes and autophagosomes, respectively. In brief, cells grown on coverslips were incubated with Lysotracker Probe for 1 h at 37 °C before fixation (4% PFA). Cells grown on bottom-glass dishes were incubated with MDC in PBS for 10 min at 37 °C and imaged in vivo in PBS.
Cells stained with LC3 antibody (0231-100/LC3-5F10, nanoTools, Teningen, Germany) were fixed with methanol, quenched with 0.2% BSA/10% FBS in PBS for 30 min and permeabilized with 0.2% TX-100 for 7 min. Cells stained with p62/SQSTM1 antibody (GP62-C, Progen, Heidelberg, Germany) and Lamp-1 (#555798, BD Biosciences Pharmingen, San Diego, CA, USA) were fixed in PFA 4% and then processed as above. Primary antibodies were detected with TRITC (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) or Alexa-488 or -546 (Life Technologies, Camarillo, CA, USA) conjugated secondary antibodies. Images were collected using a laser scanning microscope (LSM 510 META, Carl Zeiss Microimaging, Inc., Thornwood, NY, USA) equipped with a Plan Apo 63 × oil-immersion (NA 1.4) objective lens.
Quantification, morphometric analyses and co-localization analysis were carried out using LSM 510 software as previously described.60 The mean fluorescence intensities in selected regions of interest of equal size were measured. For the quantification we acquired the images, for each fluorophore, with the same setting (laser power, detector gain) as well as we kept the same threshold of fluorescence intensity in all experimental conditions (control and silenced cells). We evaluated the size of phagosomal or lysosomal compartments measuring the area occupied from each organelle marker that takes in account both the number and the dimension of these compartments.
For GFP-mRFP-LC3 assay, we counted the fluorescent puncta exhibiting green (GFP) or red (mRFP) fluorescence. The number of red-only puncta was obtained by subtracting green dots to the total red dots. The green-only puncta were excluded by quantitative analysis.
Statistical analysis
Student’s t-test was used to determine the significance for all the quantitative experiments. Error bars represent the S.D. of the average.
References
Fusco A, Fedele M . Roles of the HMGA proteins in cancer. Nat Rev Cancer 2007; 7: 899–910.
Sgarra R, Zammitti S, Lo Sardo A, Maurizio E, Arnoldo L, Pegoraro S et al. HMGA molecular network: from transcriptional regulation to chromatin remodeling. Biochim Biophys Acta 2010; 1799: 37–47.
Palmieri D, Valentino T, D'Angelo D, De Martino I, Postiglione I, Pacelli R et al. HMGA proteins promote ATM expression and enhance cancer cell resistance to genotoxic agents. Oncogene 2011; 30: 3024–3035.
D'Angelo D, Mussnich P, Rosa R, Bianco R, Tortora G, Fusco A . High mobility group A1 protein expression reduces the sensitivity of colon and thyroid cancer cells to antineoplastic drugs. BMC Cancer 2014; 14: 851.
Pallante P, Sepe R, Puca F, Fusco A . High mobility group A proteins as tumor markers. Front Med 2015; 2: 15.
Berlingieri MT, Pierantoni GM, Giancotti V, Santoro M, Fusco A . Thyroid cell transformation requires the expression of the HMGA1 proteins. Oncogene 2002; 21: 2971–2980.
Scala S, Portella G, Fedele M, Chiappetta G, Fusco A . Adenovirus-mediated suppression of HMGI(Y) protein synthesis as potential therapy of human malignant neoplasias. Proc Natl Acad Sci USA 2000; 97: 4256–4261.
Xu Y, Sumter TF, Bhattacharya R, Tesfaye A, Fuchs EJ, Wood LJ et al. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res 2004; 64: 3371–3375.
Fedele M, Pentimalli F, Baldassarre G, Battista S, Klein-Szanto AJ, Kenyon L et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 2005; 24: 3427–3435.
Belton A, Gabrovsky A, Bae YK, Reeves R, Iacobuzio-Donahue C, Huso DL et al. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS One 2012; 7: e30034.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51–64.
Levine B, Kroemer G . Autophagy in the pathogenesis of disease. Cell 2008; 132: 27–42.
Matthew R, Karantza-Wadsworth V, White E . Role of autophagy in cancer. Nat Rev Cancer 2007; 7: 961–967.
Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 2015 22: 58–73.
Gozuacik D, Kimchi A . Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004; 23: 2891–2906.
Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X . ULK1/ATG13/FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284: 12297–12305.
Liau SS, Jazag A, Ito K, Whang EE . Overexpression of HMGA1 promotes anoikis resistance and constitutive Akt activation in pancreatic adenocarcinoma cells. Br J Cancer 2007; 96: 993–1000.
Laplante M, Sabatini DM . mTOR signaling in growth control and disease. Cell 2012; 149: 274–293.
Menzies FM, Moreau K, Puri C, Renna M, Rubinsztein DC . Measurement of autophagic activity in mammalian cells. Curr Protoc Cell Biol 2012; Chapter 15: Unit 15.16.
Mizushima N, Yoshimori T . How to interpret LC3 immunoblotting. Autophagy 2007; 3: 542–545.
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12: 1–222.
Renna M, Schaffner C, Winslow AR, Menzies FM, Peden AA, Floto RA et al. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci 2011; 124: 469–482.
Munafo DB, Colombo MI . A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 2001; 114: 3619–3629.
Conte A, Kisslinger A, Procaccini C, Paladino S, Oliviero O, de Amicis F et al. Convergent effects of resveratrol and PYK2 on prostate cells. Int J Mol Sci 2016; 17. pii: E1542.
Vicinanza M, Korolchuk VI, Ashkenazi A, Puri C, Menzies FM, Clarke JH et al. PI (5)P regulates autophagosome biogenesis. Mol Cell 2015; 57: 219–234.
Mizushima N . The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22: 132–139.
Park D, Jeong H, Lee MN, Koh A, Kwon O, Yang YR et al. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep 2016; 6: 21772.
Esposito F, Tornincasa M, Chieffi P, De Martino I, Pierantoni GM, Fusco A . High-mobility group A1 proteins regulate p53-mediated transcription of Bcl-2 gene. Cancer Res 2010; 70: 5379–5388.
Puca F, Colamaio M, Federico A, Gemei M, Tosti N, Bastos AU et al. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget 2014; 5: 3234–3245.
Mussnich P, D'Angelo D, Leone V, Croce CM, Fusco A . The high mobility group A proteins contribute to thyroid cell transformation by regulating miR-603 and miR-10b expression. Mol Oncol 2013; 7: 531–542.
Tessari MA, Gostissa M, Altamura S, Sgarra R, Rustighi A, Salvagno C et al. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol Cell Biol 2003; 23: 9104–9116.
Pegoraro S, Ros G, Piazza S, Sommaggio R, Ciani Y, Rosato A et al. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013; 4: 1293–1308.
Reeves R, Edberg DD, Li Y . Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol Cell Biol 2001; 21: 575–594.
Vallone D, Battista S, Pierantoni GM, Fedele M, Casalino L, Santoro M et al. Neoplastic transformation of rat thyroid cells requires the junB and fra-1 gene induction which is dependent on the HMGI-C gene product. EMBO J 1997; 16: 5310–5321.
Pierantoni GM, Conte A, Rinaldo C, Tornincasa M, Gerlini R, Federico A et al. Deregulation of HMGA1 expression induces chromosome instability through regulation of spindle assembly checkpoint genes. Oncotarget 2015; 6: 17342–17353.
Pierantoni GM, Conte A, Rinaldo C, Tornincasa M, Gerlini R, Valente D et al. Hmga1 null mouse embryonic fibroblasts display downregulation of spindle assembly checkpoint gene expression associated to nuclear and karyotypic abnormalities. Cell Cycle 2016; 15: 812–818.
Button RW, Vincent JH, Strang CJ, Luo S . Dual PI-3 kinase/mTOR inhibition impairs autophagy flux and induces cell death independent of apoptosis and necroptosis. Oncotarget 2016; 7: 5157–5175.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31: 1095–1108.
Füllgrabe J, Ghislat G, Cho DH, Rubinsztein DC . Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci 2016; 129: 3059–3066.
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G . Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15: 81–94.
Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010; 285: 22576–22591.
Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 2011; 13: 453–460.
Renna M, Bento CF, Fleming A, Menzies FM, Siddiqi FH, Ravikumar B et al. IGF-1 receptor antagonism inhibits autophagy. Hum Mol Genet 2013; 22: 4528–4544.
Ha TK, Her NG, Lee MG, Ryu BK, Lee JH, Han J et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res 2012; 72: 4097–4109.
Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190: 881–892.
Frasca F, Rustighi A, Malaguarnera R, Altamura S, Vigneri P, Del Sal G et al. HMGA1 inhibits the function of p53 family members in thyroid cancer cells. Cancer Res 2006; 66: 2980–2989.
Gao W, Shen Z, Shang L, Wang X . Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ 2011; 18: 1598–1607.
Czarny P, Pawlowska E, Bialkowska-Warzecha J, Kaarniranta K, Blasiak J . Autophagy in DNA damage response. Int J Mol Sci 2015; 16: 2641–2662.
Joshi A, Iyengar R, Joo JH, Li-Harms XJ, Wright C, Marino R et al. Nuclear ULK1 promotes cell death in response to oxidative stress through PARP1. Cell Death Differ 2016; 23: 216–230.
Joo JH, Wang B, Frankel E, Ge L, Xu L, Iyengar R et al. The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol Cell 2016; 62: 491–506.
White E . The role of autophagy in cancer. J Clin Invest 2015; 125: 42–46.
Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4: e838.
Amaravadi R, Kimmelman AC, White E . Recent insights into the function of autophagy in cancer. Genes Dev 2016; 30: 1913–1930.
Rheinwald JG, Beckett MA . Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res 1981; 41: 1657–1663.
Pierantoni GM, Battista S, Pentimalli F, Fedele M, Visone R, Federico A et al. A truncated HMGA1 gene induces proliferation of the 3T3-L1 pre-adipocytic cells: a model of human lipomas. Carcinogenesis 2003; 24: 1861–1869.
Anzilotti S, Tornincasa M, Gerlini R, Conte A, Brancaccio P, Cuomo O et al. Genetic ablation of homeodomain-interacting protein kinase 2 selectively induces apoptosis of cerebellar Purkinje cells during adulthood and generates an ataxic-like phenotype. Cell Death Dis 2015; 6: e2004.
Federico A, Forzati F, Esposito F, Arra C, Palma G, Barbieri A et al. Hmga1/Hmga2 double knock-out mice display a ‘superpygmy’ phenotype. Biol Open 2014; 3: 372–378.
Conte A, Procaccini C, Iannelli P, Kisslinger A, De Amicis F, Pierantoni GM et al. Effects of resveratrol on p66Shc phosphorylation in cultured prostate cells. Transl Med UniSa 2016; 13: 47–58.
Pierantoni GM, Agosti V, Fedele M, Bond H, Caliendo I, Chiappetta G et al. High-mobility group A1 proteins are overexpressed in human leukaemias. Biochem J 2003; 372: 145–150.
Paladino S, Pocard T, Catino M, Zurzolo C . GPI-anchored proteins are directly targeted to the apical surface in fully polarized MDCK cells. J Cell Biol 2006; 172: 1023–1034.
Acknowledgements
This work was supported by grants (IG-11477 and MFAG-11702) from the Associazione Italiana Ricerca sul Cancro, from the Italian Ministry of Economy and Finance to the CNR for the ‘Project FaReBio di Qualità’, PNR-CNR Aging ‘Program 2012-2014’. We thank prof. David Rubinzstein for providing HeLa cells stably expressing GFP-mRFP-LC3.
Author contributions
AC, RG, GB and MT performed the cellular and molecular experiments; SP performed confocal microscopy analysis; DF performed the immunofluorescence experiments; MR contributed with materials and expertize for the autophagy experiments and data analysis; GMP, DT and AF coordinated the work and wrote the manuscript. All the authors discussed the results and revised the manuscript text.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Conte, A., Paladino, S., Bianco, G. et al. High mobility group A1 protein modulates autophagy in cancer cells. Cell Death Differ 24, 1948–1962 (2017). https://doi.org/10.1038/cdd.2017.117
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.117
This article is cited by
-
HMGA1 regulates trabectedin sensitivity in advanced soft-tissue sarcoma (STS): A Spanish Group for Research on Sarcomas (GEIS) study
Cellular and Molecular Life Sciences (2024)
-
Differential effects of the LncRNA RNF157-AS1 on epithelial ovarian cancer cells through suppression of DIRAS3- and ULK1-mediated autophagy
Cell Death & Disease (2023)
-
Definition of a Novel Immunogenic Cell Death-Relevant Gene Signature Associated with Immune Landscape in Gastric Cancer
Biochemical Genetics (2023)
-
Biological causes of immunogenic cancer cell death (ICD) and anti-tumor therapy; Combination of Oncolytic virus-based immunotherapy and CAR T-cell therapy for ICD induction
Cancer Cell International (2022)
-
DNAJC24 is a potential therapeutic target in hepatocellular carcinoma through affecting ammonia metabolism
Cell Death & Disease (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}