
Abstract
In order to establish infection, intra-macrophage parasite Leishmania donovani needs to inhibit host defense parameters like inflammatory cytokine production and apoptosis. In the present study, we demonstrate that the parasite achieves both by exploiting a single host regulator AKT for modulating its downstream transcription factors, β-catenin and FOXO-1. L. donovani-infected RAW264.7 and bone marrow-derived macrophages (BMDM) treated with AKT inhibitor or dominant negative AKT constructs showed decreased anti-inflammatory cytokine production and increased host cell apoptosis resulting in reduced parasite survival. Infection-induced activated AKT triggered phosphorylation-mediated deactivation of its downstream target, GSK-3β. Inactivated GSK-3β, in turn, could no longer sequester cytosolic β-catenin, an anti-apoptotic transcriptional regulator, as evidenced from its nuclear translocation during infection. Constitutively active GSK-3β-transfected L. donovani-infected cells mimicked the effects of AKT inhibition and siRNA-mediated silencing of β-catenin led to disruption of mitochondrial potential along with increased caspase-3 activity and IL-12 production leading to decreased parasite survival. In addition to activating anti-apoptotic β-catenin, phospho-AKT inhibits activation of FOXO-1, a pro-apoptotic transcriptional regulator. Nuclear retention of FOXO-1, inhibited during infection, was reversed when infected cells were transfected with dominant negative AKT constructs. Overexpression of FOXO-1 in infected macrophages not only documented increased apoptosis but promoted enhanced TLR4 expression and NF-κB activity along with an increase in IL-1β and decrease in IL-10 secretion. In vivo administration of AKT inhibitor significantly decreased liver and spleen parasite burden and switched cytokine balance in favor of host. In contrast, GSK-3β inhibitor did not result in any significant change in infectivity parameters. Collectively our findings revealed that L. donovani triggered AKT activation to regulate GSK-3β/β-catenin/FOXO-1 axis, thus ensuring inhibition of both host cell apoptosis and immune response essential for its intra-macrophage survival.
Similar content being viewed by others


Main
Leishmania, an intracellular protozoan parasite, resides within the macrophages by neutralizing parasiticidal effector mechanisms.1 A further challenge is to save its niche because when macrophages fail to eliminate the pathogens, they sacrifice themselves by means of initiating apoptosis.2 To understand the roles of these parameters, work have been done individually on either regulation of host cell apoptosis or immune activation during infection. But the diversifying knowledge of cell signaling network indicates the existence of several regulators known to have multiple roles thereby simultaneously modulating different interrelated parameters. We thought it worthwhile to investigate whether Leishmania could be able to manipulate both host cell apoptosis and immune balance by exploiting one such host regulatory protein.
AKT signaling is known to have vital roles in cell growth, survival, apoptosis and immune regulation.3 Earlier studies in bone marrow-derived dendritic cells (DC) showed that AKT pathway is crucial for parasite survival4 and also for inhibition of host cell apoptosis.5 Exploiting AKT pathway to inhibit host cell apoptosis has also been documented during infection with Salmonella, Anaplasma, Toxoplasma and Trypanosome.6, 7 Host immune activation essential to eliminate intracellular pathogens is also negatively modulated by AKT8 and constitutively active AKT was shown to increase the LPS-induced production of anti-inflammatory cytokine IL-10 (ref 9) and to down-regulate p65 and GSK-3β (ref 10). Two major proteins which contribute in diversifying AKT signaling are β-catenin and FOXO-1. β-catenin, a member of the Wnt signaling cascade, has a central role in transmission of peripheral signals to the nucleus. However, recent evidences suggest a role of Wnt independent regulation of β-catenin in an AKT dependent manner.11 AKT inactivates GSK-3β,12 which in turn leads to stabilization and nuclear translocation of β-catenin13 where it interacts with Tcf/Lef transcription factors to activate target anti-apoptotic genes such as Bcl-2, c-myc and cyclin D1 (ref 14). Overexpression of β-catenin has been shown to reduce apoptosis which is similar to the protection provided by inhibitors of GSK-3β15 and accumulation of β-catenin in response to GSK-3β inhibitors has been correlated with an increase in the expression of the anti-apoptotic proteins.16 FOXO-1 is a member of the Forkhead family of transcription factors, which after phosphorylation by AKT is known to get inactivated and translocated to the cytosol from nucleus.17 Activated FOXO-1, on the other hand induce apoptosis by inducing various pro-apoptotic members of mitochondria-targeting proteins.18 These regulators also have important roles in immune regulation. Sporadic evidences indicate that β-catenin inhibits inflammation in a GSK-3β-dependent manner whereas FOXO-1 has also been implicated in regulation of pro-inflammatory responses.18, 19, 20 By using constitutively active FOXO-1, its importance was shown in regulating the inflammatory responses both in the context of LPS induction or free fatty acid administration.19 Moreover, administration of siRNA targeting FOXO-1 resulted in decreased inflammatory responses in H. influenzae infection.20 β-catenin limits NF-κB mediated inflammatory responses in both Salmonella infection and TNF-α treatment by stabilizing Iκβ.21 We, therefore, were interested to determine whether these two downstream effector molecules of AKT are modulated by infection. The present paper demonstrates a mechanistic insight into how by exploiting a single host protein AKT, Leishmania inhibits both pro-inflammatory response and host cell apoptosis by activating β-catenin through inhibition of GSK-3β on one hand and inactivating FOXO-1 on the other hand. Overall, this work highlights the importance of AKT/GSK-3β/β-catenin/FOXO-1 pathway in Leishmania-mediated anti-inflammatory and anti-apoptotic responses.
Results
Role of AKT on intracellular survival of L. donovani
In order to determine the effect of AKT pathway on parasite survival, macrophages were treated with AKT inhibitor, AKTi or GFP-tagged dominant negative construct (DN-AKT) prior to infection and intracellular parasite count was determined. Transfection efficiency was determined by the expression of GFP in whole cell lysates of transfected cells (Figure 1a). Inhibition of AKT led to significantly reduced parasite survival (57.4 and 66.3% reduction for AKTi and DN-AKT, respectively, compared with infected control, P<0.001; Figure 1b). Inhibition of AKT also resulted in significant reduction of IL-10 (73.4 and 86.8% for AKTi and DN-AKT, respectively, P<0.001; Figure 1c) with simultaneous up-regulation of IL-12 (4.3- and 5.2-fold for AKTi and DN-AKT, P<0.001; Figure 1d) compared wth infected control. Phosphorylation kinetics using specific antibodies (Ser473/Thr308) revealed infection-induced activation of AKT with maximum level of phosphorylation at 6 h post infection (3.2- and 2.9-fold over control for Ser473 and Thr308, respectively, P<0.001; Figure 1e). Similar results were obtained for infected BMDM (Figure 1f). Next to study host cell apoptosis, control and 6 h infected RAW264.7 cells were subjected to H2O2 (400 μM) treatment for 1 h and apoptosis was measured by flow cytometric analysis. Compared with H2O2 control (62.5±5.7% annexin V positive cells), infected H2O2-treated macrophages showed significantly decreased apoptosis (7.5±0.8% annexin V positive cells; Figure 1g). On the contrary, treatment with AKTi prior to infection and H2O2 treatment resulted in increased apoptosis (5.2-fold, P<0.01; Figure 1g), caspase-3 activity (2.9-fold, P<0.01; Figure 1i) and mitochondrial membrane potential (4.6-fold, P<0.001; Figure 1j) over H2O2-treated infected cells. Genomic DNA fragmentation assay in DN-AKT transfected and infected H2O2-treated cells revealed higher DNA fragmentation compared with WT-AKT transfected cells (Figure 1k). A representative surface plot of the gel shows the extent of fragmentation where peaks correspond to the fragments of the genomic DNA whereas plateau denotes the integrity of the genomic DNA (Figure 1l). All these results suggest that activation of AKT signaling helps L. donovani to shift cytokine balance from pro- to anti-inflammatory response and to inhibit host cell apoptosis thereby aiding its survival within the macrophages.

Role of AKT in L. donovani infection. (a–d) RAW264.7 cells (2 × 106) were treated with either AKTi (10 μM) for 1 h or transiently transfected with WT- or DN-AKT expression plasmids for 24 h. Both these macrophages were then infected with L. donovani promastigotes (macrophage:parasite ratio, 1:10) for 24 h. Expression of GFP in transfected cells were detected by Western blotting (a) and intracellular parasite number (b) were determined by DAPI staining whereas levels of IL-10 (c) and IL-12 (d) were measured by ELISA. (e and f) Both RAW264.7 (e) and BMDM (f) were infected with L. donovani promastigotes for different time periods as indicated. Levels of phosphorylated and total AKT were then detected by Western Blotting. (g–j) RAW cells were treated with AKTi for 1 h, infected with L. donovani promastigotes for 6 h and then treated with 400 μM H2O2 for 1 h. These cells were analyzed for the extent of apoptosis by annexin V-tagged FITC-DAPI flow cytometry after washing and incubation for 24 h at 37 °C (g and h). Dual parameter dot plot of FITC fluorescence (x axis) versus DAPI fluorescence (y axis) is represented as logarithmic fluorescence intensity. Quadrants are as follows: upper left, necrotic cells; lower left, live cells; lower right, apoptotic cells; upper right, necrotic or late phase of apoptotic cells. After washing, whole cell lysate of these cells (10 μg of protein per sample) were used to determine caspase-3 activity using Ac-DEVDpNA as substrate (i). Mitochondrial integrity was measured in these AKTi treated infected-H2O2 treated cells after incubation with DiOC6 (40 nM) for an additional 30 min (j). (k) RAW264.7 cells were transiently transfected with either WT- or DN-AKT expression plasmids, infected with L. donovani promastigotes for 6 h and then treated with H2O2 (400 μM) for 1 h. DNA fragmentation profile was analyzed by agarose gel electrophoresis.(l) A representative surface plot analyzed by using ImageJ software indicates the extent of fragmentation. All experiments were repeated at least three times each and one set of representative data is shown. Bands were analyzed densitometrically and bar graphs expressing arbitrary densitometric units are presented adjacent to corresponding western blots. Error bars represent mean±S.D., n=3.**P<0.01, ***P<0.001; Student's t-test
Role of AKT-Mediated GSK-3β inactivation in L. donovani infection
Since GSK-3β is one of the important targets of activated AKT and has been implicated in regulating apoptosis and inflammatory responses,22, 23 we wanted to ascertain the role of GSK-3β in infection. Kinetic analysis with p-GSK-3βSer9 antibodies documented that like AKT, maximum phosphorylation (3.4-fold over control, P<0.001) and thereby inhibition of GSK-3β was observed at 6 h post infection (Figure 2a). BMDM also showed similar trend (Figure 2b). Moreover, macrophages inhibited with DN-AKT or AKTi followed by 6 h of infection showed a decrease in GSK-3β phosphorylation (58.1 and 59.3% reduction for AKTi and DN-AKT, respectively, compared with infected control, P<0.001; Figure 2c). In order to ascertain the role of infection-induced inactivation of GSK-3β in modulation of apoptosis, we transfected cells with constitutively active (CA)-GSK-3β, which cannot be phosphorylated thus cannot get inactivated. Transfection efficiency was determined in wild type (WT)-GSK-3β or CA-GSK-3β transfected cells by hemagglutinin (HA) expression (Figure 2d). CA-GSK-3β transfected cells showed increased apoptosis (2.1-fold, P<0.01; Figure 2e), caspase-3 activity (2.1-fold, P<0.01; Figure 2g) and mitochondrial membrane potential (2.7-fold, P<0.001; Figure 2h) compared with WT-GSK-3β transfected L. donovani-infected H2O2-treated cells. In contrast, inhibiting non-phosphorylated active GSK-3β by a competitive inhibitor SB216763 significantly decreased H2O2-induced caspase-3 activity and mitochondrial potential compared with H2O2-treated cells (data not shown). However, addition of SB216763 in infected H2O2-treated cells did not further decreased caspase-3 activity and mitochondrial potential (Figures 2g and h) indicating that infection already substantially inhibited GSK-3β. Moreover, cells expressing CA-GSK-3β showed decreased IL-10 production (55.9%, P<0.001; Figure 2i) with simultaneous stimulation of IL-12 production (3.8-fold, P<0.001) compared with WT-GSK-3β transfected L. donovani-infected cells (Figure 2j). A decrease in parasite survival was also observed in CA-GSK-3β-transfected cells (45.3%, P<0.01; Figure 2k). Collectively, these results suggest that inactivation of GSK-3β by AKT during infection results in regulating both host apoptosis and immune activation.

Role of GSK-3β in L. donovani infection. (a and b) Both RAW264.7 (a) and BMDM (b) were infected with L. donovani promastigotes (macrophage:parasite ratio, 1:10) for different time periods as indicated. Levels of phosphorylated and total GSK-3β were then detected by Western Blotting. (c) Cell lysates were prepared from AKTi treated or WT- or DN-AKT transfected infected cells and levels of phosphorylated and total GSK-3β were detected by immunoblotting. (d–h) RAW264.7 cells were either treated with SB216763 (20 μM) for 1 h or transfected with WT- or CA-GSK-3β expression plasmids, infected with L. donovani promastigotes for 6 h and then treated with 400 μM H2O2 for 1 h. Expression of Hemagglutinin (HA) in whole cell lysates were analyzed in transfected cells by Western blotting (d). Similarly, transfected cells were analyzed for the extent of apoptosis by annexin V-tagged FITC-DAPI flow cytometry (e and f), caspase-3 activity using Ac-DEVDpNA as substrate (g) and mitochondrial integrity after incubation with DiOC6 (40 nM) (h) as described in the legend of Figure 1. (i–k) Macrophages were treated with SB216763 or transfected with WT-or CA-GSK-3β expression plasmids, infected with L. donovani promastigotes for 24 h and processed for determination of either IL-10 (i) and IL-12 (j) by ELISA or checked for parasite survival (k). All experiments were repeated at least three times each and one set of representative data is shown. Bands were analyzed densitometrically and bar graphs expressing arbitrary densitometric units are presented adjacent to corresponding western blots. Error bars represent mean±S.D., n=3. NS, non-significant, **P<0.01, ***P<0.001; Student's t-test
Role of β-catenin in infection-induced inhibition of pro-inflammatory and apoptotic effects
To investigate whether β-catenin was associated with inhibition of apoptosis, siRNA-mediated knock down system was used, which resulted in 71.1% decrease of β-catenin expression compared with control siRNA treated cells (Figure 3a). When subjected to H2O2 treatment, β-catenin-silenced infected cells showed significantly increased apoptosis (2.1-fold, P<0.01; Figure 3b), caspase-3 activity (1.9-fold, P<0.01; Figure 3d) and mitochondrial membrane potential (2.2-fold, P<0.01; Figure 3e) compared with control siRNA treated infected cells. Next to ascertain the role of β-catenin on the pro-inflammatory response during infection, we measured the activity of NF-κB by luciferase reporter assay as macrophage pro-inflammatory responses are primarily regulated by NF-κB.24 For this, cells were treated with XAV939, a selective inhibitor of β-catenin25 or transiently transected with β-catenin siRNA and subjected to measurement of NF-κB luciferase activity. Significant increase in NF-κB reporter activity was observed in XAV939-treated and β-catenin-silenced infected cells (1.7- and 1.8-fold increase as compared with infected, P<0.01; Figure 3f). Moreover, XAV939 treatment or silencing of β-catenin in infected cells led to increased levels of IL-12 (3.1- and 3.2-fold over infected cells, P<0.001; Figure 3g) and decreased parasite survival (35.7 and 48.8% decrease compared with infected cells, P<0.05 and P<0.01, respectively; Figure 3h) indicating thereby the role of β-catenin in regulation of both apoptosis and pro-inflammatory response during infection.

Role of β-catenin in apoptois and inflammatory responses during infection. (a) RAW264.7 cells were transfected (24 h) with either control or β-catenin siRNA followed by infection with L. donovani promastigotes for 6 h. Expression of β-catenin was evaluated by immunoblot analysis. (b-e) Macrophages were transfected with β-catenin siRNA for 24 h, infected with L. donovani promastigotes for 6 h and then treated with 400 μM H2O2 for 1 h. These cells were then analyzed for the extent of apoptosis by annexin V-tagged FITC-DAPI flow cytometry after incubation for 24 h at 37 °C (b and c), caspase-3 activity using Ac-DEVDpNA as substrate (d) and mitochondrial integrity (e) as described in the legend of Figure 1. (f) RAW264.7 cells were treated with XAV939 (1 μM) or transfected with control or β-catenin siRNA along with pNF-κB luciferase plasmid (1 μg) and 0.5 μg of pCMV-β-gal. After 24 h of transfection, cells were infected with L. donovani promastigotes for 24 h, washed, lysed and processed for luciferase activity. (g and h) XAV939-treated or β-catenin siRNA transfected cells were infected with L. donovani promastigotes for 24 h and were processed for determination of IL-12 by ELISA (g) or estimation of extent of parasite survival (h). All experiments were repeated at least three times each and one set of representative data is shown. Bands were analyzed densitometrically and bar graphs expressing arbitrary densitometric units are presented adjacent to corresponding western blots. Error bars represent mean±S.D., n=3. *P<0.05, **P<0.01, ***P<0.001; Student's t-test
Role of AKT/GSK-3β on activation of β-catenin in infection
AKT can phosphorylate β-catenin at the Ser552 residue resulting in its activation and nuclear translocation from the cytosol.11 Kinetic analysis in infected macrophages with phospho-β-cateninSer552 antibodies showed increasing phosphorylation of β-catenin with a maximum of 3.0-fold over control at 6 h post infection (Figure 4a). This increased phosphorylation coincided with its nuclear localization (Figure 4b), thus indicating the role of AKT in β-catenin activation. β-catenin is also known to be phosphorylated by GSK-3β at another serine residue (Ser33/37) followed by ubiquitination mediated degradation.26, 27 However, no band corresponding to phospho-Ser33/37was detected when MG132 pre-treated infected macrophages was subjected to Western blot analysis with anti-phospho-Ser33/37 antibody (Figure 4c). This is in line with our earlier observation that GSK-3β remains inactive during infection (Figure 2a), thereby gets prevented from phosphorylating β-catenin. To further ascertain the individual roles of AKT and GSK-3β in nuclear localization of β-catenin, DN-AKT or CA-GSK-3β-transfected macrophages were infected with L. donovani and β-catenin level was determined in the nuclear extracts. Transfection with either constructs resulted in significant decrease in β-catenin expression (60.4 and 62.2%, respectively, compared with WT-AKT- or WT-GSK-3β-transfected L. donovani-infected cells, P<0.001) (Figure 4d). These observations suggest that either inactivation of AKT or activation of GSK-3β inhibits β-catenin activation and nuclear localization. Microscopic study using DAPI (green) as a nuclear marker and Alexa555-conjugated secondary antibody to β-catenin antibody (red) in infected cells also showed nuclear localization of β-catenin as measured by Maximum Pearson’s Coefficient (Figure 4e and 4E1). The regions showing yellow and red colors in green nuclear areas denote positive colocalization. Treatment of infected cells with either AKT inhibitor or XAV939 resulted in a decrease in nuclear localization of β-catenin (51.3 and 45.4%, respectively, P<0.01 compared with infected cells; Figure 4e and 4E1). However, GSK-3β inhibitor SB216763 did not further increase nuclear localization of β-catenin in infected cells validating that GSK-3β was already inhibited resulting in maximum β-catenin activation in infection. On the contrary, when SB216763 was added after addition of AKTi, it could significantly reverse the inhibitory effect of AKTi suggesting thereby that GSK-3β works downstream of AKT. Inhibition of AKT renders GSK-3β active which prevents nuclear localization of β-catenin. On the other hand, inhibition of GSK-3β by SB216763 after AKT inhibition fails to sequester β-catenin in cytosol thereby increasing its nuclear localization (2.0-fold, P<0.01; Figure 4e and 4E1). Similar observations were made in Western blot analysis of nuclear extracts of infected cells treated with various inhibitors (Figure 4f). These observations signify that infection promotes activation of AKT for β-catenin phosphorylation-mediated nuclear localization and at the same time renders GSK-3β inactive to prevent β-catenin degradation.

Role of AKT and GSK-3β in modulation of β-catenin localization in infection. (a) RAW264.7 cells were infected with L. donovani promastigotes (macrophage:parasite ratio, 1:10) for different time periods as indicated. Levels of phosphorylated and total β-catenin were then detected by Western Blotting using respective antibodies. (b) Nuclear fractions were isolated from RAW264.7 cells infected with L. donovani promastigotes for different times and β-catenin expression was checked by immunoblotting. (c) Infected RAW264.7 cells were lysed after different time periods and checked for the levels of phosphorylated and total β-catenin by Western Blotting using respective antibodies. (d) RAW264.7 cells were transfected (24 h) with WT-AKT, DN-AKT, WT-GSK-3β and CA-GSK-3β expression plasmids followed by infection with L. donovani promastigotes for 6 h. Nuclear fractions were isolated and expression of β-catenin was evaluated by immunoblot analysis. (e) Macrophages were treated with AKTi or SB216763 or XAV939 for 1 h followed by infection with L. donovani promastigotes for 6 h. Cells were then stained with anti-β-catenin monoclonal antibody followed by secondary Alexa555-conjugated antibody. Nuclei were stained with DAPI and cells were analyzed under microscope. Images were analyzed for colocalization using Olympus Fluoview (version 3.1a; Tokyo, Japan). For better visual aid green was used instead of blue to mark DAPI using LUT program of Fluoview. Red or yellow colors on green nucleus denotes strong colocalization, whereas green nucleus lacking any red or yellow colors denotes exclusion. (E1) Maximum Pearson’s Coeffiecient was calculated by randomly selecting at least 15 cells per field for at least three random fields per experiment using colocalization program of Olympus Fluoview (version 3.1a; Tokyo, Japan). (f) Macrophages were treated with AKTi and/or SB216763 or XAV939 and infected for 6 h. Thereafter nuclear fractions were isolated and expression of β-catenin was assessed. Lamin serves as control for the nuclear extract. All experiments were repeated at least three times each and one set of representative data is shown. Bands were analyzed densitometrically and bar graphs expressing arbitrary densitometric units are presented adjacent to corresponding western blots. Error bars represent mean±S.D., n=3. NS, non-significant, **P<0.01,***P<0.001; Student's t-test
Role of FOXO-1 in L. donovani mediated inhibition of pro-inflammatory response and apoptotic effects
As FOXO-1 is negatively regulated by β-catenin28 and is involved in transcriptional up-regulation of TLR4 (ref 19), we thought it worthwhile to study the role of FOXO-1 in apoptosis and inflammatory response in L. donovani infection and its regulation by AKT/GSK-3β/β-catenin pathway. Cells were transfected with the GFP-tagged constitutively active FOXO-1 (CA-FOXO-1) which cannot be phosphorylated and therefore remains sequestered in the nucleus.29 Microscopic studies showed that GFP-tagged wild type FOXO-1 (WT-FOXO-1) was maximally localized in cytoplasm after infection whereas expression of CA-FOXO-1 was observed in nucleus in control as well as infected cells (Figure 5a). Expression of GFP was checked in whole cell lysates to further confirm the transfection efficiency in transfected cells (Figure 5b). DNA fragmentation assay revealed that transfection of CA-FOXO-1 resulted in DNA fragmentation in infected and H2O2-treated or only H2O2-treated cells indicating an important role of FOXO-1 in apoptosis (Figure 5c). Extent of DNA fragmentation is further illustrated in the surface plot of the gel where multiple peaks correspond to the fragmented genomic DNA (Figure 5d). Transfection with CA-FOXO-1 also resulted in increased expression of TLR4 and IL-1β (2.2-fold, P<0.01 and 2.4-fold, P<0.001, respectively, compared with WT-FOXO-1 transfected L. donovani-infected cells; Figures 5e and f). As both TLR4 and IL-1β are regulated by NF-κB we wanted to understand the status of NF-κB activation in CA-FOXO-1 transfected infected cells. NF-κB luciferase activity was increased 1.7-fold in CA-FOXO-1 transfected L. donovani-infected cells over WT-FOXO-1 transfected infected cells, (P<0.01; Figure 5g). CA-FOXO-1 transfection also led to a significant decrease in IL-10 production (57.3%, P<0.01; Figure 5h) and parasite survival (45.1%, P<0.01; Figure 5i) compared with WT-FOXO-1-transfected infected cells. These results suggest an important role of FOXO-1 in infection-mediated regulation of apoptosis and inflammatory responses.

Role of FOXO-1 in apoptosis and inflammatory response during infection. (a) RAW264.7 cells were transfected with either GFP-WT-FOXO-1 or GFP-CA-FOXO-1 and infected with L. donovani promastigotes for 6 h. Cells were fixed and stained with DAPI and analyzed under microscope. (b–d) Cells were transiently transfected with WT- or CA-FOXO-1 expression plasmid (24 h) and then infected with L. donovani for 6 h. Transfection efficiency was further analyzed by checking the expression of GFP in whole cell lysates (b). Transfected and L. donovani-infected cells were treated with H2O2 for 1 h, washed and incubated for additional 24 h and the integrity of DNA was analyzed with the appearance of DNA ladders in each group via agarose electrophoresis (c). Representative surface plot indicates the extent of fragmentation (d). (e–i) Cells were transfected with the indicated constructs and infected with parasites for 24 h. This was followed by real-time PCR analysis of TLR4 levels as fold change (e) or processed for determination of IL-1β (f) and IL-10 (g) levels by ELISA. RAW264.7 cells were transfected with desired constructs along with pNF-κB luciferase plasmid (1 μg) and 0.5 μg of pCMV-β-gal. After 24 h of transfection, cells were infected with L. donovani for 24 h, lysed, and processed for luciferase activity (h). The parasite survival was determined by staining the cells with DAPI after transfection with indicated constructs and counting parasites per 100 macrophages (i). All experiments were repeated at least three times each and one set of representative data is shown. Bands were analyzed densitometrically and bar graphs expressing arbitrary densitometric units are presented adjacent to corresponding western blots. Error bars represent mean±S.D., n=3. **P<0.01, ***P<0.001; Student's t-test
AKT activation leads to cytosolic sequestration of FOXO-1 during infection
After ascertaining the modulation of FOXO-1 in establishing infection, we wanted to understand the mechanism of FOXO-1 inactivation in infection. Phosphorylation of FOXO-1 at ser256 by AKT was reported to block its activity and decrease its DNA binding.17 Likewise, an increase in phosphorylation of FOXO-1 at Ser256 residue was observed in L. donovani-infected macrophages with a maximum of 2.9-fold increase at 6 h post infection compared with uninfected control, P<0.001; Figure 6a). FOXO-1 is predominantly a nuclear protein and its phosphorylation by AKT promotes its recognition by 14-3-3 protein which results in its cytosolic translocation leading to inactivation.17 Western blot analysis revealed a decrease in nuclear FOXO-1 in infected macrophages (54.2% decrease over control, P<0.001) at 6 h post-infection (Figure 6b). To ascertain the role of AKT/GSK-3β in regulation of FOXO-1 during infection, we transfected cells with DN-AKT, which resulted in increased nuclear retention of FOXO-1 (3.2-fold compared with WT-AKT transfected infected cells, P<0.001) whereas no significant change in nuclear localization of FOXO-1 was observed in CA-GSK-3β transfected infected cells (Figure 6c). This shows that FOXO-1 is under direct regulation by AKT during infection. Microscopic analysis revealed that during infection significant decrease in nuclear FOXO-1 was seen which was markedly increased in presence of AKTi (2.8-fold, P<0.01; Figure 6d and 6D1). In contrast, no significant change was noticed in presence of GSK-3β inhibitor SB216763 further nullifying the involvement of GSK-3β in FOXO-1 regulation in infection. When SB216763 was added after addition of AKTi, it failed to trigger FOXO-1 exclusion from the nucleus thereby validating that FOXO-1 is not under the regulation by GSK-3β in infected cells (Figure 6d and 6D1). Similar observations were made in western blot analysis of nuclear extracts of AKTi and/or SB216763-treated infected cells (Figure 6e).

Role of AKT and GSK-3β in modulation of FOXO-1 localization. (a and b) RAW264.7 cells were infected for different times and phospho-FOXO-1 expression at protein level was checked by immunoblotting in whole cell lysate (a) whereas FOXO-1 level was checked in nuclear fractions (b). (c) RAW cells were transfected with WT-GSK-3β or CA-GSK-3β or WT-AKT or DN-AKT for 24 h and then infected with L. donovani promastigotes for 6 h. Cells were lysed, nuclear fractions were obtained and FOXO-1 expression at protein level was checked by immunoblotting. (d) Cells were treated with AKTi or SB216763 for 1 h followed by infection with L. donovani promastigotes for 6 h. Cells were stained with anti-FOXO-1 monoclonal antibody followed by Alexa555-conjugated secondary antibody. Nuclei were stained with DAPI, and cells were analyzed as mentioned in legends of Figure 4. (e) Macrophages were treated with AKTi and/or SB216763 and infected for 6 h. Thereafter nuclear fractions were isolated and expression of FOXO-1 was assessed. Lamin serves as control for the nuclear extract. All experiments were repeated at least three times each and one set of representative data is shown. Bands were analyzed densitometrically and bar graphs expressing arbitrary densitometric units are presented adjacent to corresponding western blots. Error bars represent mean±S.D., n=3. NS, non-significant, ***P<0.001; Student's t-test
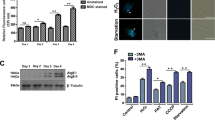
Role of AKT in parasite survival and immunomodulation in mouse model of leishmaniasis
To validate the role of AKT/β-catenin/FOXO-1 signaling in experimental mouse model of visceral leishmaniasis, we treated L. donovani-infected mice with AKTi or SB216763 (Figure 7a). Spleen parasite burden was found to be significantly reduced in mice treated with AKTi (73.9% reduction, P<0.001) compared with infected mice at 4 week post infection (Figure 7b). A significant reduction in spleen weight (66.9% reduction, P<0.001)) was observed upon treatment with AKTi as compared with infected control (Figure 7c). Treatment with AKTi also resulted in decreased levels of IL-10 (63.5% decrease over infected control, P<0.001) with a concomitant increase in IL-12 synthesis (5.8-fold increase over infected control, P<0.001) (Figures 7d and e) at 4 week post infection. However, no significant change was observed in infected and SB216783 treated mice as opposed to infected untreated mice (Figures 7c–e).

In vivo validation of the role of AKT in infection. (a) Schematic representation of the experimental protocol for treatment of L. donovani-infected mice: (i) infected and AKTi treated and (ii) infected and SB216763-treated. (b and d) Infected mice were treated as mentioned in the scheme (a) and disease progression was determined by measuring spleen parasite burden as Leishman–Donovan units (b). Spleen was isolated and spleen weight was measured (c). (d and e) Splenocytes were isolated from control, infected, and infected plus AKTi-treated or SB216763-treated mice at 4 wk post-infection and IL-10 (d) and IL-12 (e) levels were determined by ELISA. Animal experiments were done with five animals per group. Error bars represent mean±S.D., n=3. ***P<0.001; Student's t-test
Discussion
In the present study we elucidated how the intra-macrophage parasite L. donovani exploits a single host regulatory protein, AKT to limit both immune activation and macrophage apoptosis by simultaneously orchestrating β-catenin and FOXO-1 (Figure 8). Inhibition of AKT by use of inhibitor or dominant negative construct shifted the overall anti-inflammatory environment in infected cells to a pro-inflammatory milieu indicating its immunosuppressive role at least in case of Leishmania infection. Moreover, a significant increase in host cell apoptosis and mitochondrial membrane permeabilization upon AKT inactivation in H2O2-treated infected cells further emphasize on its bi-functional role which are in line with independent observations made by other groups.4, 5, 30, 31 GSK-3β was found to be inhibited by activation of AKT in L. donovani-infected condition as upon use of dominant negative AKT, phosphorylation-mediated inhibition of GSK-3β was not seen. As GSK-3β is known to inhibit or induce apoptosis depending on the stimulus,32 we transfected cells with constitutively active GSK-3β constructs which in infection resulted in significant drop in parasite burden and anti-inflammatory responses while increasing the overall pro-inflammatory responses and mitochondrial membrane permeabilization, indicating that GSK-3β works as a pro-apoptotic protein in Leishmania infection. This increase, however, was not at par with that seen in AKT-inactivated infected cells indicating the possible role of multiple targets working downstream of AKT.33

Modulation of host AKT pathway by L. donovani for successful survival. L. donovani activates host AKT by phosphorylation on serine and threonine residues, which in turn imparts a dual effect on the downstream signaling pathway: 1. Inactivation of GSK-3β by phosphorylation at Ser9 residue resulting in inhibition of Ser33/37 phosphorylation-mediated degradation of β-catenin and 2. Phosphorylation of β-catenin at Ser552 leading to its nuclear localization with concomitant inactivation of FOXO-1 by phosphorylation at Ser256 causing its cytosolic translocation. Overall these effects help parasite survival by inhibiting host cell apoptosis on one hand and modulating inflammatory responses on the other
Phosphorylation of β-catenin by AKT at Ser552 residue has been shown to enhance its transcriptional activity.11 Upon AKT inhibition in L. donovani infection a decrease in nuclear translocation of β-catenin was observed indicating a role of AKT-mediated β-catenin regulation. β-catenin is known to be degraded upon phosphorylation by GSK-3β at the Ser33/37 but no phosphorylation was observed at the Ser33/37 in MG132 treated infected cells further validating that GSK-3β gets inactivated in infection as a result of which it fails to inactivate β-catenin. Moreover, constitutive activation of GSK-3β in infected cells resulted in a decrease in nuclear β-catenin, which indicates that AKT-mediated inactivation of GSK-3β on one hand saves β-catenin from GSK-3β mediated degradation and on the other hand transactivates β-catenin by phosphorylating it at Ser552. Furthermore, XAV939 treatment or siRNA-mediated inhibition of β-catenin in infection led to an increase in NF-κB transcription and pro-inflammatory gene expression supporting a similar observation in other cell types.21, 34 Inhibition of β-catenin also resulted in increased host cell apoptosis with an overall decrease in parasite burden indicating its role downstream of AKT.
As β-catenin has a role in modulating FOXO-1 (ref 28), another protein known to regulate both apoptosis and inflammation;18, 19, 20 therefore, we studied the status of this protein in infection and found a time dependent increase in phosphorylation of FOXO-1, which corroborated with the kinetics of its cytoplasmic translocation. This confirms that phosphorylation of FOXO-1 at Ser256 residue results in its nuclear export during infection, as unphosphorylated active FOXO-1 is known to be retained in nucleus.35 Furthermore, constitutive activation of GSK-3β in infected cells did not have any impact on FOXO-1 localization indicating that FOXO-1 was not under GSK-3β regulation like β-catenin. On the contrary, dominant negative inhibition of AKT resulted in nuclear retention of FOXO-1 which suggests that AKT is able to modulate FOXO-1. There are reports which suggest that β-catenin can bind to FOXO-1 in nucleus which results in reduced transcriptional activity of β-catenin resulting in FOXO-1 mediated apoptosis.36 However, in infection as nuclear FOXO-1 content decreases due to AKT activation, AKT-GSK-3β mediated activation of β-catenin could effectively carry on its anti-apoptotic and anti-inflammatory functions. This fact was further validated when constitutively activated FOXO-1-transfected infected cells showed a significant increase in apoptosis and pro-inflammatory responses even though AKT-GSK-3β-β-catenin pathway remained unaffected.
Activation of AKT has been shown to negatively regulate the TLR pathway.37 Our previous reports demonstrate that Leishmania infection manages to dampen early macrophage pro-inflammatory responses by way of down-regulating TLR2 and TLR4 pathway.38, 39 Although we observed that AKT-mediated FOXO-1 inactivation resulted in down-regulation of TLR4 expression and IL-1β production, we also outlined the fact that it activated NF-κB, the key regulator of inflammatory response. This further resulted in increased IL-10 production which is a major anti-inflammatory cytokine up-regulated during infection. Overall this work projects AKT as a negative regulator of TLR4 pathway at least in the context of L. donovani infection. The role of AKT in parasite survival and sustenance has further been substantiated in the BALB/c mouse model of visceral leishmaniasis.
In summary, the present study elucidated for the first time the importance of key transcriptional regulators of AKT pathway which have important roles in regulation of parasite survival in L. donovani infection. These results indicate a novel regulatory mechanism adopted by the parasite to survive within the host cell by dampening pro-inflammmatory responses on one hand and inhibiting apoptosis on the other. Future work requires demonstration of the novel role of genes regulated by β-catenin and FOXO-1 as these factors have important roles in cell cycle regulation and proliferation and little is known about these in the context of leishmaniasis.
Materials and Methods
Cell culture
BMDM were isolated from femur and tibia by sacrificing 6–8 week old BALB/c mice as described previously.40 The murine macrophage cell line RAW264.7 was cultured at 37 °C with 5% CO2 in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated FBS, 100 μg/ml streptomycin and 100 U/ml penicillin.
Parasite maintenance
L. donovani (MHOM/IN/1983/AG83) parasites were cultured in medium M199 (Invitrogen) with Hanks salt containing Hepes (12 mM), l-glutamine (20 mM), 10% heat-inactivated fetal bovine serum (FBS), 50 U/ml penicillin and 50 μg/ml streptomycin (Invitrogen) as promastigotes. For in vitro infection, macrophages were infected with L. donovani promastigotes at a parasite:cell ratio of 10:1 (ref 40). For in vivo infection, 107 L. donovani promastigotes were injected via the tail vein of female BALB/c mice. Parasite burdens were ascertained by Giemsa-stained impression smears of spleen taken from infected mice. Organ parasite burden was expressed as Leishman–Donovan units (LDU) and were calculated as the number of amastigotes/1000 nucleated cells × organ weight (in grams41). Animal care and experimental procedures were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol has been approved by the Committee on the Ethics of Animal Experiments of Indian Institute of Chemical Biology (Permit Number: 147–1999).
Reagents, antibodies and constructs
Phospho-GSK-3β and GSK-3β antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Phospho-Ser33,37-β-catenin antibody and XAV939 were purchased from Abcam (Cambridge, UK). All other primary antibodies were purchased from Cell signaling (Danvers, MA, USA). HRP-conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from Sigma-Aldrich (St. Louis, MO, USA). AKTi, inhibitor of AKT and SB216763, inhibitor of GSK-3β were also purchased from Sigma. The pNF-κB luciferase plasmid containing five copies of NF-κB consensus sequences (pNF-κB-LUC) was obtained from Stratagene (La Jolla, CA, USA). WT-AKT and DN-AKT were kind gifts from Dr. Jacques Neefjes (Division of Tumor Biology, The Netherlands Cancer Institute, Amsterdam, The Netherlands).42 WT-GSK-3β (HA-GSK-3β WT pcDNA3) and CA-GSK-3β (HA-GSK-3β S9A pcDNA3) were gift from Dr Jim Woodgett (Addgene plasmid # 14753 and Addgene Plasmid 14754, respectively).43, 44 WT-FOXO-1 (GFP-FOXO-1) and CA-FOXO-1 (FOXO-1-ADA-GFP) were gift from Dr. Domenico Accili (Addgene plasmid # 17551 and Addgene Plasmid 35640, respectively).29, 45
Cytokine analysis by ELISA
ELISA was performed using a sandwich ELISA kit (Quantikine M, R&D Systems, Minneapolis, MN, USA and Abcam). The detection limit of these assays was >5.1, >2.5, <45 and <5 pg/ml for TNF-α, IL-12p70, IL-10 and IL-1β, respectively.
Apoptosis detection by Annexin V staining
RAW264.7 cells (2 × 106) were infected with L. donovani promastigotes for 6 h. One group of infected macrophages was treated with H2O2. After an hour of treatment, the culture media was replaced and cells were incubated for additional 24 h at 37 °C/5% CO2. After this the cells were washed twice with PBS and apoptosis was determined using Annexin V-FLUOS staining kit (Roche Applied Science, Indianapolis, IN, USA) as per manufacturer’s instruction. DAPI was used instead of PI. Cells were analyzed on BD FACS Verse and data were analyzed using FlowJo software.
Caspase-3 activity assay
RAW264.7 cells were washed twice with ice-cold PBS after infection and treatment and resuspended in 50 μl of ice-cold lysis buffer for 30 min (1 mM DL-dithiothreitol, 0.03% Nonidet P-40 (v/v), in 50 mM Tris pH 7.5). The lysate was centrifuged at 14 000 × g for 15 min at 4 °C. 10 μg of total protein was incubated with the caspase-3 substrate (Ac-DEVD-pNA) for 1 h at 37 °C. The absorption was measured by spectrometer at 405 nm.
Mitochondrial membrane potential assay
3,3′-Dihexyloxacarbocyanine iodide (DiOC6(3)) is a lipophilic cationic cyanine dye that is widely used to determine the mitochondrial membrane potential. After treatment and infection, RAW264.7 cells were incubated with DiOC6(3) (40 nM) for 30 min at 37 °C. The cells were then washed with ice-cold PBS and resuspended in 500 μl of PBS. Fluorescence intensities of DiOC6(3) were analyzed on a flow cytometer.
DNA Fragmentation assay
After treatment and infection, RAW264.7 cells were washed two times with PBS and lysed in 100 μl of lysis buffer (50 mM Tris, pH 8.0, 10 mM EDTA, 0.5% SDS and 1 mg/ml proteinase K) for 3 h at 56 °C. The lysate was treated with 0.5 mg/ml RNase A for another hour at 56 °C. DNA was extracted with phenol/chloroform/isoamyl alcohol (25/24/1) and mixed with loading buffer (50 mM Tris, 10 mM EDTA, 1% (w/w) low-melting point agarose, and 0.025% (w/w) bromophenol blue). This was loaded onto a pre-solidified 2% agarose gel containing 0.1 mg/ml ethidium bromide. The gels were observed and photographed under UV light.
Fluorescence microscopy
Macrophages (105) were plated onto 18 mm2 coverslips and cultured overnight. The cells were treated as mentioned and infected with L. donovani promastigotes, washed twice in PBS, and fixed with 4% formaldehyde for 30 min at room temperature. The cells were permeablized with 0.1% Triton X and incubated with blocking solution followed by primary antibody for 1 h at 4 °C. After washing, coverslips were incubated with Alexa555-conjugated secondary antibody for 1 h at room temperature. The cells were stained with DAPI (4′,6-diamidino-2-phenylindole, 1 μg/ml) in PBS plus 10 μg/ml RNase A to label the nucleus, mounted on slides and visualized under Olympus IX81 microscope equipped with a FV1000 confocal system using a 100x/60x oil immersion Plan Apo (N.A. 1.45) objectives. The images thus captured were analyzed by Olympus Fluoview (version 3.1a; Tokyo, Japan) using colocalization program and mounted using Adobe Photoshop software.
NF-κB reporter assay
1 μg NF-κB luciferase reporter vector were transfected in RAW264.7 macrophages along with 0.5 μg pCMV-β-gal in serum free medium using Lipofectamine (Invitrogen) according to manufactures instruction. Cells were harvested using reporter lysis buffer (Promega, Madison, WI, USA) and NF-κB luciferase activity was assessed by luminometer. The value of luciferase activity was normalized to transfection efficiency and this was monitored by the co-transfected β-galactosidase expression vector.
RNA-mediated interference by siRNA transfection
β-catenin specific siRNA was purchased from Santa Cruz Biotechnology. RAW264.7 cells were plated in 6-well tissue culture plates at a density of 2 × 106 cells/well in antibiotic and serum free normal growth medium, followed by transfection with siRNA as per manufacture’s instruction. Scrambled siRNA served as control.
Immunoblotting
RAW264.7 and BMDM were lysed in lysis buffer (Cell Signaling), and the concentration of proteins in the supernatants was estimated using Bradford reagent. Proteins were immunoblotted as previously described.39
Densitometry
Densitometric analyses for all experiments were carried out using ImageJ software (US NIH, Bethesda, MD, USA; http://rsb.info.nih.gov). Band intensities were quantified, and the values were normalized to endogenous control and arbitrary densitometric units were plotted in bar graphs.
Statistical analysis
All experiments were performed for at least three times. Macrophage cultures were set in triplicates and the results were expressed as the mean±S.D. Student’s t-test was employed to evaluate the statistical significances of differences among pair of data sets and P value<0.05 considered to be significant.
Abbreviations
- DN:
-
dominant negative
- WT:
-
wild type
- CA:
-
constituitively active
- BMDM:
-
bone marrow-derived macrophages
- HA:
-
haemagglutinin
- LDU:
-
Leishman–Donovan Units
- GFP:
-
green fluorescent protein
- TR:
-
texas red
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DC:
-
dendritic cells
- Ser:
-
serine
- Thr:
-
threonine
- PTEN:
-
phosphatase and tensin homolog
- DiOC6(3):
-
3,3'-dihexyloxacarbocyanine iodide
References
Bhardwaj S, Srivastava N, Sudan R, Saha B . Leishmania interferes with host cell signaling to devise a survival strategy. J Biomed Biotechnol 2010; 2010: 109189.
Cecilio P, Pérez-Cabezas B, Santarém N, Maciel J, Rodrigues V, Cordeiro da Silva A et al. Deception and manipulation: the arms of Leishmania, a successful parasite. Front Immunol 2014; 5: 480.
Zhang Y, Wang X, Yang H, Liu H, Lu Y, Han L et al. Kinase AKT controls innate immune cell development and function. Immunology 2013; 140: 143–152.
Neves BM, Silvestre R, Resende M, Ouaissi A, Cunha J, Tavares J et al. Activation of phosphatidylinositol 3-kinase/Akt and impairment of nuclear factor-kappaB: molecular mechanisms behind the arrested maturation/activation state of Leishmania infantum-infected dendritic cells. Am J Pathol 2010; 177: 2898–2911.
Ruhland A, Leal N, Kima PE . Leishmania promastigotes activate PI3K/Akt signalling to confer host cell resistance to apoptosis. Cell Microbiol 2007; 9: 84–96.
Faherty CS, Maurelli AT . Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol 2008; 16: 173–180.
Chuenkova MV, PereiraPerrin M . Trypanosoma cruzi targets Akt in host cells as an intracellular antiapoptotic strategy. Sci Signal 2009; 2: ra74.
Thi EP, Lambertz U, Reiner NE . Sleeping with the enemy: how intracellular pathogens cope with a macrophage lifestyle. PLoS Pathog 2012; 8: e1002551.
Antoniv TT, Ivashkiv LB . Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. Immunology 2011; 132: 567–577.
Guha M, Mackman N . The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 2002; 277: 32124–32132.
Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 2007; 282: 11221–11229.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA . Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995; 378: 785–789.
Monick MM, Carter AB, Robeff PK, Flaherty DM, Peterson MW, Hunninghake GW . Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J Immunol 2001; 166: 4713–4720.
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT et al. Identification of c-MYC as a target of the APC pathway. Science 1998; 281: 1509–1512.
Yuan L, Choi K, Khosla C, Zheng X, Higashikubo R, Chicoine MR et al. Tissue transglutaminase 2 inhibition promotes cell death and chemosensitivity in glioblastomas. Mol Cancer Ther 2005; 4: 1293–1302.
Kaga S, Zhan L, Altaf E, Maulik N . Glycogen synthase kinase-3beta/beta-catenin promotes angiogenic and anti-apoptotic signaling through the induction of VEGF, Bcl-2 and survivin expression in rat ischemic preconditioned myocardium. J Mol Cell Cardiol 2006; 40: 138–147.
Tzivion G, Dobson M, Ramakrishnan G . FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 2011; 1813: 1938–1945.
Wang Y, Zhou Y, Graves DT . FOXO transcription factors: their clinical significance and regulation. Biomed Res Int 2014; 2014: 925350.
Fan W, Morinaga H, Kim JJ, Bae E, Spann NJ, Heinz S et al. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J 2010; 29: 4223–4236.
Seiler F, Hellberg J, Lepper PM, Kamyschnikow A, Herr C, Bischoff M et al. FOXO transcription factors regulate innate immune mechanisms in respiratory epithelial cells. J Immunol 2013; 190: 1603–1613.
Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J . beta-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest 2007; 87: 613–624.
Beurel E, Jope RS . The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol 2006; 79: 173–189.
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 2000; 287: 1040–1046.
Lawrence T . The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 2009; 1: a001651.
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009; 461: 614–620.
Haq S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B et al. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci USA 2003; 100: 4610–4615.
Sakanaka C . Phosphorylation and regulation of beta-catenin by casein kinase I epsilon. J Biochem 2002; 132: 697–703.
Kamo N, Ke B, Busuttil RW, Kupiec-Weglinski JW . PTEN-mediated Akt/beta-catenin/Foxo1 signaling regulates innate immune responses in mouse liver ischemia/reperfusion injury. Hepatology 2013; 57: 289–298.
Qiang L, Banks AS, Accili D . Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem 2010; 285: 27396–27401.
Vazquez-Lopez R, Argueta-Donohué J, Wilkins-Rodríguez A, Escalona-Montaño A, Aguirre-García M, Gutiérrez-Kobeh L . Leishmania mexicana amastigotes inhibit p38 and JNK and activate PI3K/AKT: role in the inhibition of apoptosis of dendritic cells. Parasite Immunol 2015; 37: 579–589.
Nandan D, Camargo de Oliveira C, Moeenrezakhanlou A, Lopez M, Silverman JM, Subek J et al. Myeloid cell IL-10 production in response to Leishmania involves inactivation of glycogen synthase kinase-3beta downstream of phosphatidylinositol-3 kinase. J Immunol 2012; 188: 367–378.
Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D . GSK-3beta: A Bifunctional Role in Cell Death Pathways. Int J Cell Biol 2012; 2012: 930710.
Manning BD, Cantley LC . AKT/PKB signaling: navigating downstream. Cell 2007; 129: 1261–1274.
Orr SJ, Burg AR, Chan T, Quigley L, Jones GW, Ford JW et al. LAB/NTAL facilitates fungal/PAMP-induced IL-12 and IFN-gamma production by repressing beta-catenin activation in dendritic cells. PLoS Pathog 2013; 9: e1003357.
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004; 14: 395–403.
Hoogeboom D, Essers MA, Polderman PE, Voets E, Smits LM, Burgering BM . Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J Biol Chem 2008; 283: 9224–9230.
Fukao T, Koyasu S . PI3K and negative regulation of TLR signaling. Trends Immunol 2003; 24: 358–363.
Gupta P, Giri J, Srivastav S, Chande AG, Mukhopadhyaya R, Das PK et al. Leishmania donovani targets tumor necrosis factor receptor-associated factor (TRAF) 3 for impairing TLR4-mediated host response. FASEB J 2014; 28: 1756–1768.
Srivastav S, Kar S, Chande AG, Mukhopadhyaya R, Das PK . Leishmania donovani exploits host deubiquitinating enzyme A20, a negative regulator of TLR signaling, to subvert host immune response. J Immunol 2012; 189: 924–934.
Kar S, Ukil A, Sharma G, Das PK . MAPK-directed phosphatases preferentially regulate pro- and anti-inflammatory cytokines in experimental visceral leishmaniasis: involvement of distinct protein kinase C isoforms. J Leukoc Biol 2010; 88: 9–20.
Murray HW, Miralles GD, Stoeckle MY, McDermott DF . Role and effect of IL-2 in experimental visceral leishmaniasis. J Immunol 1993; 151: 929–938.
Kuijl C, Savage ND, Marsman M, Tuin AW, Janssen L, Egan DA et al. Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 2007; 450: 725–730.
He X, Saint-Jeannet JP, Woodgett JR, Varmus HE, Dawid IB . Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature 1995; 374: 617–622.
Stambolic V, Woodgett JR . Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J 1994; 303: 701–704.
Frescas D, Valenti L, Accili D . Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 2005; 280: 20589–20595.
Acknowledgements
This work was supported by the Department of Science and Technology (SB/SO/BB-0055/2013, SERB/F/4467/2013-14), SERB EMR/2014/000287, University Grants Commission (F. No.6-10/2016(IC)), Government of West Bengal, Department of Biotechnology, (221/BT(Estt)/RD-40/2014) and National Academy of Sciences, India (NASI). Fellowship was provided by Council of Scientific & Industrial Research (CSIR) and University Grants Commission (UGC). We thank DBT-CU-IPLS Core Facility for confocal microscope facility and CU-BD-COE for providing imaging and cytometry facility.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by JP Medema
Rights and permissions
About this article

Cite this article
Gupta, P., Srivastav, S., Saha, S. et al. Leishmania donovani inhibits macrophage apoptosis and pro-inflammatory response through AKT-mediated regulation of β-catenin and FOXO-1. Cell Death Differ 23, 1815–1826 (2016). https://doi.org/10.1038/cdd.2016.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.101
This article is cited by
-
Apoptosis and its pathways as targets for intracellular pathogens to persist in cells
Parasitology Research (2024)
-
Gasdermin-D activation promotes NLRP3 activation and host resistance to Leishmania infection
Nature Communications (2023)
-
Delayed IL-12 production by macrophages during Toxoplasma gondii infection is regulated by miR-187
Parasitology Research (2020)
-
MSC-secreted TGF-β regulates lipopolysaccharide-stimulated macrophage M2-like polarization via the Akt/FoxO1 pathway
Stem Cell Research & Therapy (2019)
-
Differential immune response modulation in early Leishmania amazonensis infection of BALB/c and C57BL/6 macrophages based on transcriptome profiles
Scientific Reports (2019)
