
Abstract
The influenza virus infects millions of people each year and can result in severe complications. Understanding virus recognition and host responses to influenza infection will enable future development of more effective anti-viral therapies. Previous research has revealed diverse yet important roles for the annexin family of proteins in modulating the course of influenza A virus (IAV) infection. However, the role of Annexin-A1 (ANXA1) in IAV infection has not been addressed. Here, we show that ANXA1 deficient mice exhibit a survival advantage, and lower viral titers after infection. This was accompanied with enhanced inflammatory cell infiltration during IAV infection. ANXA1 expression is increased during influenza infection clinically, in vivo and in vitro. The presence of ANXA1 enhances viral replication, influences virus binding, and enhances endosomal trafficking of the virus to the nucleus. ANXA1 colocalizes with early and late endosomes near the nucleus, and enhances nuclear accumulation of viral nucleoprotein. In addition, ANXA1 enhances IAV-mediated apoptosis. Overall, our study demonstrates that ANXA1 plays an important role in influenza virus replication and propagation through various mechanisms and that we predict that the regulation of ANXA1 expression during IAV infection may be a viral strategy to enhance its infectivity.
Similar content being viewed by others

Main
Frequently occurring epidemics of influenza A virus (IAV) infection are major causes of morbidity and mortality, particularly in young children, elderly and pregnant individuals.1 A total of 3–5 million cases of severe influenza cases are reported annually worldwide, and between 250 000–500 000 people across the globe die each year as a direct result of influenza infection.2 The particular strain, H1N1 caused a devastating pandemic in 1918, which lasted for more than a year. It is estimated that one-third of the world’s population was infected due to this outbreak, which killed up to 50 million people in the first 25 weeks.3 The three major pandemic outbreaks of influenza in the past 10 years have caused millions of deaths, and future pandemics are expected to exert a similarly devastating impact on human society.4
The annexins are a family of Ca2+ and phospholipid binding proteins structurally comprised of a tightly packed hydrophilic core domain made of α-helices.5 The C terminal is highly conserved but N terminal is unique for each annexin.6 The annexins have been shown to play a role in both enhancing susceptibility and protecting against a number of infectious diseases, such as tuberculosis,7 Hepatitis C8 and IAV. Annexin-A5 was identified as a receptor for IAV9 and assists the entry of IAV into cells.10 Annexin-A2 supports viral replication when incorporated into IAV by converting plasminogen into plasmin.9, 11 However, annexin-A6 binds viral M2 protein (Ma et al. 2012)12 and increases late endosomal cholesterol levels13 to inhibit IAV replication. The role of ANXA1 in IAV is unclear, but ANXA1 levels are upregulated in porcine monocytes infected with swine flu virus.14 ANXA1 is an immune-modulatory protein that plays an important role in various cellular processes including the inhibition of cellular infiltration and inflammation in many models of disease.15
Apoptosis is the intrinsically-controlled-programmed cell death that occurs during physiological and pathological conditions. IAV has been shown to induce apoptosis in many cell types16, 17 and three of the 12 genes of the IAV have been implicated to be involved in apoptosis.18 Apoptosis was originally considered to be an anti-viral host defense, while later research demonstrated that overexpression of anti-apoptotic protein, Bcl-2, lead to lowered viral titers.19 ANXA1 has been reported to be a pro-apoptotic protein, where cellular ANXA1 expression induces spontaneous apoptosis20 and enhanced apoptosis in response to apoptotic stimuli such as TNF-α or TRAIL.21, 22 ANXA1 is shown to mediate caspase-3 activation, BAD de-phosphorylation, which induces BAD translocation to the mitochondria.21, 22
Here, we examined the importance of ANXA1 in H1N1 IAV infection, replication and apoptosis in vitro and in vivo.
Results
ANXA1 deficient mice are protected against IAV infection
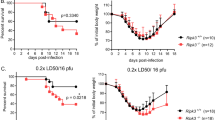
WT and ANXA1 deficient (−/−) mice infected with a lethal dose (25 pfu/g) of A/Puerto Rico/8/1934(H1N1) Influenza A virus (A/PR8) intra-tracheally. Mice were euthanized after they had lost more than 25% of initial body weight as an assessment of infection severity. ANXA1−/− mice exhibited significantly lower weight loss following A/PR8 infection (Figure 1a) which resulted in more survival of ANXA1−/− mice (Figure 1b). Mice were euthanised 3 and 5 days post-infection (DPI) with A/PR8 and lungs were processed for viral titer determination by plaque assay. At 5 DPI, significantly lower virus titers were observed in ANXA1−/− mice (Figure 1c, P<0.05). Significantly lower expression of viral genes (quantified by qPCR using M gene and NS1) were observed in ANXA1−/− lungs at 5 DPI compared to WT lungs (Figures 1d and e). As 25 pfu/g is a high dose of virus, the levels of virus at 3 DPI may overwhelm the host, even in the absence of ANXA1. Therefore, we determined if infected mice with a sub-lethal dose of A/PR8 (12.5 pfu/g). A significant reduction in body weight was observed in WT but not ANXA1−/− mice at 3 DPI (Figure 1f), which was mirrored with significantly lower viral gene expression in the lungs of ANXA1−/− mice (Figures 1g and h). These results indicate that loss of ANXA1 reduces virus titers during IAV infection, and ANXA1 is an important factor in IAV-induced morbidities.

Annexin-A1 reduces survival and enhances morbidity and virus replication after influenza virus A/PR/8/34 infection. Wild-type and Annexin-A1 (ANXA1) −/− mice were infected intra-tracheally with 25 pfu/g influenza A/PR/8/34 and survival was monitored up to 12 days post infection. (a) Weight loss was significantly reduced in ANXA1−/− (n=10) mice compared with wild-type mice (n=10). (b) ANXA1−/− mice (n=10) exhibited significantly reduced mortality compared with the wild type mice (n=10) (p=0.0084, log rank). (c) Lungs were extracted at day 5 post-infection and assessed for viral titers using plaque assays on MDCK cells. (d) M viral gene expression or (e) NS1 gene expression was assessed at indicated days post-infection using qPCR. CT values were converted to viral titers using a standard curve for M gene or NS1 expression. (f) Mice were infected with a sub-lethal dose of IAV (12.5 pfu/g). Weight loss was significantly reduced in ANXA1−/− (n=4) mice compared with wild-type mice (n=4) at day 3 post-infection. (g) Lungs were extracted at day 3 post-infection and assessed for viral titers using plaque assays on MDCK cells. (h) NS1 viral gene expression was assessed at day 3 post-infection using qPCR. CT values were converted to viral titers using a standard curve for M gene or NS1 expression. *P<0.05, **P<0.01
ANXA1 deficiency does not result in differences in cytokine production
WT and ANXA1−/− mice were infected with 25 pfu/g A/PR8 and bronchoalveolar lavage fluid (BAL) was collected and cytokines were measured using ELISA. The pro-inflammatory cytokines IL-1β and IL-12 peaked at 1DPI, similar to the anti-inflammatory cytokines IL-10 and TGF-β, while TNF-α peaked at day 3. Similarly, the anti-viral cytokines IFN α/β reached maximal levels at 3 DPI. IL-6 and IFNγ increased in a time-dependent manner from 1 to 7 DPI, maximal at day 7, similar to the time-course of albumin leakage into the lung, indicating lung damage (Figure 2a, Supplementary Figure S1). The only observable and significant difference was seen with TNF-α at 3 DPI, where ANXA1−/− mice exhibited higher levels of TNF-α, with no differences in other cytokines tested.

ANXA1 deficiency does not affect cytokine production in the lungs, but enhances leukocyte infiltration wild-type and Annexin-A1 (ANXA1)−/− mice were infected intra-tracheally with influenza A/PR/8/34. (a–c) Cell free bronchoalveolar lavage fluids were analyzed at indicated days post-infection for the indicated cytokines. **P<0.01 versus WT on the same day. (d) Total cells were counted using trypan blue staining. (e–i) Differential leukocyte quantification was performed using multicolor flow cytometry with the indicated antibodies. *P<0.05; **P<0.01 versus UI control. ξP<0.05; ξξP<0.01 versus WT on the same day. (j) Lungs were harvested 5 days and 10 days post-infection from WT and ANXA1−/− mice, respectively, and stained with haematoxylin and Eosin. (k) Histological scoring of representative lung sections. Results shown are of n=4–5 mice per group, repeated at least twice
ANXA1 deficiency results in enhanced leukocyte recruitment to the lungs following IAV infection
We next determined the role of ANXA1 in inflammatory cell recruitment into the lungs after IAV infection. The total number of leukocytes in the BAL was higher in ANXA1−/− mice at 7 DPI (Figure 2d). Using multi-color flow cytometry staining for CD45+ cells (Figure 2e), an accumulation of neutrophils (Ly6G+SiglecF−), alveolar macrophages (Ly6G-SiglecF+), eosinophils (Ly6G+SiglecF+) and lymphocytes (Ly6G-SiglecF-CD11c-CD3/CD19+) were observed at 3 DPI while lymphocyte infiltration peaked at 7 DPI (Figures 2f–i). A significant increase in neutrophils at 3 and 7 DPI, and lymphocytes at 7 DPI was observed in ANXA1−/− mice. To confirm this, histopathology of uninfected and infected lungs using H&E staining was performed (Figure 2j). No significant pathology was observed in uninfected lungs. By 5 DPI, mild inflammatory cell infiltrate was observed in the peribronchiolar compartment and red blood cells were also detected in the conducting airways and the alveolar air spaces, indicative of pulmonary hemorrhage. By 10 DPI, inflammation was moderate and did not extend to the alveolar parenchyma in the WT lungs, while severe inflammatory cell infiltrate of the peribronchiolar space with a greater magnitude of inflammatory foci was observed in the alveolar parenchyma of ANXA1−/− lungs. The degree of inflammation was scored and showed that ANXA1−/− lungs display greater tissue inflammation compared with WT BALB/c mice 10 DPI (Figure 2k).
ANXA1 is increased and cleaved upon infection
A significant increase in ANXA1 mRNA was observed at 1 DPI (Figure 3a). This translated to lung protein levels, which were increased at 3 DPI. In addition, cleavage of ANXA1 into a 33 kD fragment was also evident at 3 DPI (Figure 3b). In vitro, A549 human lung epithelial cells were infected with 1 and 10 MOI A/PR8 and ANXA1 mRNA levels increased in a biphasic manner, significant as early as 3 h post-infection after infection (Figure 3c). This biphasic increase in ANXA1 was also observed at protein levels. By 24 h, ANXA1 is seen to be cleaved with maximum levels at 48 h post-infection (Figure 3d).

ANXA1 is induced and cleaved upon IAV infection. (a and b) Wild-type and Annexin-A1 (ANXA1)−/− deficient mice were infected intra-tracheally with influenza A/PR/8/34. Lungs were extracted at indicated days post-infection and assessed for ANXA1 expression using qPCR (mean±S.E. of 3 mice) or Western blotting. (c and d) A549 lung epithelial cells were infected with 1 MOI of A/PR8. ANXA1 expression was measured at indicated time points using qPCR (mean±S.E. of at least 3 independent experiments,*P<0.05, **P<0.01 versus UI) or Western blotting. (e) ANXA1 expression in nasal swabs from healthy controls or influenza A infected patients was measured using ELISA. *P<0.05 in 12 controls and 11 patients
To determine if this could be translated clinically, nasopharyngeal swab fluid was obtained from healthy participants or patients hospitalized with acute respiratory tract infections and PCR+ for influenza A H1N1 or H3N2 virus.23 Higher levels of ANXA1 were observed in influenza infected patients compared with fluid from healthy controls (Figure 3e).
ANXA1 enhances virus replication in vitro
A549 lung epithelial cells were transfected with scrambled or ANXA1-shRNA prior to infection with IAV for 24 and 48 h. M gene and NS1 gene expression was observed to be 2 logs lower in ANXA1-shRNA supernatants 24 and 48 h PI (Figures 4a and b). Silencing of ANXA1 decreased virus load using a plaque assay by 1 log in the supernatants of infected cells at 24 and 48 h PI (Figure 4c). Further, overexpression of ANXA1 induced significantly higher virus titers (Figure 4d).

ANXA1 promotes virus replication in vitro. (a and b) Scramble or ANXA1-shRNA transfected A549 lung epithelial cells were infected with 1 MOI of influenza A/PR8 for the indicated time points. Virus was quantified in supernatants using qPCR for M gene expression or NS1 expression. CT values were converted to viral particles using a standard curve for M1 or NS1 expression (mean±S.D. of duplicates with at least three independent repeats). (c) Viral titers were measured in the supernatants using plaque assays (mean±S.D. of duplicates with at least three independent repeats). (d) A549 cells transfected with empty vector (EV) or ANXA1 pcDNA3.1-V5 (ANXA1-V5) were infected with 1 MOI of influenza A/PR8. Virus was quantified in supernatants using qPCR for M1 gene expression. CT values were converted to viral particles using a standard curve for M1 expression (mean±S.E. of three independent experiments. *P<0.05; **P<0.01 versus Scr-sh or EV at the same time points
ANXA1 affects virus binding at the host cell surface
To systematically dissect the time course and location where ANXA1 may affect IAV replication and subsequent propagation, we performed confocal microscopy of IAV nucleoprotein (NP). First, to determine if ANXA1 modulates the binding of the virus to the host cell, IAV (1 MOI) was added to scr-shRNA or ANXA1-shRNA A549 cells on ice, which prevents endocytosis. After the indicated time points, excess virus particles were washed away and the cells were stained with ANXA1 and viral NP antibodies. Control shRNA cells contained more viral particles per cell while ANXA1-shRNA cells contained significantly less NP-expressing viral particles per cell (Figures 5a and b). ANXA1 affects viral binding through a calcium-dependent manner, as ANXA1 is found in the membrane as a calcium binding protein, and this membrane pool of ANXA1 can be removed by EDTA. Treatment of cells with EDTA resulted in lower viral particles per cell when infected with 5 MOI of IAV even after 15 min of binding (Figures 5c and d).

ANXA1 enhances IAV binding and nuclear infection. (a and b) Scramble or ANXA1-shRNA transfected A549 lung epithelial cells were infected with 1 MOI of influenza A/PR8 for the indicated time points on ice. (c and d). A549 cells were washed with PBS or PBS+3 mM EDTA for 30 min prior to infection with 1 MOI of influenza A/PR8 for 15 min on ice. (e–h) Scramble or ANXA1-shRNA transfected A549 lung epithelial cells were infected with 1 MOI of influenza A/PR8 for the indicated time points at 37 °C. Viral NP (red) and ANXA1 (green) were visualized using confocal microscopy. For a–f, the number of viral particles were quantified per cell. For g and h, the per cent of cells with infected nuclei were counted per field
Silencing ANXA1 reduces viral uptake at early time points
Scr-shRNA and ANXA1-shRNA cells were infected with IAV for 1, 2 and 4 h and the number of viral particles per cell was assessed using confocal microscopy. A significant reduction (by 2 fold) in the number of viral particles was observed in ANXA1-shRNA cells infected for 4 h (Figures 5e and f, Supplementary Figure S2). The fluorescence intensity of ANXA1 was approximately 50% less in ANXA1-shRNA cells compared with scr-shRNA cells, confirming the silencing of ANXA1.
Silencing ANXA1 reduces viral transport to the nucleus
Scr-shRNA and ANXA1-shRNA cells were infected with IAV for longer time points of 8, 12, 24 and 36 h. The number of cells with NP in the nucleus was lower in ANXA1-shRNA cells at 4 h of infection, as well as at later time points of infection (Figures 5g and h, Supplementary Figures S3 and S4). Therefore, we determined if viral transport in the endosomes to the nucleus was modulated by ANXA1 by examining the percentage of viruses in early endosomes (EE) and late endosomes (LE) after IAV infection. As EE are cycling endosomes, control uninfected cells also contain EE and LE (Supplementary Figures S5 and S6). Upon IAV infection, colocalization of EE and viral NP proteins was lower in ANXA1-shRNA cells after 15 min and 30 min of infection (Figures 6a, c and d, Supplementary Figure S5). This was not a consequence of less EE in ANXA1-shRNA cells, as both Scr-sh and ANXA1-sh cells contained similar EE content when uninfected or infected (Figure 6b). After 45 min of infection, the number of virus particles colocalizing with LE was also lower in ANXA1-shRNA cells compared with scr-shRNA control cells (Figures 6e, g and h,Supplementary Figure S6), without affecting the LE content once again (Figure 6f). To determine if this is due to ANXA1 localization in the EE and LE, we assessed the colocalization of ANXA1 with EE and LE. Figure 6i shows that ANXA1 is colocalized and recruited to EE specifically around the nucleus in uninfected, but more in IAV infected cells. Similarly, ANXA1 can be found colocalized with LE around in the nucleus in both uninfected and IAV infected cells.

ANXA1 enhances early and late endosome trafficking of IAV. Scramble or ANXA1-shRNA transfected A549 lung epithelial cells were infected with 1 MOI of influenza A/PR8 for the indicated time points. (a) Percentage of virus colocalized in early endosomes was quantified. (b) Number of early endosomes per cell was quantified. (c and d) Early endosomes were stained with Rab5a (green), viral NP (red) and DAPI (blue). (e) Percentage of virus colocalized in late endosomes was quantified. (f) Number of late endosomes per cell was quantified. (g and h) Late endosomes were stained with Rab7a (green), viral NP (red) and DAPI (blue). (i) Colocalization of ANXA (red) and early (Rab5a)/late (Rab7a) endosomes (green)
ANXA1 promotes virus-induced apoptosis and caspase-3 activation
IAV has been shown to promote caspase-3 dependent apoptosis for virus propagation, and previous studies have shown that ANXA1 can promote apoptosis. Apoptosis, analyzed through the cleavage of caspase 3 and 7 in the lungs of mice 3 DPI with A/PR8 was increased in WT but not ANXA1−/− mice (Figure 7a). When lung epithelial cells were cultured from WT and ANXA1−/− mice and subjected to infection ex vivo, ANXV-PI staining showed that apoptosis more evident in WT (23.9% ANXV+PI−) than ANXA1−/− cells (6.2% ANXV+PI−) (Figure 7b). In vitro, silencing ANXA1 reduced cell viability in uninfected cells (Figure 7c), and resulted in significantly enhanced cell viability after 24 h IAV infection (Figure 7d). Further, IAV infection induced caspase-3/7 activity, cleaved caspase-3 and cleaved PARP, which was significantly inhibited when ANXA1 was silenced (Figures 7e–g). Similarly, BAX translocation to the mitochondria and AIF translocation from the mitochondria to the cytosol and cytochrome C release was lower in ANXA1-shRNA transfected cells after IAV infection (Figures 7h and i). Collectively, these data establish that ANXA1 deficiency inhibits IAV-induced apoptosis, which may lead to lower virus replication.

ANXA1 enhances IAV-induced apoptosis. Wild-type and Annexin-A1 (ANXA1)−/− deficient mice were infected intra-tracheally with influenza A/PR/8/34. Lungs were extracted at 3 days post-infection and apoptosis was assessed using (a) caspase 3 or 7 cleavage or (b) Annexin-V/PI staining. Tubulin was used as a loading control. (c and d) A549 lung epithelial cells (control or shRNA silenced for ANXA1 (ANXA1-sh) were infected with 1 MOI of influenza A/PR8. Cell viability was assessed after 24 h using crystal violet staining. (e) Caspase-3 activity was measured in control or ANXA1-siRNA A549 cells uninfected or infected with 1 MOI of influenza A/PR/8/34 for indicated times. *P<0.05; **P<0.01 versus 0. (f) A549 cells were infected with media (M) or indicated MOI of influenza A/PR8 for 24 h. Cell lysates were subjected to immunoblot analysis for the indicated antibodies. GADPH was used as a loading control. (g and h) Scramble and ANXA1-shRNA transfected A549 cells were infected with 1 MOI of influenza A/PR8 and cells were lysed at the indicated time points and subjected to immunoblot analysis for the indicated antibodies. (i) Scramble and ANXA1-shRNA transfected A549 cells were infected with 5 MOI of influenza A/PR8 and cells were fractionated into mitochondrial and cytosolic fractions at the indicated time points and subjected to immunoblot analysis for the indicated antibodies. AIF was used as a mitochondrial loading control and GAPDH was used as a cytosolic loading control
Role of NF-κB and AKT in ANXA1-dependent apoptosis
NF-κB has been widely reported as a pro-survival gene and ANXA1 has previously been shown to regulate NF-κB, either positively or negatively, depending on the cell type and stimulus used. Inactivation of NF-κB by ANXA1 upon infection might lead to higher apoptosis and, hence, higher viral loads. After 4 h of IAV infection, cells overexpressing ANXA1 exhibited lower NF-κB promoter activity (Figures 8a and b). This was not due to an effect on viability, as cells overexpressing ANXA1 exhibited higher viability, while in contrast, cells silenced for ANXA1 exhibited lower viability (Supplementary Figure S7). Figure 8b shows that Akt phosphorylation increases in a time-dependant manner upon influenza infection and is more activated in the absence of ANXA1. To determine if Akt regulates NF-κB and downstream apoptotic proteins, cells were pretreated with an Akt1 inhibitor A674563 before IAV infection. AKT phosphorylation increases with IAV infection, and is not inhibited by A674563, as this inhibitor works by blocking the activation of downstream targets (Figure 8c). ANXA1 silenced cells exhibited higher p65 and IkBα phosphorylation, which is Akt dependent (Figure 8d). Finally, we discovered that inhibitor of apoptosis cIAP2, but not cIAP1 or xIAP, is increased upon IAV infection and more highly expressed when ANXA1 is silenced.

ANXA1 inhibits IAV-induced NF-κB activation and enhances AKT activation. (a and b) Scramble and ANXA1-shRNA transfected A549 cells, or EV and ANXA1-V5 cells were transfected with pNF-κB-luc reporter and renilla-plasmids for 48 h and infected with 1 MOI of influenza A/PR/8/34 for 4 h. Luciferase activity was measured after 4 h of infection. Luciferase activity was calculated by normalizing with the pGL-luc reporter controls (mean±S.E. of at least 3 independent experiments,**P<0.01 versus Scr-sh infected cells). (c and d) Scramble and ANXA1-shRNA transfected A549 cells were infected with 1 MOI of influenza A/PR/8/34 and cells were lysed at the indicated time points and subjected to immunoblot analysis for the indicated antibodies
ANXA1 associates with NS1
A549 cells were infected with PR8 virus and an immunoprecipitation was done with NS1 and M2. After 4 and 8 h of infection, ANXA1 can associate with NS1, but not M2 (Figure 9a). Further, a co-immunoprecipitation was performed with ANXA1 plasmids cotransfected with plasmids to various IAV proteins such as NS1, M2 and PB1-F2. Once again, ANXA1 is seen to associate with NS1, which may suggest a possible physical association between ANXA1 and NS1 (Figure 9b).

ANXA1 associates with NS1 viral protein. (a) A549 lung epithelial cells were infected with 1 MOI of A/PR8 and immunoprecipitation was performed with mouse IgG or mouse anti-ANXA1 antibody and probed with the antibodies to the viral proteins. (b) 293T cells were transfected with the indicated plasmids for 36 h and co-immunoprecipitation was performed by pulling down with Flag-beads and immunostained with the indicated antibodies. Results are representative of two independent experiments
In summary, our results show that ANXA1−/− mice are protected against influenza infection and this is related to lower viral replication and propagation in lung epithelial cells. ANXA1 is involved in the binding, uptake and trafficking of the virus to the nucleus. In addition, we show that IAV can induce apoptosis, which is dependent on the presence of ANXA1. Therefore, the presence of ANXA1 enhances apoptosis, which can lead to greater virus replication (Figure 10).

Summary figure. During the IAV lifecycle, IAV binds to the host cell and is endocytosed into early and late endosomes. Uncoating occurs and the viral membrane fuses to the endosomal membrane, which allows viral RNP to enter the cytoplasm and nucleus. Transcription of viral RNA occurs, and translation of viral protein occurs through host machinery. Nuclear export of viral RNA and budding then occur. (a) ANXA1 is shown in this study to enhance binding of the virus to the host cell, and increases endocytosis of the virus. (b) ANXA1 is found in early and late endosomes near the nucleus, and silencing of ANXA1 results in lower nuclear infection. (c) More virus replication is observed in ANXA1 expressing cells. (d) In addition, IAV induces AKT phosphorylation to activate NF-kB and IKB phosphorylation and degradation. cIAP2 is enhanced by IAV, which is AKT dependent. (e) ANXA1 is shown to inhibit AKT activation and NF-kB activity. This may result in increased apoptosis. (f) ANXA1 promotes the release of cytochrome C from the mitochondria, enhances caspase 3/7 activation and PARP cleavage. (g) This leads to apoptosis and enhanced virus replication
Discussion
Our present findings support an important role for ANXA1 in a mouse model of IAV infection. The loss of this gene in an in vivo model results in protection against flu infection with decreased weight loss and increased survival as IAV was unable to propagate efficiently in ANXA1−/− mice. Indeed, the presence of ANXA1 leads to lower inflammation and higher IAV replication through the enhancement of viral transport into the nucleus. This leads to more apoptosis and increased viral replication. Moreover, ANXA1 is a virus-inducible protein in vivo and in vitro, which indicates that the induction of ANXA1 during infection may be a strategy for the virus to enhance its infectivity.
Our in vivo results show that high levels of ANXA1 is detrimental to the host, resulting in more weight loss and IAV-induced morbidity, due to higher viral titers and viral propagation. ANXA1 has been shown to be an anti-inflammatory mediator, modulating cytokine production in response to zymosan24, 25 or LPS24 and in an asthma mouse model.26, 27, 28, 29 However, ANXA1 may also promote anti-viral cytokine production in response to poly(I:C),30 and ANXA1−/− dendritic cells exhibit a mature phenotype and produced less pro-inflammatory cytokines.26 Therefore it may be possible that no difference in cytokine production was observed in the bronchoalveolar fluid due to the crossing out of functions of ANXA1, that is, in some cell types, ANXA1 inhibits cytokine production, while in others, ANXA1 promotes cytokine production. We also showed that ANXA1−/− mice exhibited markedly higher leukocyte infiltration in the lungs after infection. This is similar to previous studies where ANXA1 inhibited leukocyte recruitment in various models of inflammation.28, 31, 32 ANXA1 may modulate leukocyte migration by inhibiting adhesion to the endothelium,33 after being secreted by neutrophils upon adhesion.34
We have shown that ANXA1 is a virus-regulated host protein, which is induced and cleaved at later time points. This could be a host response, or result of an interplay between the host cell and the proteins of the virus such as NS1 that could modulate ANXA1 expression. These results are similar to other studies where ANXA1 levels were upregulated in porcine monocytes infected with swine flu virus.14 Similar cleavage of ANXA1 was reported in tuberculosis infection,7 and allergen induced asthma.27 This cleavage is thought to be a result of proteolytic action of enzymes such as elastase or calpain and cleaves at the N-terminal of the protein. In addition, ANXA1 is cleaved at its C-terminus by serine proteases and it has been hypothesized that this cleaved form is pro-inflammatory,35 while the N-terminal peptide has long been known to be anti-inflammatory. This 33 kDa cleaved form devoid of the N-terminus is present in high amounts in bronchoalveolar lavage from cystic fibrosis patients and correlates with amount of neutrophil infiltration.36 In addition, a cleavage-resistant ANXA1 exhibited stronger anti-inflammatory effects.35, 37 This indicates that the cleavage of ANXA1 may result in the inactivation of ANXA1’s anti-inflammatory functions or that the cleaved ANXA1 may have pro-inflammatory functions.
We also have demonstrated that ANXA1 can bind to NS1. NS1 is a non-essential viral gene that has been implicated in a number of strategies, which viruses use to evade the host immune response,38 such as the inhibition of IFNα/β production, which results in uncontrolled virus replication. In addition, NS1 can activate PI3K which is upstream of AKT,39 and furthermore, NS1 was shown to inhibit apoptosis through the activation of PI3K.39, 40 Interestingly, we show here that ANXA1 can enhance IAV-induced apoptosis through the inhibition of AKT activation. Therefore, ANXA1 may induce apoptosis in response to IAV infection through the inhibition of NS1 activity.
Through loss and gain of function studies, we have confirmed that ANXA1 plays a role in the binding and entry, and these lower viral titers after silencing ANXA1 are also reflected after budding into the supernatant. We also show that ANXA1 is important in the trafficking of the virus to the nucleus, where ANXA1 expressing A549 cells exhibit higher nuclear viral NP. Further, ANXA1 may regulate the endosomal trafficking of the virus to the nucleus. Although ANXA1 has not been implicated in the binding of virus to the host cell, other cell surface receptors such as EGF-R have been shown to enhance binding of other viruses to the host cell. Interestingly, ANXA1 is a substrate of the EGF-R tyrosine kinase.41 The N-terminus of ANXA1 has been shown to be associated with endosomal membranes,42 and ANXA1 can regulate EGF-induced inward vesiculation by accumulating on the internal vesicles of multivesicular endosomes.43
Apoptosis plays an important role in influenza virus propagation44 and ANXA1 is a pro-apoptotic molecule,20, 44 which may assist the propagation of the virus and hence lower survival of the mice. ANXA1 may induce dephosphorylation of BAD upon infection and allow its transportation to the nucleus for induction of apoptosis through intrinsic pathway45 as IAV has also been shown to induce apoptosis via BAD-mediated mitochondrial dysregulation.46 Our results show that ANXA1 enhances BAX translocation to, and cytochrome C release from the mitochondria, which in turn can result in the activation of caspase 3/7 and the induction of apoptosis. IAV has previously been shown to induce caspase-3-dependent apoptosis,18 and our results extend a new arm to the current dogma, which shows that ANXA1 is upstream of caspase-3 as well as AKT.44 In our study, ANXA1 was found to inhibit NF-κB activity induced by IAV infection. Previous studies in cancer cells demonstrated that ANXA1 can regulate other signaling pathways important in the regulation of NF-κB, such as ERK.47, 48 In addition, ANXA1 can regulate NF-κB activation, either positively47, 49 or negatively50, 51 depending on the stimulus and cell type used. Active NF-κB signaling is required for virus infection,52 and NF-κB has been shown to be important in the synthesis of viral RNA.53 Therefore, ANXA1 may enhance the expression and activity of NF-kB during IAV infection and promote virus propagation.
In summary, our study shows that ANXA1 plays a critical role during IAV, which involves promoting virus inward trafficking to the nucleus (Figure 10) and IAV-induced apoptosis. As apoptosis is one of the mechanisms through which IAV promotes its propagation, it is likely that it is responsible for the reduced viral titers observed in the absence of ANXA1. ANXA1 mRNA expression is increased during infection, and is also cleaved, which may indicate that it is a host factor that may be modulated by the virus.
Materials and Methods
Ethics statement
All experiments were performed in strict accordance with the guidelines of the National Advisory Committee for Laboratory Animal Research (NACLAR) and approved Institutional Animal Care and Use Committee (IACUC) protocols at the National University of Singapore. For clinical samples, the collection and recruitment was approved by the National Healthcare Group’s Domain Specific Institutional Review Board (NUS IRB Ref 09/641).
Patient recruitment
Written informed consent forms were collected from healthy volunteers and patients with PCR+Influenza A H3N2 or H1N1 patients within a period of 14 days from the onset of the infection for the study, prior to collection of the nasal swabs as previously described.24 This was approved by the National Healthcare Group’s Domain Specific Institutional Review Board (NUS IRB Ref 09/641).
Mice
BALB/c ANXA1−/− mice were a kind gift from Prof. Roderick Flower, QMUL, UK. Mice were age matched and BALB/c mice were used as control mice for each experiment. All mice were maintained under pathogen-free conditions in the animal housing unit and were transferred to the ABSL2 facility for experiments involving infection with IAV.
Viruses
For viral propagation, 1HAU virus was injected into 10–12 day incubated chicken eggs and further incubated for 3 days. On day 3, the eggs were chilled at −80 °C to euthanize the embryo and the allantoic fluid was collected. The fluid was spun in 100 000 MWCO concentrators to concentrate the virus and viral plaque assays were performed to quantify the viral titers.
Infection
Mice were infected intra-tracheally (rather than intranasally, as intra-tracheal infection has less variability) with either 25 pfu/g or 12.5 pfu/g of H1N1/PR8 virus in 20 μl of PBS. Their body weight was measured daily and they were euthanized when a loss of 25% of initial body weight was observed. A549 cells were cultured in F-12K media (Sigma-Aldrich, Singapore) supplemented with 10% FBS and 1% penicillin–streptomycin solution. Cells were washed once with PBS and then infected with indicated MOIs in serum free F-12K media for 1 h, after which cells were washed with PBS and replaced with fresh media.
Plaque assay
Serial ten-fold dilutions of lung homogenates were allowed to adsorb onto confluent monolayers of Madin Darby Canine Kidney cells (MDCK) on a 24-well plate for 1 h. The supernatant was then removed and replaced with 1% agarose supplemented with serum free Dulbecco’s Modified Eagle Medium (DMEM) (Hyclone, Logan, Utah, USA) and 2 μg/ml TPCK (L(tosylamido-2-phenyl) ethyl chloromethyl ketone) treated Trypsin. Plates were then incubated for 3–4 days at 37 °C in 5% CO2. Subsequently, agarose overlays were removed and the plaques were visualized after addition of crystal violet stain.
Real time PCR
Lungs were harvested from infected mice and small pieces were cut and stored in RNAlater immediately. Total RNA was extracted from the lungs using an RNeasy kit (Qiagen, Limburg, Netherlands). Viral RNA from supernatants was isolated using GeneJet viral DNA/RNA purification kit (Life Technologies, Carlsbad, CA, USA). A total of 500 ng of total RNA was reverse transcribed using MuMLV reverse transcriptase (Promega, Madison, WI, USA). For quantification of ANXA1, a SYBR green-based real-time PCR method was used, and β-actin mRNA was quantified to normalize the total RNA concentration between different samples. Primers for ANXA1 used: F: 5′-AAGGTGTGGATGAAGCAACC-3′; R: 5′-TGCATCAAACTGAGCTGGAG-3′ and β-actin were F: 5′-CCCAAGGCCAACCGCGAGAAGAT-3′ and R: 5′-GTCCCGGCCAGCCAGGTCCAG-3′. The primers for viral M1/M2 gene detection were F: 5′-GGACTGCAGCGTTAGACGCTT-3′; R:5′-CATCCTGTTGTATATGAGGCCCAT-3′. A standard curve of 101–108 PFU/ml was performed and all data from viral M1/M2 PCR was converted to viral particles (PFU/ml). A reaction mix of 20 μl was composed of 1 μl of each gene-specific primer at 10 μM, 10 μl of SYBR green master mix, 2 μl of 10-fold-diluted cDNA, and 6 μl of distilled water. The amplification program was as follows: 95 °C for 5 min followed by 40 cycles of 95 °C for 10 s, 60 °C for 10 s and 72 °C for 10 s. The specificity of the assay was confirmed by melting curve analysis at the end of the amplification program.
Cytokine production
Bronchoalveolar lavage fluid was obtained from uninfected and infected mice at 1,3 and 7 days post-infection. Fluids were spun down to remove contaminating leukocytes and supernatants were analyzed for IFN-γ, IL-6, IL-12p70, IL-10, IL-1β, TNFα, TGF-β, IFNα, (Ebioscience, San Diego, CA, USA) and IFNβ (Biolegend, San Diego, CA, USA) following the manufacturer’s instructions. ANXA1 levels in nasal swab fluid was analyzed using a homemade ELISA. Briefly, Nunc Maxisorb (NUNC) 96 well plates were coated with 50 μl of 1 μg/ml goat anti-ANXA1 antibody (Santa Cruz) in PBS overnight at 4 °C. Plates were washed and blocked with 1% BSA for 1 h. Plates were washed again and samples were added to the wells overnight at 4 °C. Wells were aspirated and washed and 50 μl of 0.5 μg/ml rabbit anti-ANXA1 antibody (Invitrogen, Carlsbad CA, USA) was added for 1 h at room temperature, followed with washing and addition of 50 μl of 1 μg/ml donkey anti-rabbit horse radish peroxidase (Santa Cruz, Dallas, TX, USA) for 1 h. Wells were washed and substrate was added for up to 15 min in the dark. Plates were read in a plate reader at 450 nm.
Cellular infiltration
Bronchoalveolar lavage fluid was obtained from uninfected and infected mice 7 days post infection and was spun to separate the cells and lavage. The red blood cells were removed using a lysis buffer and cells were counted using a hemacytometer. These cells were then stained with CD45, Cd11c, CD3/B220, Ly6-G and Siglec F to determine the cellular composition.
Eosinophils—CD45+ SigF+ Ly6G+ F4/80− CD11clow
Neutrophils (PMNs)—CD45+ SigF− Ly6G+
Alveolar macrophages—CD45+ Sig F+ Ly6Gint CD11c+ F480+
Lymphocytes—CD45+ SigF− Ly6G− F4/80− CD14− CD3/CD19+
Histology
Lungs were harvested and fixed in 4% paraformaldehyde. To evaluate inflammation processed and sliced tissues were stained with haematoxylin and eosin. Lung sections were scored blinded on a score of 0–3, no inflammation; 1, mild, inflammatory cell infiltrate of the perivascular/peribronchiolar compartment; 2, moderate, inflammatory cell infiltrate of the perivascular/peribronchiolar space with modest extension into the alveolar parenchyma; 3. severe, inflammatory cell infiltrate of the perivascular/peribronchiolar space with a greater magnitude of inflammatory foci found in the alveolar parenchyma.
Western blot
Cells were harvested and lysed, supernatant was collected and stored at −80 °C until evaluation by SDS-PAGE analysis. Equal amounts of protein from each sample were subjected to 10% SDS-PAGE at a constant voltage of 125 V. The proteins were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). Proteins were determined by Western blotting with specific antibodies, and expression signals were obtained by enhanced chemiluminescence. Protein expression was normalized to GAPDH or tubulin levels.
Confocal microscopy
Cells were seeded onto sterilized glass coverslips contained in 24-well plates. Cells were infected with indicated MOI of influenza virus either on ice or incubated at 37 °C for indicated times. After indicated times of infection, cells were washed with cold phosphate-buffered saline and fixed with cold 3% paraformaldehyde in PBS for 30 min at 4 °C. Cells were then washed and incubated with anti-rabbit ANXA1or influenza A NP antibody diluted in 2% (v/v) fetal bovine serum, 2% (w/v) bovine serum albumin in PBS, for 1 h at room temperature. After which, the coverslip was washed and cells were next incubated with fluorophore-conjugated secondary antibody Alexa488 anti-rabbit IgG (Invitrogen). After the final wash, each coverslip was prepared for confocal analysis on a Zeiss confocal microscope. Images were processed using Imaris software (Zurich, Switzerland).
Statistical analysis
Statistical significance was determined by the Student t test or analysis of variance for multiple comparisons of normal distributions (post-hoc tests using the Bonferroni test), with a value of P<0.05 considered to be statistically significant. For survival curves, log rank (mantel cox) test was used.
Abbreviations
- −/− :
-
Deficient
- A/PR8:
-
A/Puerto Rico/8/1934
- ANXA1:
-
Annexin-A1
- BAD:
-
Bcl-2-associated death promoter
- DPI:
-
Days post-infection
- FPR:
-
formyl peptide receptor
- IAV:
-
Influenza A virus-
- MDCK:
-
Madin Darby Canine Kidney cells
- NS:
-
Non-structural
- RNP:
-
Ribonucleoprotein
- shRNA:
-
short hairpin RNA
- TRAIL:
-
TNF-related apoptosis inducing ligand
- WT:
-
Wildtype
References
Kumar D, Michaels MG, Morris MI, Green M, Avery RK, Liu C et al. Outcomes from pandemic influenza A H1N1 infection in recipients of solid-organ transplants: a multicentre cohort study. Lancet Infect Dis 2010; 10: 521–526.
Pandemic Influenza Preparedness and Response: A WHO Guidance Document. World Health Organization: Geneva, 2013.
Taubenberger JK, Morens DM . 1918 Influenza: the Mother of All Pandemics. Emerg Infect Dis 2006; 12: 15–22.
Yewdell J, García-Sastre A . Influenza virus still surprises. Curr Opin Microbiol 2002; 5: 414–418.
Gerke V, Moss SE . Annexins: from structure to function. Physiol Rev 2002; 82: 331–371.
Raynal P, Pollard HB . Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim Biophys Acta 1994; 1197: 63–93.
Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG . Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol 2008; 9: 1189–1197.
Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U et al. Role of Annexin A2 in the production of infectious hepatitis C virus particles. J Virol 2010; 84: 5775–5789.
Huang RTC, Beate L, Oliver R . Involvement of annexin V in the entry of influenza viruses and role of phospholipids in infection. FEBS Lett 1996; 392: 59–62.
Otto M, Matthias O, Anna G, Hua F, Oliver R, Huang RTC . Identification of annexin 33 kDa in cultured cells as a binding protein of influenza viruses. FEBS Lett 1994; 356: 125–129.
LeBouder F, Lina B, Rimmelzwaan GF, Riteau B . Plasminogen promotes influenza A virus replication through an annexin 2-dependent pathway in the absence of neuraminidase. J Gen Virol 2010; 91: 2753–2761.
Ma H, Kien F, Manière M, Zhang Y, Lagarde N, Tse KS et al. Human annexin A6 interacts with influenza a virus protein M2 and negatively modulates infection. J Virol 2012; 86: 1789–1801.
Musiol A, Gran S, Ehrhardt C, Ludwig S, Grewal T, Gerke V et al. Annexin A6-balanced late endosomal cholesterol controls influenza A replication and propagation. MBio 2013; 4: e00608–e00613.
Katoh N . Detection of annexins I and IV in bronchoalveolar lavage fluids from calves inoculated with bovine herpes virus-1. J Vet Med Sci 2000; 62: 37–41.
Lim LHK, Pervaiz S . Annexin 1: the new face of an old molecule. FASEB J 2007; 21: 968–975.
Stray SJ, Air GM . Apoptosis by influenza viruses correlates with efficiency of viral mRNA synthesis. Virus Res 2001; 77: 3–17.
Fesq H, Heike F, Michael B, Marianne N, Diethard G . Programmed cell death (apoptosis) in human monocytes infected by influenza A virus. Immunobiology 1994; 190: 175–182.
Herold S, Ludwig S, Pleschka S, Wolff T . Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J Leukoc Biol 2012; 92: 75–82.
Olsen CW, Kehren JC, Dybdahl-Sissoko NR, Hinshaw VS . bcl-2 alters influenza virus yield, spread, and hemagglutinin glycosylation. J Virol 1996; 70: 663–666.
Solito E, de Coupade C, Canaider S, Goulding NJ, Perretti M . Transfection of annexin 1 in monocytic cells produces a high degree of spontaneous and stimulated apoptosis associated with caspase-3 activation. Br J Pharmacol 2001; 133: 217–228.
Petrella A, Festa M, Ercolino SF, Zerilli M, Stassi G, Solito E et al. Induction of annexin-1 during TRAIL-induced apoptosis in thyroid carcinoma cells. Cell Death Differ 2005; 12: 1358–1360.
Canaider S, Solito E, de Coupade C, Flower RJ, Russo-Marie F, Goulding NJ et al. Increased apoptosis in U937 cells over-expressing lipocortin 1 (annexin I). Life Sci 2000; 66: PL265–PL270.
Tambyah PA, Sepramaniam S, Mohamed Ali J, Chai SC, Swaminathan P, Armugam A et al. microRNAs in circulation are altered in response to influenza A virus infection in humans. PLoS One 2013; 8: e76811.
Damazo AS, Yona S, D’Acquisto F, Flower RJ, Oliani SM, Perretti M . Critical protective role for annexin 1 gene expression in the endotoxemic murine microcirculation. Am J Pathol 2005; 166: 1607–1617.
Getting SJ, Flower RJ, Perretti M . Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br J Pharmacol 1997; 120: 1075–1082.
Huggins A, Paschalidis N, Flower RJ, Perretti M, D’Acquisto F . Annexin-1-deficient dendritic cells acquire a mature phenotype during differentiation. FASEB J 2009; 23: 985–996.
Chung YW, Oh HY, Kim JY, Kim J-H, Kim IY . Allergen-induced proteolytic cleavage of annexin-1 and activation of cytosolic phospholipase A2 in the lungs of a mouse model of asthma. Proteomics 2004; 4: 3328–3334.
Ng FSP, Wong KY, Guan SP, Mustafa FB, Kajiji TS, Bist P et al. Annexin-1-deficient mice exhibit spontaneous airway hyperresponsiveness and exacerbated allergen-specific antibody responses in a mouse model of asthma. Clin Exp Allergy 2011; 41: 1793–1803.
Lee SH, Kim DW, Kim HR, Woo SJ, Kim SM, Jo HS et al. Anti-inflammatory effects of Tat-Annexin protein on ovalbumin-induced airway inflammation in a mouse model of asthma. Biochem Biophys Res Commun 2012; 417: 1024–1029.
Bist P, Shu S, Lee H, Arora S, Nair S, Lim JY et al. Annexin-A1 regulates TLR-mediated IFN-β production through an interaction with TANK-binding kinase 1. J Immunol 2013; 191: 4375–4382.
Guido BC, Zanatelli M, Tavares-de-Lima W, Oliani SM, Damazo AS . Annexin-A1 peptide down-regulates the leukocyte recruitment and up-regulates interleukin-10 release into lung after intestinal ischemia-reperfusion in mice. J Inflamm 2013; 10: 10.
Drechsler M, de Jong R, Rossaint J, Viola JR, Leoni G, Wang JM et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ Res 2015; 116: 827–835.
Lim LHK, Solito E, Russo-Marie F, Flower RJ, Perretti M . Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: Effect of lipocortin 1. Proc Natl Acad Sci 1998; 95: 14535–14539.
Perretti M, Croxtall JD, Wheller SK, Goulding NJ, Hannon R, Flower RJ . Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat Med 1996; 2: 1259–1262.
Williams SL, Milne IR, Bagley CJ, Gamble JR, Vadas MA, Pitson SM et al. A proinflammatory role for proteolytically cleaved annexin A1 in neutrophil transendothelial migration. J Immunol 2010; 185: 3057–3063.
Tsao FH, Meyer KC, Chen X, Rosenthal NS, Hu J . Degradation of annexin I in bronchoalveolar lavage fluid from patients with cystic fibrosis. Am J Respir Cell Mol Biol 1998; 18: 120–128.
Pederzoli-Ribeil M, Maione F, Cooper D, Al-Kashi A, Dalli J, Perretti M et al. Design and characterization of a cleavage-resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood 2010; 116: 4288–4296.
Hale BG, Randall RE, Ortín J, Jackson D . The multifunctional NS1 protein of influenza A viruses. J Gen Virol 2008; 89: 2359–2376.
Shin Y-K, Y-K S, Liu Q, Tikoo SK, Babiuk LA, Zhou Y . Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J Gen Virol 2007; 88: 13–18.
Ehrhardt C, Wolff T, Pleschka S, Planz O, Beermann W, Bode JG et al. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol 2007; 81: 3058–3067.
Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 2011; 17: 589–595.
Seemann J, Weber K, Osborn M, Parton RG, Gerke V . The association of annexin I with early endosomes is regulated by Ca2+ and requires an intact N-terminal domain. Mol Biol Cell 1996; 7: 1359–1374.
White IJ, Bailey LM, Aghakhani MR, Moss SE, Futter CE . EGF stimulates annexin 1-dependent inward vesiculation in a multivesicular endosome subpopulation. EMBO J 2006; 25: 1–12.
Wurzer WJ, Ehrhardt C, Pleschka S, Berberich-Siebelt F, Wolff T, Walczak H et al. NF-κB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem 2004; 279: 30931–30937.
Solito E, Kamal A, Russo-Marie F, Buckingham JC, Marullo S, Perretti M . A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J 2003; 17: 1544–1546.
Tran AT, Cortens JP, Du Q, Wilkins JA, Coombs KM . Influenza virus induces apoptosis via BAD-mediated mitochondrial dysregulation. J Virol 2013; 87: 1049–1060.
Bist P, Leow SC, Phua QH, Shu S, Zhuang Q, Loh WT et al. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-κB: implication in breast cancer metastasis. Oncogene 2011; 30: 3174–3185.
Alldridge LC, Harris HJ, Plevin R, Hannon R, Bryant CE . The annexin protein lipocortin 1 regulates the MAPK/ERK pathway. J Biol Chem 1999; 274: 37620–37628.
Jia Y, Morand EF, Song W, Cheng Q, Stewart A, Yang YH . Regulation of lung fibroblast activation by annexin A1. J Cell Physiol 2013; 228: 476–484.
Ouyang N, Zhu C, Zhou D, Nie T, Go MF, Richards RJ et al. MC-12, an annexin A1-based peptide, is effective in the treatment of experimental colitis. PLoS One 2012; 7: e41585.
Zhang Z, Huang L, Zhao W, Rigas B . Annexin 1 induced by anti-inflammatory drugs binds to NF-kappaB and inhibits its activation: anticancer effects in vitro and in vivo. Cancer Res 2010; 70: 2379–2388.
Nimmerjahn F, Dudziak D, Dirmeier U, Hobom G, Riedel A, Schlee M et al. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J Gen Virol 2004; 85: 2347–2356.
Kumar N, Xin Z-T, Liang Y, Ly H, Liang Y . NF-kappaB signaling differentially regulates influenza virus RNA synthesis. J Virol 2008; 82: 9880–9889.
Acknowledgements
This study was funded through grants from NUHS (R-185-000-239-750) and NMRC (NMRC/CBRG/056/2014) to LHKL. ANXA1−/− mice and pcDNA3.1-V5-Annexin-A1 plasmids were a kind gift from Professor Roderick Flower from the William Harvey Research Institute, London. The authors would like to thank Dr. Esther Koh, Advanced Imaging Lab, Life Sciences Institute; Dr. Paul Hutchinson, Immunology Flow Cytometry Core, Life Sciences Institute and Mr. Benson Chua, Transgenic Mouse Core, Life Sciences Institute for their help and assistance.
Author contributions
S. Arora, S. Alonso and LHKL designed the experiments and analysed the data. S. Arora, WL, PB, RPS, LHM, LBW, LF, BY performed the experiments and analyzed the data. GS, PT, AMF provided materials. S Arora and LHKL wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Silke
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Arora, S., Lim, W., Bist, P. et al. Influenza A virus enhances its propagation through the modulation of Annexin-A1 dependent endosomal trafficking and apoptosis. Cell Death Differ 23, 1243–1256 (2016). https://doi.org/10.1038/cdd.2016.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}