
Abstract
We previously reported that the combination of two safe proteostasis regulators, cysteamine and epigallocatechin gallate (EGCG), can be used to improve deficient expression of the cystic fibrosis transmembrane conductance regulator (CFTR) in patients homozygous for the CFTR Phe508del mutation. Here we provide the proof-of-concept that this combination treatment restored CFTR function and reduced lung inflammation (P<0.001) in Phe508del/Phe508del or Phe508del/null-Cftr (but not in Cftr-null mice), provided that such mice were autophagy-competent. Primary nasal cells from patients bearing different class II CFTR mutations, either in homozygous or compound heterozygous form, responded to the treatment in vitro. We assessed individual responses to cysteamine plus EGCG in a single-centre, open-label phase-2 trial. The combination treatment decreased sweat chloride from baseline, increased both CFTR protein and function in nasal cells, restored autophagy in such cells, decreased CXCL8 and TNF-α in the sputum, and tended to improve respiratory function. These positive effects were particularly strong in patients carrying Phe508del CFTR mutations in homozygosity or heterozygosity. However, a fraction of patients bearing other CFTR mutations failed to respond to therapy. Importantly, the same patients whose primary nasal brushed cells did not respond to cysteamine plus EGCG in vitro also exhibited deficient therapeutic responses in vivo. Altogether, these results suggest that the combination treatment of cysteamine plus EGCG acts ‘on-target’ because it can only rescue CFTR function when autophagy is functional (in mice) and improves CFTR function when a rescuable protein is expressed (in mice and men). These results should spur the further clinical development of the combination treatment.
Similar content being viewed by others
Main
Cystic fibrosis (CF), the most common lethal recessive disease in Caucasians, affects ~70 000 subjects worldwide and results from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR).1, 2 Mutation-specific CFTR-repairing therapies with a channel-potentiator (Ivacaftor) are available for ~5% of CF patients bearing membrane-resident class III mutants.3 In contrast, for the vast majority of CF patients bearing the most common F508del-CFTR mutation, an FDA-approved combined treatment with the corrector Lumacaftor (that promotes ER to plasma membrane (PM) traffic) and Ivacaftor is marginally effective.4, 5, 6, 7 F508del-CFTR is retained in the endoplasmic reticulum (ER) and prematurely degraded before it reaches the PM.8 Besides a gating defect,8 F508del is unstable and rapidly dismissed from the PM even upon treatment with correctors. When Ivacaftor is combined in vitro with CFTR correctors, the former counteracts the latter by further decreasing F508del-CFTR PM stability after rescue.9, 10 This might explain the marginal clinical effects of this combination in F508del-CFTR patients4 as it fails to reverse sweat test, a surrogate marker of disease reversal,11, 12 consistent with putative ‘off-target’ effects.
Discordance in therapeutic response rate complicates mutation-specific approaches. Existing candidate drugs, as Lumacaftor, fail to act on other class II mutants,13, 14 show poor efficacy in vitro and lack of benefit in vivo5 in CF patients bearing only one copy of F508del. This indicates that a different rationale is needed based on mechanistic rather than mutation-specific approaches, and suggests strategies focusing on individual and not average responses to therapy.13
Recent results suggest that CFTR orchestrates a proteostatic network that influences multiple cellular functions by acting as a hub protein.15 This hub-dysfunction model proposes that the proteostasis network is widely deranged, both in transgenic CF mice and in primary epithelial cells from F508del-CFTR homozygous patients, at two levels. First, autophagy, the major mechanism determining cytoplasmic protein turnover, is blocked owing to tissue transglutaminase (TG2)-mediated depletion of the essential autophagy-related protein Beclin 1 (BECN1), leading to secondary accumulation of the autophagic substrate SQSTM1/p62.16 Second, peptide fragments released from proteolytically cleaved F508del-CFTR provoke an overactivation of the pleiotropic protein kinase CK2, which in turn contributes to F508del-CFTR degradation.17 Combined inhibition of both TG2 by the repurposed cysteamine, FDA-approved for the treatment of cystinosis,18, 19 (which reestablishes autophagy) and over-active CK2 by the over-the-counter green-tea flavonoid epigallocatechin gallate (EGCG) synergize in vitro and thereby rescue and stabilize, respectively, a functional F508del-CFTR protein at the PM, both in mice and in primary nasal cells from F508del-CFTR homozygotes.20 This prompted a pilot trial combining cysteamine and EGCG in ten F508del-CFTR homozygotes, showing that the combination treatment was well tolerated, reverted sweat chloride toward normal and significantly attenuated biomarkers of airway inflammation.20
Here, we provide new insights from newly generated transgenic mice and patients primary cells and investigate in a phase-2 clinical trial the individual patient responsiveness to a combination of cysteamine plus EGCG in CF patients bearing different classes of CFTR mutation. Moreover, we evaluate the feasibility of using both functional and mechanistic biomarkers as a prediction test of patient responsiveness to facilitate a future personalized approach to precision CF therapy.
Results
Proof-of-concept studies in mice
To provide the proof-of-concept for the use of cysteamine plus EGCG in CF patients bearing one copy of F508del-CFTR and another severe CFTR mutation with negligible residual CFTR activity, F508del-CFTR homozygous mice (CftrF508del/F508del) were backcrossed with transgenic knockout (KO) Cftr mice (Cftr−/−) to obtain F508del/null CFTR heterozygous mice (CftrF508del/−). Mice were gavaged with cysteamine or EGCG alone or cysteamine plus EGCG for 5 days followed by EGCG alone or vehicle for further 2 weeks.20 The primary end point of this mouse trial (Supplementary Figure S1) was the rescue of CFTR function and protein. Secondary end points were the restoration of BECN120, 21, 22 and the reduction of Cxcl2 (chemokine[C-X-C motif]-ligand 2) and Tnf-α (tumor necrosis factor) transcripts in lung homogenates.
Cysteamine alone or combined with EGCG (but not EGCG alone) restored rectal potential difference (RPD) responses to forskolin in vivo to mean 79.4% (95% confidence interval (CI): 68.1–90.8%) of WT controls in CftrF508del/F508del mice and 84.7% (77.5–91.8%) in CftrF508del/− mice (P<0.001) (Figure 1a and b). The treatment significantly improved the forskolin-induced increase in short-circuit current (Isc) in mouse ileum mounted in Ussing chambers to 130% (123.6–136.4%) and 138.7%, (130.4–147.0%) of WT controls (P<0.001), respectively, which was partially reverted by a selective CFTR inhibitor, CFTRinh172 (Figure 1d and e), indicating that CFTR function was restored by the treatment. The inability of Cftr−/− mice to mount a similar response to cysteamine (Figure 1c and f) indicated that mutated CFTR protein is indispensable for therapeutic effect. No effects of cysteamine or EGCG on CFTR function were observed in WT mice (Figure 1h and Supplementary Figure S2). Cysteamine alone or combined with EGCG, but not EGCG alone, rescued mature CFTR protein (band C) expression to >50% of WT values and restored BECN1 expression in intestinal homogenates from either CftrF508del/F508del and CftrF508del/− (Figure 2a), whereas no CFTR protein was detected in intestine from Cftr−/− mice, regardless of treatment (Figure 2b).

Proof-of-concept studies in mice: effects of treatment on CFTR function in CftrF508del/F508del, CftrF508del/−, Cftr−/− and CftrF508del/F508delBecn1+/– mice. Effects on CFTR function of oral administration of different combinations of cysteamine and EGCG in CftrF508del/F508del (a and d), CftrF508del/− (b and e) and Cftr−/− (c and f) mice. The different treatment schedules are indicated. (a–c) Rectal potential difference (RPD) in vivo in response to 20 μM forskolin (Fsk) (mean±S.D.); (d–f) CFTR-dependent Cl− secretion measured by means of forskolin-induced increase of the chloride current (Isc (μA/cm2)) ex vivo in the ileum mounted in Ussing chambers and effects of CFTR inhibition (CFTRinh-172, abbreviated Inh) and amiloride (abbreviated Am) for each group of treatment (#P<0.001 versus vehicle-treated mice). (g–i) Effects of treatment on CFTR function in Cftr+/+Becn1+/– (g), FVB/129Cftr+/+ (h) and CftrF508del/F508delBecn1+/– (i) mice. The different treatment schedules are indicated. CFTR-dependent Cl− secretion measured as by means of forskolin-induced increase of the chloride current (Isc (μA/cm2)) ex vivo in the ileum mounted in Ussing chambers and effects of CFTR inhibition (CFTRinh-172, abbreviated Inh) and amiloride (abbreviated Am) for each group of treatment (#P<0.001 versus vehicle-treated mice). Eight-week-old mice, n=10 per group of treatment. Treatments: Cys, cysteamine; EGCG, epigallocatechin gallate; Cys+EGCG, 5 days treatment with cysteamine and EGCG; Cys+Wo+EGCG, 5 days treatment with cysteamine and EGCG followed by 2 weeks of cysteamine washout in the presence of EGCG

Proof-of concept studies in mice: effects of treatment on rescue of functional CFTR protein and lung inflammation in CftrF508del/− mice. (a) Representative immunoblot with anti-CFTR (clone CF3 Abcam) and BECN1 (clone Ab55878) (top left) and densitometric measurement as fold increase respect to vehicle-treated mouse normalized to β-actin levels (bottom left) in one CftrF508del/− mouse per treatment group; (right) assessment of CFTR-dependent Cl− secretion measured by means of forskolin-induced increase of the chloride current (Isc (μA/cm2)) ex vivo in the ileum mounted in Ussing chambers and effects of CFTR inhibition (CFTRinh-172, abbreviated Inh) and amiloride (abbreviated Am) (right) in one mouse per treatment group. (b) Representative immunoblot with anti-CFTR (clone CF3 Abcam) (left) and assessment of CFTR-dependent Cl− secretion (right) in one Cftr−/− mouse per treatment group. (c) Representative immunoblot with anti-CFTR (clone CF3 Abcam) (top) and densitometric measurement (bottom) in one CftrF508del/F508delBecn1+/– mouse per treatment group mean±S.D. of independent measurements; #P<0.001 versus vehicle-treated mice (ANOVA). (d) Effects of oral administration of cysteamine and EGCG on cytokine expression in lung homogenates from CftrF508del/− mice. The different treatment schedules are indicated. Cxcl2 (left) and Tnf (right) mRNA levels in lung homogenates from 8-week-old mice (n=10 per group of treatment). (*P<0.01, #P<0.001 versus untreated mice). Cys, cysteamine; EGCG, epigallocatechin gallate; Cys+EGCG, 5 days treatment with cysteamine and EGCG; Cys+Wo+EGCG, 5 days treatment with cysteamine and EGCG followed by 2 weeks of cysteamine washout in the presence of EGCG; Cys+EGCG+Wo, 5 days treatment with cysteamine and EGCG followed by 2 weeks of washout of both cysteamine and EGCG
To determine whether the effects of cysteamine on F508del-CFTR rescue were linked to its capacity to restore autophagy, we obtained CftrF508del/F508del mice in Becn1 haploinsufficient background (CftrF508del/F508del/Becn1+/−). Cysteamine lost its therapeutic efficacy in CftrF508del/F508del Becn1+/– mice (Figure 1g and i and Figure 2c), supporting the importance of BECN1-dependent autophagy for drug effects.
The combination treatment significantly reduced Cxcl2 and Tnf-α mRNA levels in lung homogenates from CftrF508del/F508del and CftrF508del/− mice (Figure 2d). All the responses observed in these mice were maintained for 2 weeks beyond cysteamine withdrawal, provided that EGCG treatment was maintained during this period (P<0.001 versus mice treated with vehicle during cysteamine withdrawal) (Figures 1 and 2).
Taken together, proof-of-concept data supported the feasibility of treating autophagy-competent mice expressing at least one F508del-CFTR allele by a combination of cysteamine and EGCG.
Human studies
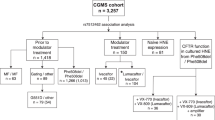
To translate preclinical data in humans, 52 consenting CF patients were sequentially enrolled at Regional Cystic Fibrosis Care Center, University of Naples Federico II, in a phase-2 clinical trial. Patients were F508del homozygous or compound heterozygous for (i) F508del or another class II mutation on one allele and a severe CFTR mutation with minimal residual CFTR activity (class I or II) on the other allele or (ii) class I mutations on both alleles (negligible CFTR synthesized). Schedule of treatment comprised oral cysteamine alone for 8 weeks, followed by cysteamine plus EGCG for further 4 weeks (or 8 weeks in a subgroup of 10 F508del-CFTR homozygous patients) followed by EGCG alone for additional 8 weeks (Figure 3). Throughout, all subjects continued their pre-study medication. Forty-two of 52 subjects who accepted the treatment schedule were assigned to three different cohorts by genotype, whereas 10 who declined treatment schedule, all bearing F508del or other class II CFTR mutations, were allocated to the untreated ‘standard-of-care’ control group (cohort 4) to follow the natural history of disease during an overlapping period. Baseline characteristics of study population and scheduled visits are reported in Supplementary Tables S1–S5.

Study profile and study design. Other*=Class I or II mutations. § Patients (Subgroup 1A, N=10) had already participated to the first part of the study and were enrolled after at least 4 months of washout when all laboratory and clinical parameters returned at the values registered prior study. Dosage: cysteamine: <12 years: 900 mg/die in four doses; >12 years: 1200 mg/die in four doses. EGCG: 270 mg/die in one dose. AE, adverse events; Cys, cysteamine; EGCG, epigallocathecin gallate
In vitro studies before in vivo treatment
Before in vivo treatment, primary nasal epithelial cells freshly collected by nasal brushing at baseline were challenged in vitro with cysteamine and/or EGCG, and then kept in culture with medium alone or EGCG up to 48 h.20 The combination treatment in vitro restored both CFTR function and CFTR band C protein in PM fractions up to >50% of non-CF controls in all but one patient bearing at least one class II mutation. F508del homozygous patients were responsive, as described,20 whereas all subjects bearing two class I mutations were unresponsive. The effects of combination treatment in vitro in class II patients persisted up to 48 h following cysteamine washout, provided that EGCG was maintained in the culture medium (Figure 4, Supplementary Table S6).

In vitro test of patients responsiveness to treatment in nasal cells freshly collected at week 0 from patients enrolled in the clinical study. (a) Analysis of CFTR function and (b) residual CFTR protein at PM in nasal epithelial cells from 45/52 CF subjects enrolled in the clinical study at baseline and cultured for 18 h with or without cysteamine (250 μM) or with cysteamine plus EGCG (80 μM) and then kept for 48 h in medium or medium added with EGCG. Individual values of patients in different cohorts. (a) Rate of halide efflux, in CF subject of different cohorts expressed as percent of values of five non-CF healthy controls (considered as 100% of value). The analysis was performed on at least 50 cells per sample and per experiment. (b) Individual values of residual CFTR protein at the PM of CF patients in different cohorts. The values are expressed as percent of five non-CF healthy control. Mean values of independent experiments. Black line represents the mean value. Cys, cysteamine; EGCG, epigallocatechin gallate; Wo, washout. Cys+EGCG, treatment with cysteamine and EGCG; Cys+EGCG+Wo, treatment with cysteamine and EGCG followed by 48 h of culture with medium alone (washout of both cysteamine and EGCG); Cys+Wo+EGCG, treatment with cysteamine and EGCG followed by 48 h of cysteamine washout in the presence of EGCG
In vivo treatment
In total, 88.5% of patients completed the study. Mean adherence to treatment was 93.8%. Given the short half-life of cysteamine, data from two patients with adherence <60% were excluded. The pharmacokinetic profile of cysteamine was as described in our pilot trial (Supplementary Figure S3).20 The incidence of adverse events was comparable to that observed in cysteamine-treated cystinosis (Supplementary Tables S7–S11).
Treatment efficacy. The primary end point was the change in CFTR function, measured by a two-site assessment: decrease of sweat chloride values from baseline of >15% coupled to increase of CFTR function and CFTR band C protein in nasal epithelial cells of >15% of non-CF controls. Secondary end points were (i) restoration of autophagy in nasal brushing (mechanistic end point biomarkers); (ii) reduction from baseline of inflammatory cytokines in both nasal cells and sputum (biomarker of organ inflammation); (iii) absolute change from baseline in percentage of predicted forced expiratory volume at 1 s (ppFEV1) (marker of clinical efficacy).
The study met the primary end point of efficacy. In the 34 treated patients who completed the study the combined therapy reduced sweat chloride concentrations by mean −18.0 mmol/liter (range, −66 to 21; 95% CI, −23.7, −12.3 mmol/l), corresponding to mean −19.8% (−50 to 24.4%; 95% CI, −25.5; −14.1%) as compared with baseline (P<0.0001, t-test treatment versus baseline) (Figure 5a,Supplementary Figure S4) and increased CFTR function in nasal cells from mean (95% CI) 5.8% (4.8, 6.7%) at week 0 up to mean 20.9% (18.7, 23.1%) (expressed as percentage of non-CF controls) after combination treatment (P<0.0001 versus baseline) (Figure 5b). Accordingly, the abundance of band C CFTR protein increased from mean 9.9% (95% CI, 9.4, 10.5%) of non-CF control values to 60.5% (55.4, 65.7%) after combined treatment (P<0.0001 versus baseline) (Figure 5c,Supplementary Figure S5a). The increase percentages of CFTR function and band C protein in nasal brushing are strongly correlated (Pearson's correlation coefficient=0.61 with the associate P=0.0001). Notably, the percentage change of sweat chloride is negatively correlated with change in CFTR function in nasal cells (Pearson’s correlation coefficient=−0.47 with the associated P=0.005, Spearman’s correlation coefficient=−0.44 with P=0.009) (Figure 5d). Changes of primary end points were negligible in the untreated cohort 4 throughout the study (P>0.5 versus baseline) (Figure 5a–c).

Treatment efficacy on primary end point in study population. (a) Mean change in sweat chloride concentrations (mmol/liter) from baseline during the whole study period in treated (blue line) (weeks 8, 12, 20, 24 of treatment schedule) and untreated (red line) groups (week 12, 24 of observation). In treated cohorts, weeks 16, 24 and 28 (italic) indicate the corresponding time points in the subgroup of patients who received combination treatment for 8 weeks. Data from all cohorts of treated patients at the end of each treatment period are pooled for the analysis. Bars are 95% CI. #P<0.0001. (b–c) Mean values of CFTR function (b) and CFTR band C protein (c) expressed as percent of non-CF control subjects in treated (left) and untreated (right) patients during the whole study period. Bars are 95% CI. #P <0.001 versus baseline. (d) Scatter plot of nasal and sweat measure of CFTR function. The red line represents the estimated linear regression model, with a regression coefficient of −1.1 and its associated P-value of 0.0046. Wk=week. (e) Individual sweat chloride values (mmol/liter) in all study cohorts. Black lines indicate mean values in different cohorts of treated patients (weeks 0, 8, 12, 20, 24 of treatment schedule). Red lines indicate mean values of untreated patients (cohort 4) (week 0, 12, 24 of observation). Red dashed lines indicate the pathological sweat test cutoff value
Regarding the secondary end points, the combination treatment increased BECN1 levels in nasal cells from mean (95% CI) 22.4% (21.9, 22.8%) of non-CF controls at the baseline, up to 53.1% (49.6, 56.6%) and decreased SQSTM1/p62 from mean 276.3% (274.1, 278.6%) of controls to 134.0% (119.5, 148.6%) (P<0.0001 versus baseline) (Supplementary Figure S5b). Moreover, in treated patients, CXCL8 (chemokine[C-X-C motif]-ligand 8) and TNF-α mRNA levels in nasal brushing and protein levels in the sputum significantly decreased after combination treatment respect to baseline (P<0.001) with a mean percentage (95% CI) decrease of CXCL8 and TNF-α of −57.7% (−45.5, −69.9%) and −55.8% (−45.5, −66.2%), respectively, in nasal cells and −45.6% (−34.5, −56.7%) and −26.3% (−14.8, −37.9%) in the sputum. Notably, cytokine expression remained attenuated after 4 weeks following drug discontinuation (Figure 6a, d and e,Supplementary Figure S6). Changes in either autophagy markers or cytokine expression (Figure 6a) were negligible in the untreated cohort 4 throughout the study.

Treatment efficacy on secondary outcomes in study population. (a) Percentage change in CXCL8 and TNF-α transcripts in nasal cells (left) and CXCL8 and TNF-α protein levels in the sputum (right) throughout the study in all treated (blue lines) (weeks 12, 24 of treatment schedule) and untreated (red lines) cohorts (week 12, 24 of observation). (b) Absolute change in ppFEV1 throughout the study in all treated (blue lines) (weeks 12, 24 of treatment schedule) and untreated (red line) (week 12, 24 of observation) cohorts. (c) Absolute change in ppFEV1 throughout the study in treated patients who received 4 (dashed line) or 8 (solid line) weeks of combination of cysteamine plus EGCG. Bars are 95% CI. Wk, week. Week 12 and 24 or Week 16 and 28 (italic) indicate the corresponding time points in patients who received the combination treatment for 4 or 8 weeks, respectively. (d and e) Individual value of percentage change from baseline of CXCL8 (left) and TNF-α (right) transcript levels in brushed nasal cells (top) and CXCL8 and TNF-α protein levels in the sputum (bottom) throughout the study. Black lines indicate mean values of treated patients; red lines indicate mean values of untreated patients. In treated cohorts, Week 12 and 24 or Week 16 and 28 (italic) indicate the corresponding time points in the subgroup of patients who received combination treatment for 4 or 8 weeks, respectively
In this short-term clinical trial (the effective combination treatment covers 4 or 8 weeks of the 24-week study) a mean absolute change from baseline, although not significant, of 3.2 in ppFEV1 (95% CI, 0.2, 6.3) was observed after combination treatment and tended to increase at the post-treatment visit (Figure 6b). The increase was greater (6.2 (95% CI, 1.0, 11.4)) in the subgroup of patients (subgroup A of cohort 1) who received combination treatment for 8 weeks (Figure 6c). By contrast, negligible mean changes of –0.7 (95% CI, −4.7, 2.8) were observed in the untreated group (Figure 6b).
Individual responsiveness to treatment. Given the well-known inter- and intra-individual variability of either ppFEV123 or sweat chloride concentrations,24 and the discordance in response rate to treatment even in patients with the same genotype,25, 26 we assessed the individual patient responsiveness to treatment by analyzing the proportion of patients who met primary end point criteria. We pre-specified that two criteria must be simultaneously fulfilled to define responsiveness: (1) decrease of sweat chloride concentrations to values below −99% CI from mean individual values during the preceding 24 months, including week – 0 value; 2) increase of both CFTR function and band C protein in nasal cells to >15% of non-CF controls. Next, we analyzed the proportion of participants who had fulfilled the primary end point (referred to as responders), who also show: (i) autophagy rescue in nasal cells (BECN1 >50% of non-CF controls and SQSTM1/p62 levels <50% from baseline); (ii) decrease of CXCL8 and/or TNF-α levels in nasal cells and/or sputum by >30% from baseline; (iii) absolute change from baseline in ppFEV1. Individual values are shown in Figure 5e, and Figures 6d and 7, Supplementary Figure S7 and Supplementary Tables S12–13.

Individual values of biomarkers measured in brushed nasal cells in all study cohorts throughout the study: CFTR function (a) CFTR band C protein (b) and BECN1 (c). Values are expressed as percentage of non-CF controls (considered as 100% value). Black lines indicate mean values in different cohorts. Red dashed lines indicate 15% (a) and 50% (b and c) of values of non-CF control subjects. Wk, week; Cys, cysteamine; EGCG, epigallocatechin gallate. In treated cohorts Wk 12–20 and Wk 16–24 (italic) indicate the corresponding time points in patients who received the combination treatment for 4 or 8 weeks, respectively. In untreated cohort Wk 12 and Wk 24 indicate the corresponding observational time points throughout the study
Among 34 treated participants, 26 (76.5%) fulfilled composite primary end point criteria after combination treatment. Sweat chloride fell below 60 mmol/liter in eight subjects bearing at least one class II mutation (Figure 5e). Patients bearing class I mutations on both alleles were non-responders. None of untreated participants met the composite primary end point criteria throughout the study (Fisher's test P<0.001 versus treated subjects). Only three responders (all bearing F508del) met the criteria after the first 8 weeks of treatment with cysteamine alone (Fisher's test P=0.5 versus untreated patients). Autophagy in nasal cells was restored in all responders after combination treatment but in none of untreated participants throughout the study (Fisher test P<0.001).
In 23 of 26 responders, at least two of either nasal or sputum CXCL8 and/or TNF-α levels decreased by >30% from baseline after combination treatment. None of untreated subjects manifested a similar decrease versus baseline (Fisher's test, P <0.01 versus treated subjects) (Figure 6d and e).
In 13 out of 34 treated patients (but in none of the untreated patients, Fisher's test P=0.021) the increase in ppFEV1>4 (relative to baseline) achieved at the end of the combination treatment, was conserved at the post-treatment visit (Table 1). Ten of them belong to the 26 responders, and other 2 (no. 12 and 27, Table 1) met all the sequential steps of responsiveness, except for the sweat test, i.e., restoration of functional CFTR protein and autophagy coupled to stable decrease of inflammatory biomarkers. Six of them (No. 1, 2, 5, 8, 9, 10 of Table 1) received combination treatment for 8 instead of 4 weeks (subgroup A of cohort 1). This is consistent with preclinical data in mice demonstrating that the beneficial effects of treatment are maintained for several weeks after drug withdrawal. Notably, the occasional (at one single visit) changes in ppFEV1 in untreated cohort were never coupled to a decrease of inflammatory cytokines nor to restoration of CFTR function or autophagy (Table 1). To look at causal relationship between improvement in ppFEV1 at the end of combination treatment and at the last post-treatment visit, a linear regression model was inferred. A positive regression coefficient was obtained (0.8) with an associated P=0.000135.
Predictive value of biomarkers
Personalized approaches to treatment require biomarkers as either (i) predictors of putative individual responsiveness before treatment or (ii) early detectors of efficacy during treatment before clinical benefits become evident.
Analysis of biomarkers in vitro before treatment
In all but one treated patients, in vitro challenge of nasal cells at week 0 with cysteamine plus EGCG restored both CFTR function and band C protein in PM fractions up to >50% of non-CF controls. All responders to the treatment in vivo manifested a rescue to >50% of CFTR function and protein in vitro (sensitivity: 100%), whereas 25/29 (86.2%) patients who responded in this way in vitro were also responders in vivo. Thus, rescuing >50% of functional CFTR protein in vitro could support the clinical decision to treat in the future with a negative predictive value of 100% and a positive predictive value of 86.2% (sensitivity 100%, specificity 50%). Therefore, a biomarker for treatment response in vivo could include absolute changes in CFTR protein levels (Supplementary Figure S8, see ROC curve).
Analysis of biomarkers during in vivo treatment
The restoration of CFTR function, as measured ex vivo during in vivo treatment, could be a useful tool to monitor the therapeutic responsiveness before stable beneficial effects on lung inflammation or clinical parameters become evident. The restoration of CFTR function was present in 12/13 (99.3%) patients who manifested a stable control of inflammation coupled to an absolute increase of ppFEV1>4 percentage points that is maintained at the end of the study.
Discussion
Our results challenge the currently accepted model that CF is a mere channelopathy that must be treated with agents directly acting on CFTR. The data provide the first evidence that targeting the abnormal proteostasis network results in CFTR repair coupled to clinical benefit in CF patients bearing at least one copy of class II CFTR mutation, including F508del, whereas it is not effective in subjects carrying two class I mutations. This effect can be obtained by two proteostasis regulators, the repurposed drug cysteamine, and the safe nutraceutical EGCG, which target two key nodes of mismanaged proteostasis in CF, disabled autophagy and CK2 overactivation.
The effects of treatment are consistent with our mechanistic animal data showing that this combination treatment is not effective in Cftr-null mice (no protein rescuable), whereas it is similarly effective in mice bearing one or two copies of F508del-CFTR, provided that F508del-CFTR mice are autophagy-competent (no effect in CftrF508del/F508del/Becn1+/− mice). In addition, cell-based pre-treatment data in patient's primary nasal cells support the use of combination treatment in patients bearing other severe class II mutations, including N1303K or G85E.
The primary efficacy end point of our clinical study was the restoration of CFTR function in two organs (airways and sweat duct). A short-term treatment with cysteamine plus EGCG, but not with either alone, decreased baseline sweat chloride concentrations by ~20% while restoring functional CFTR protein up to ~20% of non-CF controls in nasal cells. The combination treatment reversed the pathogenic cascade of disease as it re-established autophagy in nasal cells, indicating that drugs act ‘on-target’, in agreement with the hypothesis underpinning our drug design. The restoration of a functional CFTR protein is clinically relevant, as it significantly reduces inflammation markers and increases FEV1 predicted from baseline, even beyond the treatment period.
Personalized medicine is in vogue27, 28, 29, 30 and CF is a paradigm of heterogeneity in patient response to treatments.25 Pooled analysis in large-scale trials might not consider such variability, thus entailing the need of patient-centered approaches to assess drug efficacy. Testing patient's responsiveness to treatment using surrogate markers may help to predict individual responsiveness at the clinical level. In total, 76.5% of treated patients were responders to the combination treatment, whereas none of untreated participants met these criteria throughout the study. The genotype determined the likelihood of response as all responders carried at least one copy of F508del-CFTR or other class II mutations, but none of them carried two class I CFTR mutants.
CF is characterized by unresolved inflammation.31 After the combination treatment, 88.4% of responders showed >30% decrease of inflammatory cytokines that was retained at the last post-treatment visit, in agreement with mouse data. The prolonged control of lung inflammation may pave the way for the effect of treatment on lung function. Six of 10 patients who received the combination treatment for 8 weeks, retained major clinical benefit until the last post-treatment visit, suggesting a clinical benefit increasing over time within a stably un-inflamed environment.
Testing putative individual responsiveness to treatment by appropriate biomarkers before in vivo therapy should support the decision to treat. We show that restoring CFTR function in vitro in nasal cells in response to cysteamine plus EGCG is highly predictive of whether the combination treatment will restore CFTR function in vivo. Biomarkers of autophagy may add value to monitoring treatment efficacy in patients, according to the supposed mechanism of action. Hence, this in vitro assay may constitute a tool to guide the clinical development of CF treatments, allowing to identify patients who may profit from therapeutic options as the one that we detail here.
Our data indicate sweat test as feasible surrogate biomarker of CFTR function, provided that standardized procedures are respected to reduce intra-individual variability. Sweat chloride changes exhibit significant inverse correlation with CFTR function in nasal cells. Although more variable than CFTR measurements in nasal cells, sweat test should be recommended in clinical trials aimed at CFTR repair.
Our novel therapy results from long-term academic research aiming at refining disease-relevant targets for the action of safe/repurposed drugs. Our combination has another major advantage over existing channel-centered therapies for F508del-CFTR as it is well tolerated and affordable, potentially costing >10-fold less than currently approved drugs. Thus, our affordable safe drug-repurposing strategy will likely set the bar for future clinical trials aiming at repairing CFTR defect.
Our study has some limits mainly related to the small sample size. To compensate for the small number, we selected objective stringent biomarkers of efficacy that likely would not be affected by randomization. The short duration of treatment does not allow to determine the long-term clinical benefit, although patients who had a persistent better clinical benefit had received a longer period of combination treatment. Further placebo-controlled trials on a larger population sample will be required to assess the long-term effects of this combination therapy, especially with respect to pulmonary function, physical and cognitive performance and subjective well being.
Materials and methods
Mouse studies
Mice and treatments
CF mice homozygous for the F508del-CFTR in the FVB/129 outbred background (Cftrtm1EUR, F508del, FVB/129, abbreviated CftrF508del/F508del, were obtained from Bob Scholte, Erasmus Medical Center Rotterdam, The Netherlands, CF coordinated action program EU FP6 LSHM-CT-2005-018932).32 Transgenic KO Cftr mice (B6.129P2-KOCftrtm1UNC, abbreviated Cftr−/−), were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA).33 The heterozygous CftrF508del/+ males were backcrossed with the heterozygous Cftr+/− females to obtain F508del/null CFTR heterozygous mice (abbreviated CftrF508del/−). CftrF508del/+ female mice were backcrossed to the C57BL/6J background Becn1+/– male mice (generous gift from Beth Levine, Center for Autophagy Research, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, USA and Francesco Cecconi, University of Tor Vergata, Rome, Italy)34 to obtain at the first generation Becn1 haploinsufficient F508del heterozygous mice (abbreviated CftrF508del/+/Becn1+/−). These mice CftrF508del/+/Becn1+/− were crossbred to obtain Becn1 haploinsufficient F508del homozygous mice (abbreviated CftrF508del/F508del/Becn1+/−). Mice for the study were aged 8-week-old.
Mice were gavaged with vehicle or cysteamine (60 μg/kg in 100 μl saline/day) or EGCG (150 μg/kg in 100 μl saline/day) alone for 5 days or with cysteamine plus EGCG for 5 days followed by EGCG alone or vehicle for further 2 weeks, as reported.20
At the end of the treatment, mice were anesthetized with Avertine (tribromoethanol, 250 mg/kg, Sigma Aldrich, Milan, Italy, T48402) and a segment of tail was collected for genotyping. Mice were then killed and lungs and intestines collected for analysis. All the procedures in mice were approved by the local Ethics Committee for Animal Welfare (IACUC No. 553 and 582) and were carried out in strict respect of European and National regulations.
Genotyping of the new mouse models
The newly generated CftrF508del/− and the CftrF508del/F508del/Becn1+/–were housed at the San Raffaele Scientific Institute SOPF animal house (Milan, Italy). These mice were provided with a special food, consisting of an equal mixture of SRM-A (Arie Blok, Woerden, The Netherlands) and Teklad 2019 (Harlan Laboratories, San Pietro al Natisone, Udine, Italy) and water acidified to pH 2.0 with HCl and containing 60 g/l PEG 3350, 1.46 g/l NaCl, 0.745 g/l KCl, 1.68 g/l NaHCO3 and 5.68 g/l Na2SO4. Newborn mice were genotyped by cutting a small piece of tail 12 days after birth. DNA was extracted by digesting tails with Direct PCR Lysis Reagent (Viagen, CA, USA) and 1 mg/ml Proteinase K overnight at 56 °C. For CftrF508del/− two PCR reactions were performed. For the CftrF508del/F508del mutation, thermocycling consisted of an initial polymerase activation step at 95 °C for 5 min, amplification was performed with 30 cycles of 95 °C for 1 min, 52 °C for 1 min and 72 °C for 1 min with a final extension at 72 °C for 2 min; for the Cftr−−/− mutation thermocycling consisted of an initial polymerase activation step at 94 °C for 3 min, amplification was performed with 30 cycles of 94 °C for 30 s, 57 °C for 30 s and 72 °C for 30 s with a final extension at 72 °C for 10 min. For CftrF508del/F508del/Becn1+/–, after analyzing the CftrF508del/F508del mutation, the Becn1+/– thermocycling consisted of an initial polymerase activation step at 95 °C for 4 min, amplification was performed with 30 cycles of 95 °C for 1 min, 62 °C for 1 min and 72 °C for 1.30 min with a final extension at 72 °C for 10 min. The sequences of PCR primers are reported in Supplementary Table S15.
Procedures
Measurement of RPD. RPD was measured as described.20 In brief, RPD was measured with a protocol similar to that for nasal potential difference measurements in humans, as described.35, 36 Mice were anesthetized with Avertine (250 mg/kg). RPD was sensed with a digital volthometer inserted for ~2 cm in the rectum. Potentials were measured with respect to a subcutaneous 1 M NaCl-filled needle. A second rectal tube was used for continuous perfusion of drug administration into the rectum. The Cl− containing solution had the following composition: 145 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 0.1 mM amiloride, at a final pH 7.4. The Cl−-free solution had the following composition: 145 mM sodium gluconate, 4 mM potassium gluconate, 4 mM calcium gluconate, 1 mM magnesium gluconate, 10 mM HEPES, 0.1 mM amiloride, at a final pH 7.4. To assess CFTR function in vivo, we evaluated the response of the RPD to the presence of forskolin and luminal amiloride (100 μM, to block Na+-dependent RPD) in Cl−-free solutions, as previously described.20
Ussing chamber. Chambers for mounting tissue biopsy were obtained from Physiologic Instruments (model P2300, San Diego, CA, USA). Chamber solution was buffered by bubbling with a mixture of 95% O2 and 5% CO2. Tissues were short circuited using Ag/AgCl agar electrodes. Short-circuit current and resistance were acquired or calculated using the VCC-600 transepithelial clamp from Physiologic Instruments and the Acquire &Analyze2·3 software for data acquisition (Physiologic Instruments), as previously described.37, 38 A basolateral-to-apical chloride gradient was established by replacing NaCl with Na-gluconate in the apical (luminal) compartment to create a driving force for CFTR-dependent Cl− secretion. CFTR channels present at the apical surface of the epithelium (lumen side of the tissue) were activated.
Real time and reverse transcript PCR analysis. The analysis was performed as previously described.20, 39, 40 The specifications of primers are reported in Supplementary Table S15.
Immunoblot analysis. Western blot analysis was performed as previously described16, 20, 21, 22 with antibodies against the following proteins: BECN1, CFTR, β-actin, SQSTM1/p62. The densitometric analysis was performed by Image J software and each data point was expressed as the mean±S.D. of independent experiments. The antibodies specifications are reported in Supplementary Table S16.
Human studies
Patients and study design
An open-label phase-2 clinical trial was conducted between 12 March 2014 and 9 January 2015 at the Department of Translational Medical Sciences, Regional Cystic Fibrosis Care Center, University of Naples Federico II. The protocol of this study is an amendment of the study registered with primary registry EU-CT, Eudract Number #2013-001258- 82 in order to include CF patients bearing different CFTR mutations for the treatment with cysteamine and EGCG. This amended study was approved by the Italian Agency of Drug and independent institutional local Ethics Committee and was done in compliance with the Good Clinical Practice Guidelines, the amended Declaration of Helsinki, and regulatory requirements. Written informed consent was obtained from all patients and/or their legal guardians.
Among 74 eligible patients in regular follow-up at the at the Department of Translational Medical Sciences, Regional Cystic Fibrosis Care Center, University of Naples Federico II, 52 consenting patients were sequentially enrolled.
Inclusion criteria were as follows: (i) diagnosis of CF with sweat chloride ⩾60 mmol/l and two CF causing mutations at CFTR gene molecular analysis; (ii) age ⩾6 years; (iii) genotype characterized by (a) F508del on both alleles; (b) F508del or another class II mutation on one allele and a severe CFTR mutation with minimal residual CFTR activity (class I or II) on the other allele or (c) class I mutations on both CFTR alleles; (iv) FEV1 at least of 40% of predicted value for persons of their age, sex and height.
Exclusion criteria were as follows: (i) treatment with glucocorticoids per os or via inhalation at screening or within 4 weeks before screening visit; (ii) treatment with oxygen; (iii) treatment with other experimental drugs; (iv) referred hypersensitivity (local or general) to cysteamine or penicillamine prior study modifications of therapeutic regimens in patients assuming macrolides, antiasthmatic, mucolytic drugs, Dornase alfa and/or NSAIDs within 28 days before screening visit; (v) organ transplantation; (vi) kidney (creatinine 2 × upper normal limit) or hepatic alterations (ALT and/or AST 5 × upper normal limit) at screening visit; (vii) pregnancy or nursing at screening visit; (viii) refusing to employ contraceptive methods during the study; (ix) psychiatric pathologies or neurological diseases.
After enrollment, safety and efficacy were assessed by clinical and laboratory tests, including nasal brushing, at baseline and at the end of each treatment schedule (Figure 3). Additional assessments were undertaken 4 weeks after drug discontinuation.
We recorded adverse events and clinical and laboratory results throughout the study. Severe adverse events were registered. Pill counts were undertaken at several time points during treatment to evaluate adherence.
Outcomes
Primary end point: (i) sweat chloride values <15% from baseline coupled to (ii) CFTR function and CFTR band C protein in brushed nasal epithelial cells >15% of non-CF controls.
Secondary end points: (i) restoration of autophagy in nasal cells (BECN1 protein levels >50% of non-CF controls and SQSTM1/p62 <50% from baseline; (ii) reduction from baseline of inflammatory cytokines in both nasal cells and sputum; (iii) absolute change from baseline in the ppFEV1.
Composite criteria to define individual responsiveness to treatment:
Pre-specified criteria that must be simultaneously fulfilled to define individual responsiveness to treatment:
(1) sweat chloride concentrations to values <−99% CI from the mean of the individual values measured within the preceding 24 months and including the value at week 0;
(2) band C CFTR protein and CFTR channel function in brushed nasal epithelial cells >15% of healthy non-CF control values.
Schedule of treatment
Active treatment comprised oral cysteamine (cysteamine bitartrate, trade name Cystagon, Orphan Europe) with or without EGCG (trade name Epinerve, SIFI Pharmaceuticals). Initially, Cystagon (900 mg/die for patients ⩽12 years and 1200 mg/die for patients>12 years in four divided doses) was given alone for 8 weeks, followed by a combination of Cystagon (same dose) with Epinerve (270 mg once daily) for further 4 weeks (or for 8 weeks in a subgroup of 10 F508del-CFTR homozygous patients) followed by Epinerve alone for additional 8 weeks (Figure 3). Throughout, all subjects continued their pre-study medication.
Therapeutic formulation and dose administration of cysteamine and EGCG
Cysteamine: each hard capsule contains 150 mg fixed of cysteamine (as mercaptamine bitartrate). The other ingredients are microcrystalline cellulose, starch, pregelatined, magnesium stearate/sodium lauryl sulfate, colloidal silicon dioxide, croscarmellose sodium; capsule shells: gelatin, titanium dioxide, black ink on hard capsules. The prescribed dose was taken four times a day, every 6 h, just after or with food.
EGCG: each tablet contains 135 mg of EGCG. The other ingredients are microcrystalline cellulose, calcium phosphate dibasic, silicon dioxide, magnesium stearate, mono-diglyceride of fatty acids. The prescribed dose was taken fasting.
Pharmacokinetic analysis
Plasma cysteamine concentrations were measured in six F508del/F508del participants at week 8 before and 2, 4 and 6 h after a single oral dose of cysteamine (300 mg). The methodologies of the analyses for plasma cysteamine levels are described in Supplementary Materials.
Nasal brushing
Freshly isolated brushed nasal epithelial cells were obtained by nasal brushing, as previously described,20, 22 from enrolled CF patients and from five non-CF consenting healthy volunteers. Brushes with cells were rapidly transferred in 15-ml sterilized tubes containing RPMI1640 medium (Invitrogen, Carlsbad, CA, USA) with 1% penicillin-streptomycin (Lonza Group LTD, Basel, Switzerland, 17-602E). The tubes were incubated at 37 °C for 2 h on a thermal shaker, to remove all cells from brushes. Cells were centrifuged at 800 × g (2000 rpm) for 20 min. The supernatant fractions were discarded and the cell pellet treated with 150 ml of trypsin-versene (EDTA) solution (Lonza, 17–161) for 4 min at 37 °C to disaggregate possible cell clusters. Cells were placed in CELLC T-25 flasks (Sarstedt Ltd, CS300) with BEGM medium after centrifugation at 800 × g (2000 rpm) for 10 min. Non-specific epithelial cells were removed with daily medium changes.
In vitro studies before in vivo treatment
Before in vivo treatment (week 0) nasal epithelial cells were freshly collected by nasal brushing from the patients enrolled in the study. Nasal brushing from 45 of 52 patients provided a sufficient number of cells for in vitro evaluation. Nasal cells were cultured in vitro for 18 h with 250 μM cysteamine (Sigma Aldrich, M9768) or 80 μM EGCG (Sigma Aldrich, E4143) or cysteamine plus EGCG, and then kept in culture with medium alone up to 48 h, in the presence or absence of EGCG, as described.20 We tested the rescue of CFTR function by measuring the rate of halide efflux, and the amount of mature bend C CFTR protein by surface biotinylation and western blot as previously described.20
Ex vivo analysis during in vivo treatment
Samples collected at week 0 and at each point through the study were immediately used to assess CFTR function and protein levels, BECN1 and SQSTM1/p62 proteins and cytokine expression. The values were compared with those from five non-CF healthy volunteers.20
Procedures
Halide efflux analysis
The analysis of halide efflux was performed in nasal cells, by the iodide-sensitive fluorescent indicator, SPQ (Molecular Probes/Invitrogen, Carlsbad, CA, USA, M440), as previously described.20 In brief, the cells were incubated for 20 min at 37 °C in a humidified chamber with 5% CO2 with the iodide-sensitive fluorescent indicator SPQ, as described (REF). The SPQ-loaded cells were mounted on a LSM510 Meta confocal microscope (Zeiss, Milan, Italy) with a 37 °C heated stage and perfused with iodide buffer for 5–8 min. Changes in CFTR-mediated SPQ fluorescence were observed at the 445 nm wavelength in response to excitation at 340 nm during perfusion at 37 °C in nitrate buffer replaced with 130 mM NaNO3 (Sigma Aldrich, S8170) with 20 μM forskolin (Fsk; Sigma Aldrich, F6886) plus 100 μM IBMX (Sigma Aldrich, I5879) and fluorescence intensity measured for a further 10–12 min. Signals were recorded at 30-s interval. For each minute the average of the fluorescence intensity was measured from 50 cells for population per coverslip and the peak of halide efflux rate (usually after Fsk plus IBMX adding) of cells was calculated in accordance with the Stern-Volmer relationship.41 The rates were calculated using SigmaPlot Version 7.1 for each mean fluorescence trace for each time point generated from the 50 cells examined per population per coverslip.41, 42, 43, 44, 45
Immunoblot analysis, cell surface biotinylation assay and membrane fractionation
The proteins were obtained from nasal epithelial cells. To detect CFTR protein, a large amount (120 μg) of protein was loaded. Western blot was performed as previously described20 with antibodies against the following proteins: BECN1, CFTR, β actin, SQSTM1/p62. The antibodies specifications are reported in Supplementary Table S16.
Insoluble pellet was obtained by centrifugation at 9000 × g at 4 °C for 20 min after lysis and dissolved 5 times in sample buffer, boiled at 95 °C for 5 min and resolved on a polyacrylamide gel for western blot analysis of SQSTM1/p62, as described.20
The densitometric analysis was performed by Image J software and each data point was expressed as the mean±S.D. of independent experiments.
Cell-surface proteins were biotinylated and the PM fraction was collected to detect mature CFTR band C at the PM. Sulfosuccinimidyl-6-(biotinamido) hexanoate (sulfo-NHS-LC-Biotin, Pierce, Rockford, IL, USA, 21335), was dissolved at 1 mg/ml in PBS (Gibco/Thermo Scientific Milan, Italy, 18912–014, pH 8.2), as described.22 After homogenization with a Potter-Elvehjem pestle, the cells were centrifuged at 2300 × g for 15 min at 4 °C. Supernatant fractions that contain the cytoplasmic and PM fractions were centrifuged 1 h at 16 000 × g at 4 °C; the pellet that contains the intact membrane, was solubilized in Buffer A (20 mM Tris-HCl, pH 7.4, 2 mM EDTA, 20 mM 2-mercaptoethanol, 1 × PMSF, 1 μg/ml inhibitor protease cocktail (Sigma Aldrich, P8340)+1% Triton X-100 (Sigma Aldrich, X- 100-RS) and centrifuged 1 h at 60 000 × g in the ultracentrifuge. The supernatants were representative of PM fraction. Equivalent amounts of protein (500 μg) were used for streptavidin-agarose affinity isolation (Pierce, Rockford, IL, USA, 20349). Biotinylated proteins of PM immunoblotted against CFTR or FLOT1. The densitometric analysis was performed by Image J software and each data point was expressed as the mean±S.D. of independent experiments.
Measure of TNF-α and CXCL8 expression levels in nasal brushing
RNA extraction was performed as detailed above. For human samples, thermocycling consisted of an initial polymerase activation step at 95 °C for 5 min, amplification was performed with 40 cycles of 95 °C for 15 s, 60 °C for 10 s and 72 °C for 20 s with data acquisition at this stage and the reaction finished by the built in melt curve. Expression levels of genes were normalized to the housekeeping gene GAPDH in the same sample. The specifications of primers are reported in Supplementary Table S15.
Measure of TNF-α and CXCL8 levels by ELISA in sputum
Sputum samples were diluted 1 : 2 with PBS and digested with 1 U/μl of DNAse for 4 h at 37 °C. The supernatants were collected and stored at −80 °C until usage. The samples were then centrifuged at 800 × g for 10 min and the supernatants used to measure TNF-α and CXCL8 levels by means of standard ELISA kits (R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s instructions, as reported.16, 46 Samples were read in triplicate at 450 nm in Microplate Reader (BioRad, Milan, Italy) using Microplate Manager 5.2.1 software. Values were normalized to protein concentration evaluated by Bradford analysis.
Statistical analysis
Categorical variables are presented as proportion (%) and continuous variables as mean (and S.D.) when normally distributed or median (and IQR) when not.
Mice
All laboratory tests on mice were performed at least in triplicate. Between-group comparisons were evaluated by one-way analysis of variance, applied to mean (or median) values of continuous variables; post hoc comparisons were made using Bonferroni correction, when appropriate. We set the level of significance at P<0.05.
Humans
An ethical committee (including statisticians) validated the plan of the study. Normality of data distribution was tested and the effects of treatment in CF patients on either sweat chloride or nasal CFTR function were analyzed by repeated ANOVA, comparing the means of variables measured at different times of treatment, and during the 4-week follow-up period, as previously described.20 For brushed nasal cells all laboratory tests on cells were performed at least in triplicate. According to Šidák correction for multiple comparisons, each comparison47 was considered significant if P-values (for two-tailed test) were < , where α is the overall type I error probability and n is the number of comparisons. Values registered at the end of combination treatment, either 4-week or 8-week treatment, with cysteamine plus EGCG were pooled for the analysis. The Fisher's test was used to assess the proportion of subjects who fulfilled the primary end point between treated (at the end of the combination treatment) and untreated (during an observational period of the same duration) groups for variation of CFTR function (see the exact definition of primary end point above).
, where α is the overall type I error probability and n is the number of comparisons. Values registered at the end of combination treatment, either 4-week or 8-week treatment, with cysteamine plus EGCG were pooled for the analysis. The Fisher's test was used to assess the proportion of subjects who fulfilled the primary end point between treated (at the end of the combination treatment) and untreated (during an observational period of the same duration) groups for variation of CFTR function (see the exact definition of primary end point above).
For secondary end points a paired T-test was used to evaluate differences between two time points in the same group of patients. We used a Fisher's test to evaluate differences between groups (treated versus untreated, as detailed above) for rescue of autophagy, cytokine values in both nasal brushing and sputum, and absolute change in ppFEV1. For prediction tests and biomarkers, unpaired T-tests were uses to compare two groups of quantitative data, correlation tests (Pearson’s and Spearman’s) were used to search for relationship between quantitative data, linear model were used to search for causal relationship between quantitative data. ROC curve analysis was applied to analyze potential biomarker for treatment response. Data was processed through EXCEL (versions 2010). All data processing and analyses were carried out with SAS statistical software (version 9.2; SAS Institute, Cary, NC, USA) and R software (version 3.0.2).
Abbreviations
- CF:
-
cystic fibrosis
- CFTR:
-
cystic fibrosis transmembrane conductance regulator
- EGCG:
-
epigallocatechin gallate
- ER:
-
endoplasmic reticulum
- PM:
-
plasma membrane
- TG2:
-
tissue transglutaminase
- BECN1/Beclin 1:
-
autophagy-related protein Beclin 1
- SQSTM1:
-
sequestosome 1
- CSNK2:
-
casein kinase 2
- KO:
-
knockout
- RPD:
-
rectal potential difference
- CXCL2:
-
chemochine (C-X-C motif) ligand 2
- TNF:
-
tumor necrosis factor
- WT:
-
wild type
- CXCL8:
-
chemokine (C-X-C motif) ligand 8
- FEV1:
-
forced expiratory volume at first second
References
Ratjen F, Döring G . Cystic fibrosis. Lancet 2003; 361: 681–689.
De Boeck K, Zolin A, Cuppens H, Olesen HV, Viviani L . The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros 2014; 13: 403–409.
Ramsey BW, Davies J, McElvaney NG, Bell SC, Drevinek P, Griese M et al. VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365: 1663–1672.
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M et alTRAFFIC and TRANSPORT Study Groups. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015; 373: 220–231.
Boyle A MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E et al. VX09-809-102 study group. CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2014; 2: 527–538.
Davis PB . Another beginning for cystic fibrosis therapy. N Engl J Med 2015; 373: 274–276.
Jones AM, Barry PJ . Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: should we curb our enthusiasm? Thorax 2015; 70: 615–616.
Amaral MD, Farinha CM . Rescuing mutant CFTR: a multi-task approach to a better outcome in treating cystic fibrosis. Curr Pharm Des 2013; 19: 3497–3508.
Cholon DM, Quinney NL, Fulcher ML, Esther CR Jr, Das J, Dokholyan NV et al. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med 2014; 6: 246ra96.
Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM et al. Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci Transl Med 2014; 6: 246ra97.
Accurso FJ, Van Goor F, Zha J, Stone AJ, Dong Q, Ordonez CL et al. Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. J Cyst Fibros 2014; 13: 139–147.
De Boeck K, Kent L, Davies J, Derichs N, Amaral M, Rowe SM et alEuropean Cystic Fibrosis Society Clinical Trial Network Standardisation Committee. CFTR biomarkers: time for promotion to surrogate end-point. Eur Respir J 2013; 41: 203–216.
Amaral MD . Novel personalized therapies for cystic fibrosis: treating the basic defect in all patients. Intern Med 2015; 277: 155–166.
Awatade NT, Uliyakina I, Farinha CM, Clarke LA, Mendes K, Sole A et al. Measurements of functional responses in human primary lung cells as a basis for personalized therapy for cystic fibrosis. EBioMedicine 2014; 2: 147–153.
Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006; 127: 803–815.
Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 2010; 12: 863–875.
Venerando A, Franchin C, Cant N, Cozza G, Pagano MA, Tosoni K et al. Detection of phospho-sites generated by protein kinase CK2 in CFTR: mechanistic aspects of Thr1471 phosphorylation. PloS One 2013; 8: e74232.
Gahl WA . Early oral cysteamine therapy for nephropathic cystinosis. Eur J Pediatr 2003; 162: S38–S41.
Emma F, Nesterova G, Langman C, Labbè A, Cherqui S, Goodyer P et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant 2014; 29: 87–94.
De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 2014; 10: 2053–2074.
Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on DF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012; 8: 1657–1672.
Villella VR, Esposito S, Bruscia EM, Vicinanza M, Cenci S, Guido S et al. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ 2013; 20: 1101–1115.
Taylor-Robinson D, Whitehead M, Diderichsen F, Olesen HV, Pressler T, Smyth RL et al. Understanding the natural progression in %FEV1 decline in patients with cystic fibrosis: a longitudinal study. Thorax 2012; 67: 860–866.
DeMarco ML, Dietzen DJ, Brown SM . Sweating the small stuff: Adequacy and accuracy in sweat chloride determination. Clin Biochem 2014; 48: 443–447.
Corvol H, Thompson KE, Tabary O, le Rouzic P, Guillot L . Translating the genetics of cystic fibrosis to personalized medicine. Transl Res 2015; 168: 40–49.
Vanscoy LL, Blackman SM, Collaco JM, Bowers A, Lai T, Naughton K et al. Heritability of lung disease severity in cystic fibrosis. Am J Respir Crit Care Med 2007; 175: 1036–1043.
Schork NJ . Personalized medicine: time for one-person trials. Nature 2015; 520: 609–611.
Jameson JL, Longo DL . Precision medicine-personalized, problematic, and promising. N Engl J Med 2015; 372: 2229–2234.
Bilton D . Personalised medicine in cystic fibrosis must be made affordable. Paediatr Respir Rev 2014; 155: 6–7.
Bosch B, De Boeck K . Searching for a cure for cystic fibrosis. A 25-year quest in a nutshell. Eur J Pediatr 2015; 175: 1–8.
Cantin AM, Hartl D, Konstan MW, Chmiel JF . Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros 2015; 14: 419–430.
vanDoorninck JH, French PJ, Verbeek E, Peters RH, Morreau H, Bijman J et al. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 1995; 14: 4403–4411.
Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O et al. An animal model for cystic fibrosis made by gene targeting. Science 1992; 257: 1083–1088.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112: 1809–1820.
Fischer H, Fukuda N, Barbry P, Illek B, Sartori C, Matthay MA . Partial restoration of defective chloride conductance in DF508 CF mice by trimethylamine oxide. Am J Physiol Lung Cell Mol Physiol 2001; 281: L52–L57.
Illek B, Fischer H . Flavonoids stimulate Cl conductance of human airway epithelium in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol 1998; 275: L902–L910.
Marchelletta RR, Gareau MG, McCole DF, Okamoto S, Roel E, Klinkenberg R et al. Altered expression and localization of ion transporters contribute to diarrhea in mice with Salmonella-induced enteritis. Gastroenterol 2013; 145: 1358–1368.
Gondzik V, Awayda MS . Methods for stable recording of short-circuit current in a Na+-transporting epithelium. Am J Physiol Cell Physiol 2011; 301: C162–C170.
Maiuri L, Luciani A, Giardino I, Raia V, Villella VR, D’Apolito M et al. Tissue transglutaminase activation modulates inflammation in cystic fibrosis via PPAR gamma down-regulation. J Immunol 2008; 180: 7697–7705.
Amano H, Yamamoto H, Senba M, Oishi k, Suzuki S, Fukushima K et al. Impairment of endotoxin-induced macrophage inflammatory protein 2 gene expression in alveolar macrophages in streptozotocin-induced diabetes in mice. Infect Immun 2000; 68: 2925–2929.
Stern M, Munkonge FM, Caplen NJ, Sorgi F, Huang L, Geddes DM et al. Quantitative fluorescence measurements of chloride secretion in native airway epithelium from CF and non-CF subjects. Gene Ther 1995; 2: 766–774.
Verkman AS, Galietta LJV . Chloride channels as drug targets. Nat Rev Drug Discov 2009; 8: 153–171.
Jayaraman S, Teitler L, Skalski B, Verkman A . Longwavelength iodide-sensitive fluorescent indicators for measurement of functional CFTR expression in cells. Am J Physiol Cell Physiol 1999; 277: C1008–C1018.
Munkonge F, Alton EW, Andersson C, Davidson H, Dragomir A, Edelman A et al. Measurement of halide efflux from cultured and primary airway epithelial cells using fluorescence indicators. J Cyst Fibros 2004; 3: 171–176.
Mansoura MK, Biwersi J, Ashlock MA, Verkman A . Fluorescent chloride indicators to assess the efficacy of CFTR cDNA delivery. Hum Gene Ther 1999; 10: 861–875.
Paine R, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL et al. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med 2012; 40: 90–97.
Abdi H . Bonferroni and Sidak corrections for multiple comparison. In: Salkind NJ (eds). Encyclopedia of Measurement and Statistics. Sage: Thousand Oaks, CA, USA, 2007, pp 103–107.
Acknowledgements
We thank the participating patients and their families. We thank Dr. Bob Scholte, Erasmus Medical Center Rotterdam, The Netherlands, who provided Cftrtm1EUR (F508del (FVB/129) mice (European Economic Community European Coordination Action for Research in Cystic Fibrosis program EU FP6 SHM-CT-2005-018932), Dr. Beth Levine, Center for Autophagy Research, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, USA and Dr. Francesco Cecconi, University of Tor Vergata, Rome, Italy, who provided C57BL/6J background Becn1+/– mice, Dr. Valeria R Villella, Dr. Romina Monzani and Dr. Candida Bonelli, European Institute for Research in Cystic Fibrosis, for technical assistance. This study was supported by The European Institute for Research in Cystic Fibrosis (IERFC) non-profit foundation, Italian Cystic Fibrosis Association (LIFC) and Regional Cystic Fibrosis Associations of Campania, Sicilia, Lazio, Puglia (to Dr. L Maiuri, Dr. Raia), E-Rare (Rescue CFTR preclinic) (to Dr. L Maiuri and Dr. Kroemer); Telethon (#GGP12128) (to Dr. L Maiuri, Dr. Raia, Dr. MC Maiuri), Agence National de la Recherche (ANR) – Projetsblancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI) (all to Dr. Kroemer). The funders of the study had no role in study design, data collection, data analysis, data interpretation or writing of the report.
Author contributions
VR, LM and GK conceived, designed and analyzed the overall study. VR, LM, GK, AM and AT wrote the manuscript. VR and AT designed and wrote the clinical trial protocol. AT, FDG, AS identified patients, executed the trial procedures, dispensed study drugs, collected data and followed up the study participants, under the supervision of VR. AT and FDG coordinated the collaborative work between clinical and laboratory teams and the biochemical work-up of clinical trial samples under the supervision of VR and LM. AT, FDG and AS obtained patient consent. LS performed sweat test analyses. PB and ADP did spirometric analyses. RG and CAL performed nasal brushings. SE did western blot analysis and assessment of CFTR function in nasal brushings, did in vitro cultures on patients nasal cells. DDS, IS and EF did all preclinical experiments on mice, did cross-breading and genotyping of mice, performed mouse treatments, assessment of CFTR function, protein and cytokine assessment and together with MCM analyzed mice data under the supervision of LM. GDR and SL analyzed the pharmacokinetic profile of cysteamine. MCM, AM, GB, SG contributed to data analysis and discussion. GS did statistical analysis. All authors have seen and approved the final version.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
LM, VR and GK are listed as inventors on a patent application (No. 13/895741) owned by the No-profit Foundation European Institute for Research in Cystic Fibrosis, describing the use of cysteamine for the treatment of CF. The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tosco, A., De Gregorio, F., Esposito, S. et al. A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ 23, 1380–1393 (2016). https://doi.org/10.1038/cdd.2016.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.22
This article is cited by
-
Protein kinase CK2: a potential therapeutic target for diverse human diseases
Signal Transduction and Targeted Therapy (2021)
-
Bioactive Thymosin Alpha-1 Does Not Influence F508del-CFTR Maturation and Activity
Scientific Reports (2019)
-
Luigi Maiuri: un Grande Uomo - a Great Spirit
Cell Death & Disease (2019)
-
Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis
Cell Death & Disease (2019)
-
Defective proteostasis in celiac disease as a new therapeutic target
Cell Death & Disease (2019)
