
Abstract
Liposarcoma (LPS) is a type of soft tissue sarcoma that mostly occurs in adults, and in humans is characterized by amplifications of MDM2 and CDK4. The molecular pathogenesis of this malignancy is still poorly understood and, therefore, we developed a mouse model with conditional inactivation of PTEN and p53 to investigate these pathways in the progression of the disease. We show that deletion of these two tumor suppressors cooperate in the formation of multiple subtypes of LPS (from well-differentiated LPS to pleomorphic LPS). In addition, progression of the tumors is further characterized by the expression of D cyclins and CDK4/6, which allow for continued cell division. Microarray analysis also revealed novel genes that are differentially expressed between different subtypes of LPS, which could aid in understanding the disease and to unravel potential new therapeutic targets.
Similar content being viewed by others

Main
Liposarcoma (LPS) is a neoplasm that arises from adipose tissue and accounts for ~20% of the soft tissue and bone sarcoma that develop in the United States.1 There are four main histological subtypes of LPSs that are classified morphologically and consist of well-differentiated LPS (WDLPS), dedifferentiated LPS (DDLPS), myxoid/round cell LPS (MLPS) and pleomorphic LPS (PLPS). The subtypes differ in terms of biological behavior and metastatic potential, with WDLPS having a low recurrence rate and a lower tendency to metastasize than MLPS or PLPS.2 The treatment options are mainly surgery and radiation with chemotherapy for systemic disease. Though depending upon the subtype there is variation in terms of chemotherapy sensitivity, and few options exist for aggressive local or metastatic disease.3
Chromosomal amplifications of the 12q13–15 region, which include the MDM2, CDK4 and HMGA2 genes, among others, are a characteristic feature of LPSs.4, 5, 6 MDM2 amplification occurs in several cancers and is thus of importance in tumorigenesis.7 MDM2, which is a negative regulator of p53, blocks p53-dependent transcription and recruitment of transcription coactivators, by binding within its N-terminal hydrophobic pocket. MDM2 also has been reported to have p53-independent functions that control proliferation, apoptosis, epithelial-to-mesenchymal transition and the initiation of adipocyte differentiation.7, 8 In WDLPS, amplification of MDM2 and p53 mutations seem to be mutually exclusive and p53 mutations have been associated with the dedifferentiation process from WDLPS to DDLPS. Also the location of LPSs in the body appears to be an important factor. In retroperitoneal WDLPS/DDLPS tumors MDM2 amplifications and p53 mutations were mutually exclusive but in non-retroperitoneal DDLPS tumors p53 mutations occur in the presence of MDM2 amplifications.9, 10 The latter implicates a role for mutant p53 in the dedifferentiation process. Interestingly, some of the most common cancers found in p53 heterozygote Li-Fraumeni patients are soft tissue sarcomas such as LPSs, fibrosarcomas, and rhabdomyosarcomas.11, 12
In addition to the p53 pathway, the PI3K-AKT pathway is commonly dysregulated in human cancer. Its downstream effects include protein synthesis, genetic stability, cellular proliferation and survival. Mutations in PTEN, which normally regulate the PI3K-AKT pathway, have been found in multiple lipomas (benign adipocytic neoplasms)13 and AKT activation has been found in human LPSs.14 Therefore, the role that this pathway has in adipocyte transformation predicts it as a potential target pathway for therapeutics.
In this study, we sought to understand the combined role of p53 and Pten deletions in a mouse model of LPS. Accumulating evidence from zebrafish and xenograph models have begun to address their combined importance in this tumor type.15, 16 Using conditional inactivation of tumor suppressors p53 and PTEN in adipose tissue, we show that deletion of p53 and Pten together results in tumor formation in ~85% of mice with tumors representing all four major subtypes of LPS observed in humans. A gene expression microarray analysis of these tumor types indicates many differences in gene expression and unravels a new possible method of diagnosis of these tumors. This mouse model also demonstrates a role for the MDM2 protein in the formation of some LPS cell types even in the absence of the p53 functions. Once p53 and PTEN functions are eliminated, the next step in the transformation pathway of LPSs is the enhanced production of some D cyclins and CDK4/6, leading to continual signaling for cell division. Interestingly this is the same pathway observed when p53 knockout mice produce T-cell lymphomas.17
Results
High penetrance of LPS formation initiated by PTEN and p53 deficiency
Accumulating evidence points to important roles for the p53 and PI3K/PTEN pathway in the development of LPSs. In this study, we examined the consequences of deletion of Pten and p53 in adipose tissue of mice. Adenovirus-cre was either injected into the adipose tissue surrounding the ovary (gonadal fat pad) or surrounding the testes resulting in either deletion of exons 2–10 in p53flox/flox mice,18 or exon 5 in the Ptenflox/flox mice.19 Injection of adenovirus-cre into adipose tissue of either p53flox/flox, Pten+/+ mice or p53+/+, Ptenflox/flox mice led to no tumor formation, whereas injection of p53flox/flox, Ptenflox/flox mice led to >85% of mice with tumor formation (Figure 1). Tumors in the p53flox/flox, Ptenflox/flox mice were detected by palpation as early as 81 days post injection and were 50% penetrant at 153 days. Thus, deletion of both alleles of p53 and Pten, in combination, results in highly penetrant LPS development in this mouse model.

Survival curve of p53flox/flox, Ptenflox/flox and Ptenflox/flox; p53flox/flox mice. Adenovirus-cre injection into adipose tissue of male and female p53-floxed, and Pten-floxed allele mice in combination leads to liposarcoma at the indicated time points. p53flox/flox (n= 8), Ptenflox/flox (n= 8) and Ptenflox/flox; p53flox/flox (n= 22)
Development of multiple subtypes of LPS in p53flox/flox; Ptenflox/flox mice
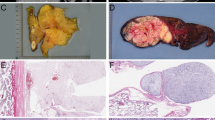
Histological analysis of the LPSs, which formed in the p53flox/lox; Ptenflox/flox mice, showed that all four main subtypes, WDPLS, DDPLS, MLPS and PLPS, were represented (Figure 2a). Interestingly, within the same tumor one or two different subtypes could be found. Evidence points to mesenchymal stem cells (MSCs) being the target cell for the development of sarcomas and LPSs from which multiple types of these tumors are possible.20 Approximately, 80% of the tumors examined had a dedifferentiated subtype component, whereas the pleomorphic subtype represented the least number of cases (Figure 2b). Marker analysis to verify the histologically identified subtypes showed that WDLPS and MLPS, which have been classified as being more mature adipocytic sarcomas, were characterized by high expression of adipocytic markers (LPL) when compared with DDLPS and PLPS (Figure 2c, upper panels). In the contrary, DDLPS and PLPS subtypes, which are classified as being more immature LPSs, display stronger and more diffuse expression of MSC markers (HGF) than WDLPS and MLPS (Figure 2c, lower panels, and data not shown).21

Classification of liposarcomas generated in the Ptenflox/flox; p53flox/flox mouse model. (a) Representative hematoxylin and eosin (H&E) of the four major subtypes of liposarcoma identified; well-differentiated liposarcoma (WDPLS), dedifferentiated liposarcoma (DDLPS), myxoid liposarcoma (MPLS) and pleomorphic liposarcoma (PLPS). (b) Bar diagram graph represents the percentage of the each of the subtypes of liposarcoma. A small percentage of other subtypes of sarcomas were identified, including rhabdomyosarcoma and osteosarcoma. (c) Different subtypes of liposarcoma are characterized by a unique lipocytic phenotype. Lipoprotein lipase (LPL) is an adipocytic marker and stains more mature adipocytic sarcomas such as WDLPS and MLPS. Hepatocyte growth factor (HGF) is a mesenchymal stem cell marker and stains immature liposarcomas such as DDLPS and PLPS. Scale bar corresponds to 50 μm
Expression of cell cycle regulation genes in multiple subtypes of p53flox/flox; Ptenflox/flox LPS tumors
Cell cycle regulation in human cancer is often deregulated resulting in unscheduled proliferation, genomic instability and chromosomal instability leading to aneuplody. Cyclin-dependent kinases (CDK) and cyclins are the main driving forces of cell cycle regulation and they are in turn regulated by p53 and PTEN. Therefore, we wanted to investigate some of the molecular consequences of p53 and PTEN loss on these proteins in LPS. Here we found that there is an aberration in the G1–S checkpoint with the upregulation of CDK4, CDK6, Cyclin D1 and Cyclin D3 in all four of the subtypes of LPS (WDLPS, DDLPS, MLPS and PLPS) (Figure 3 and data not shown). The available evidence suggests that Cyclin D1 and Cyclin D3 can be regulated by PTEN,22, 23 however, this is the first time it has been shown that they are overexpressed in the absence of both p53 and PTEN in a mouse model of LPS. These results suggest that alterations of this pathway are essential for the oncogenic process of the different subtypes of LPS.

Immunohistochemical analysis of cell cycle markers in WDLPS and DDLPS subtypes. There is a deregulation in the G1–S checkpoint with the upregulation of CDK4, Cyclin D1 and Cyclin D3. Scale bar corresponds to 50 μm
MDM2 expression in subtypes of mouse LPSs
Most LPSs have amplifications of the MDM2 gene with a smaller number of LPSs having loss or mutation of the TP53 gene. It is assumed that MDM2 amplification would be similar to a p53 loss or mutation on affecting downstream pathways. However, with increasing evidence that MDM2 has p53-independent roles we, therefore, wanted to investigate the status of MDM2 in tissue sections of p53flox/flox; Ptenflox/flox LPS tumors.7, 8 We investigated MDM2 RNA levels by real-time PCR (Taqman) in normal versus tumor tissue but did not see an appreciable difference between normal fat and tumors (1.5- to twofold, data not shown) as seen in human LPS samples.24 Therefore, we examined the protein expression of MDM2 by immunohistochemistry. Interestingly, MDM2 protein is expressed at very high levels in WDLPS and DDLPS sections in areas with clear lipoblasts (Figure 4, panels a and b) and absent from DDLPS sections lacking lipoblasts and MLPS (Figure 4, panels c and d). Similarly, this has been seen in the case of CDK4 where it is not amplified in the well-differentiated component of a tumor and amplified in the dedifferentiated component.25

Immunohistochemical analysis of MDM2 in WDLPS, DDLPS and MPLS subtypes. MDM2 protein is expressed at very high levels in WDLPS and DDLPS sections in areas with lipoblasts (a and b) and absent in DDLPS sections lacking lipoblasts (c) and MLPS (d)
Given MDM2’s p53-independent role in initiation of adipocyte differentiation,8 MDM2 expressing areas might be undergoing a dedifferentiation process that is controlled by MDM2. We performed IHC analysis in serial sections using MDM2 and C/EBP delta, which has been shown to be important for the dedifferentiation process. Interestingly, MDM2 and C/EBP delta showed similar patterns of expression around lipoblasts (Supplementary Figure 1), which allows for a potential explanation for the restricted MDM2 expression pattern and dedifferentiation from WDLPS to DDLPS.
Bioinformatics analysis of differentially expressed genes in two of the biological types of LPS
To understand the possible progression of WDLPS to DDLPS, we analyzed gene expression profiles by microarray to identify pathways and genes that might be differentially expressed. This would aid in understanding the molecular consequences for the loss of p53 and PTEN pathways in different subtypes of LPS. The differentially expressed genes between WDPLS and DDLPS are shown in a Heat Map (Figure 5, expression data for all samples in Supplementary file 2). While this list of genes is not statistically significant due to limited number of samples and multiple hypotheses testing, it suggests candidate genes in differentiation between the two subtypes as well as potential new markers of tumorigenesis and therapeutic targets. Table 1 shows the upregulated genes in each subtype compared with the other subtype that have a greater than threefold change in expression and P value <0.01 (genes that have a twofold change but of particular importance are marked in the table). Many of the genes that are of interest, in distinction of the two subtypes, relate to the control of lipid, fatty acid, amino acid and carbohydrate metabolism (Fabp4, Lpl, Pgm5, Ldlr, Got1, Ech1), insulin-like growth factor pathway (Igf2, Igf1,Foxo1), transcription factors (Foxc2, Sox6, Pparg, Irx5, Meis2, Ebf1, Sox17), obesity-related genes (Lep, Ntrk2), Wnt signaling (Nkd2, Fzd4) or genes related to cell proliferation and apoptosis (Vegfa, G0s2). Two genes that are p53 regulated or regulate p53, Got1 and Aurka, respectively, might help to explain targets that are of interest in the formation of LPSs in the absence of p53. These data suggest potentially novel genes that can be used to distinguish between WDPLS and DDLPS subtypes and can also provide insight into LPS pathogenesis and progression.

Heat Map of differentially expressed genes between WDPLS and DDLPS. The heat map was generated using genes with P-value ≤0.01 (uncorrected) and fold change >3
Disscussion
High-level amplification of the MDM2 and CDK4 genes have become markers for studying human LPSs, implicating deregulation of both the p53 pathway and cell cycle regulation as necessary aspects for tumor progression. However, researchers have also seen that other points in the p53 pathway can be deregulated such as p53 itself with or without MDM2 amplification.9, 10 In addition, another widely mutated gene in cancer, PTEN, a regulator of the PI3K-AKT pathway, has been found in multiple lipomas (benign adipocytic neoplasms)13 and AKT activation has been found in human LPSs.14 This finding is of significance owing to the many drugs that have been found to target the PI3K-AKT-mTOR pathway and the lack of drugs currently effective for the treatment of LPSs or that are still in the clinical trial period.26 In this study, we developed a mouse model of LPS that demonstrates the combined effect of p53 and PTEN deletion in the generation of these tumors and its potential utility in the development of novel therapeutics.
The interaction of the p53 and PI3K/AKT/PTEN pathways is well documented. Likewise, combinatorial deletion or inactivation of members from each pathway is also very common. In this mouse model, we observe that when either alleles of p53 or Pten were deleted individually it did not result in any tumor formation but in combination several different subtypes of LPS were formed. It could be that inactivation of both pathways in a stem cell or progenitor type cell could lead to the formation of the different subtypes of LPS (WDLPS, DDLPS, MLPS and PLPS). Two previous studies, have suggested this possibility as well as a way for the progression of tumorigenesis.21, 27
In addition, one of the mechanisms that could be the driving force for tumor progression, in the absence of p53 and PTEN, is cell cycle regulation. In this model of LPS, the loss of p53 and PTEN promotes the abnormal expression of D cyclins and CDK4/6. CDKs are bound by regulatory subunits called cyclins that are in turn made at certain times during the cell cycle and regulate enzymatic activity. Mutations that deregulate these CDK-cyclin complexes lead to continued proliferation or inappropriate re-entry into the cell cycle. In this model, altered expression of these cell cycle regulation proteins could be one of the driving forces of uncontrolled growth and LPS tumorigenesis. Specifically, the increased expression of the G1–S phase regulators CDK4 and cyclin E would serve to enhance the G1–S transition.
It is of some interest that p53 knockout mice develop thymic lymphomas. These tumors select for a common series of mutations in a specific order. After the gatekeeper mutation of p53, then Pten deletions are commonly selected for and this is followed by Cyclin D1, D2, D3 and Cdk-6 amplifications and overexpression. The LPSs described here have demonstrated an identical series of events, suggesting a common pathway to diverse cancers following a p53 mutation.
Other mechanisms involved in this tumor model could be regulating the dedifferentiation of tumor subtypes. MDM2 has been shown to exert a p53-independent role on the initiation of adipocyte differentiation through controlling CREB-dependent transactivation, as well as the determination of cell fate of MSCs, another p53-independent function. Adipocytes, osteoblasts, myocytes and chondrocytes all differentiate from MSCs.8 In the current study, MDM2 expression was found in areas of lipocytes in both WDLPS and DDLPS p53flox/flox; Ptenflox/flox tumors. Therefore, in this model MDM2 might be directing these cells toward a more dedifferentiated cell type, a hypothesis that still needs to be further tested. Another potential way that MDM2 might direct cells toward a dedifferentiated phenotype is through epigenetic reprogramming. MDM2-dependent degradation of RB increased levels and activity of DNA methyltransferase DNMT3A28 and thus silencing of tumor suppressor genes. Similarly, MDM2 interacts with the histone methyltransferase SUV39H1 and can in certain instances lead to repression of p53 target genes29, 30 and potentially epigenetic reprogramming.31
To begin to understand how WDLPS and DDLPS differ molecularly in this mouse model in the presence of p53 and PTEN deletions we ran gene expression microarray analysis to compare the two different LPS tumor types. Our investigation would hopefully lead to the discovery of novel deregulated pathways, potential new therapeutic targets and pathways involved in the dedifferentiation process of the subtypes. Some deregulated genes and pathways were similar to a previous human study of WDLPS and DDLPS32 (lipid, fatty acid and carbohydrate metabolism: Fabp4, Lpl, transcription factors: Fox and Sox family members), however some novel ones were also found (insulin-like growth factor pathway: Igf1, Igf2). The expression of the insulin-like growth factor pathway genes is most likely due to the interaction of both the p53 and PTEN pathways being deregulated. The insulin-like growth factor pathway has been an attractive target for therapies and its importance in LPS tumorigenesis is becoming more evident. Therefore, it is interesting that IGF2 has recently been identified as a potential therapeutic target and in esophageal cancers has been shown to be suppressive toward tumor growth and metastasis.33 Another pathway, Wnt signaling, which is highly involved in development and adipogenesis could be another key pathway that should be investigated in LPS.34
We also have some clues as to transcription factors that might be working to induce dedifferentiation through epigenetic mechanisms. SOX6 is an important developmental gene as well as involved in differentiation. It is also known to suppress cyclin D1 activities by interacting with beta-catenin and HDAC1, thus, leading to a decrease in acetylated H3 and H4 at the cyclin D1 promoter.35 SOX6 is misexpressed in the tumor subtypes though it is unknown if it works in conjunction with MDM2 to promote these processes.
Genes that are p53 regulated or regulate p53 might aid in the investigation of mechanisms responsible for LPS formation when p53 and PTEN are deleted in adipose tissue. Got1 and Aurka were found to be upregulated in DDLPS. Through Got1, p53 might be a novel regulator of glucose production and inhibit the use of glucose in pathways that promote tumorigenesis.36 Likewise, Aurka has been shown to be important not only in the p53 pathway but also in the Pten/Akt pathway. Specifically, Aurka regulates ESC pluripotency through phosphorylation-mediated inhibition of p53-directed gene expression.37 Inhibition of Aurka in the absence of p53 made cells more susceptible to mitotic arrest and slippage.38 Similarly, Aurka inhibition can downregulate Akt and promotes significant cell death.39
In summary, we have shown that combined deletion of p53 and PTEN leads to the formation of several subtypes of LPS in a mouse model of the disease. Molecular mechanisms involved in the progression include deregulation of cell cycle genes leading to uncontrolled cell proliferation as well as upregulation of MDM2, which might promote dedifferentiation and epigenetic reprogramming. Finally, gene expression microarray analysis has given us not previously known genes that can be further investigated in exploring these hypotheses.
Materials and Methods
Animal model
All animal study protocols were approved by the University of Medicine and Dentistry of New Jersey Institutional Animal Care and Use Committee review board. Conditional alleles for Pten (PtenTm1Hwu) (Lesche et al.19) and p53 (Trp53Tm1Brn, Jonkers et al.18) mice were purchased from Jackson Laboratories, Bar Harbor, ME, USA. In all, 8–10-week old mice were used for injection of an adenovirus expressing cre recombinase (Ad5CMVCre, University of Iowa's Vector Core Facility (http://www.uiowa.edu/~gene) into adipose tissue. Concentrated virus (25 μl; 4 X1011PFU/ml) was mixed with 20 μl of Dulbecco’s modified eagle medium (D-MEM). Mice were monitored for tumor development by palpitation at weekly intervals. Kaplan–Meier analysis was generated using GraphPad software.
Histological analysis
Adipose tissue and tumor samples were harvested and fixed in 10% formalin overnight. The samples were embedded in paraffin and sectioned. The sections were subjected to hemotoxylin and eosin staining or immunohistochemical staining following the standard avidin-biotin protocol. The following antibodies were used: MDM2 (clone SMP14 Abcam, Cambridge, MA, USA), LPL (Abcam ab21356), HGF (Novus Biologicals NBP1-19182, Littleton, CO, USA), CDK4 (Abcam ab7955), CDK6 (Novus Biologicals NB100-91722), Cyclin D1 (Cell Signaling 2978, Danvers, MA, USA), Cyclin D3 (Cell Signaling 2936), C/EBP delta (antibodies-online, Atlanta, GA, USA; ABIN233849).
Gene expression microarray
Total RNA was isolated from frozen tissue using TRIzol (Life Technologies) followed by clean-up with the Qiagen RNeasy kit and subjected to the 2100 Bioanalyzer from Agilent Technologies. cDNA samples were generated and labeled using Affymetrix 1-cycle expression kit (Affymetrix, Santa Clara, CA, USA) and hybridized to Affymetrix GeneChip Mouse Genome 430 2.0 Array using Affymetrix hybridization kit materials according to manufacturer’s instruction. The raw Affymetrix. CEL files were imported into GeneSpring GX11 software (Silicon Genetics, Redwood City, CA, USA). Quality control was performed to remove bad-quality probes. Normalized expression values were calculated by the GC Robust Multi-array Average algorithm and subjected to mean transformation to collapsed all probe sets to respective genes in R. Collapsed gene expression values from all samples were used to find differentially expressed genes. The mean gene expression of each liposarcoma subtype was used to calculate the fold change between subtypes. We used gene expression fold change threshold of 3 and uncorrected T-test P-value of less than 0.01 to define a list of differentially expressed genes between the subtypes. Bonferroni correction was not used due to limited number of samples.
Abbreviations
- LPS:
-
liposarcoma
- WDLPS:
-
well-differentiated liposarcoma
- DDLPS:
-
dedifferentiated liposarcoma
- MLPS:
-
myxoid/round cell liposarcoma
- PLPS:
-
pleomorphic liposarcoma
- MSCs:
-
mesenchymal stem cells
- IHC:
-
immunohistochemistry
- LPL:
-
lipoprotein lipase
- HGF:
-
hepatocyte growth factor
- MDM2 :
-
mouse double minute 2 homolog
- p53 :
-
tumor protein p53
- Pten :
-
phosphatase and tensin homolog
- PI3K:
-
phosphatidylinositol-3 kinase
- AKT:
-
protein kinase B
- mTOR:
-
mammalian target of rapamycin
- CDK:
-
cyclin-dependent kinases
- CDK4:
-
cyclin-dependent kinase 4
- CDK6:
-
cyclin-dependent kinase 6
- C/EBP delta:
-
CCAAT/enhancer-binding protein delta
- Fabp4 :
-
fatty acid binding protein 4, adipocyte
- Lpl :
-
lipoprotein lipase
- Pgm5 :
-
phosphoglucomutase 5
- Ldlr :
-
low-density lipoprotein receptor
- Got1 :
-
glutamic-oxaloacetic transaminase 1
- Ech1 :
-
enoyl CoA hydratase 1
- Igf2 :
-
insulin-like growth factor 2
- Igf1 :
-
insulin-like growth factor 1
- Foxo1 :
-
forkhead box O1
- Foxc2 :
-
forkhead box C2
- Sox6 :
-
SRY (sex determining region Y)-box 6
- Pparg :
-
peroxisome proliferator-activated receptor gamma
- Irx5 :
-
iroquois homeobox 5
- Meis2 :
-
meis homeobox 2
- Ebf1 :
-
early B-cell factor 1
- Sox17 :
-
SRY (sex determining region Y)-Box 17
- Lep :
-
leptin (murine obesity homolog)
- Ntrk2 :
-
neurotrophic tyrosine kinase, receptor, type 2
- Nkd2 :
-
naked cuticle homolog 2
- Fzd4 :
-
frizzled class receptor 4
- Vegfa:
-
vascular endothelial growth factor A
- G0s2 :
-
G0/G1 switch 2
- Got1 :
-
glutamic-oxaloacetic transaminase 1
- Aurka :
-
aurora Kinase A
- DNMT3A:
-
DNA (cytosine-5)-methyltransferase 3A
- SUV39H1:
-
suppressor of variegation 3–9 homolog 1
- CREB:
-
cAMP response element-binding protein
References
Conyers R, Young S, Thomas DM . Liposarcoma: molecular genetics and therapeutics. Sarcoma 2011; 2011: 483154.
Rieker RJ, Weitz J, Lehner B, Egerer G, Mueller A, Kasper B et al. Genomic profiling reveals subsets of dedifferentiated liposarcoma to follow separate molecular pathways. Virchows Arch 2010; 456: 277–285.
Jones RL, Fisher C, Al-Muderis O, Judson IR . Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer 2005; 41: 2853–2860.
Forus A, Weghuis DO, Smeets D, Fodstad O, Myklebost O, van Kessel AG . Comparative genomic hybridization analysis of human sarcomas: I. Occurrence of genomic imbalances and identification of a novel major amplicon at 1q21-q22 in soft tissue sarcomas. Genes Chromosomes Cancer 1995; 14: 8–14.
Pedeutour F, Forus A, Coindre JM, Berner JM, Nicolo G, Michiels JF et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer 1999; 24: 30–41.
Szymanska J, Virolainen M, Tarkkanen M, Wiklund T, Asko-Seljavaara S, Tukiainen E et al. Overrepresentation of 1q21-23 and 12q13-21 in lipoma-like liposarcomas but not in benign lipomas: a comparative genomic hybridization study. Cancer Genet Cytogenet 1997; 99: 14–18.
Manfredi JJ . The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev 2010; 24: 1580–1589.
Hallenborg P, Feddersen S, Francoz S, Murano I, Sundekilde U, Petersen RK et al. Mdm2 controls CREB-dependent transactivation and initiation of adipocyte differentiation. Cell Death Differ 2012; 19: 1381–1389.
Pilotti S, Della Torre G, Lavarino C, Di Palma S, Sozzi G, Minoletti F et al. Distinct mdm2/p53 expression patterns in liposarcoma subgroups: implications for different pathogenetic mechanisms. J Pathol 1997; 181: 14–24.
Schneider-Stock R, Walter H, Radig K, Rys J, Bosse A, Kuhnen C et al. MDM2 amplification and loss of heterozygosity at Rb and p53 genes: no simultaneous alterations in the oncogenesis of liposarcomas. J Cancer Res Clin Oncol 1998; 124: 532–540.
Li FP, Fraumeni JF Jr . Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 1969; 71: 747–752.
Malkin D, Friend SH, Li FP, Strong LC . Germ-line mutations of the p53 tumor-suppressor gene in children and young adults with second malignant neoplasms. N Engl J Med 1997; 336: 734.
Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet 1999; 8: 1461–1472.
Hernando E, Charytonowicz E, Dudas ME, Menendez S, Matushansky I, Mills J et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med 2007; 13: 748–753.
Gutierrez A, Snyder EL, Marino-Enriquez A, Zhang YX, Sioletic S, Kozakewich E et al. Aberrant AKT activation drives well-differentiated liposarcoma. Proc Natl Acad Sci USA 2011; 108: 16386–16391.
Smith KB, Tran LM, Tam BM, Shurell EM, Li Y, Braas D et al. Novel dedifferentiated liposarcoma xenograft models reveal PTEN down-regulation as a malignant signature and response to PI3K pathway inhibition. Am J Pathol 2013; 182: 1400–1411.
Dudgeon C, Chan C, Kang W, Sun Y, Emerson R, Robins H et al. The evolution of thymic lymphomas in p53 knockout mice. Genes Dev 2014; 28: 2613–2620.
Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A . Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001; 29: 418–425.
Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 2002; 32: 148–149.
Rodriguez R, Rubio R, Gutierrez-Aranda I, Melen GJ, Elosua C, Garcia-Castro J et al. FUS-CHOP fusion protein expression coupled to p53 deficiency induces liposarcoma in mouse but not in human adipose-derived mesenchymal stem/stromal cells. Stem Cells 2011; 29: 179–192.
Matushansky I, Hernando E, Socci ND, Matos T, Mills J, Edgar MA et al. A developmental model of sarcomagenesis defines a differentiation-based classification for liposarcomas. Am J Pathol 2008; 172: 1069–1080.
Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM . PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol 2003; 23: 6139–6149.
Zhu X, Kwon CH, Schlosshauer PW, Ellenson LH, Baker SJ . PTEN induces G(1) cell cycle arrest and decreases cyclin D3 levels in endometrial carcinoma cells. Cancer Res 2001; 61: 4569–4575.
Sasaki T, Ogose A, Kawashima H, Hotta T, Hatano H, Ariizumi T et al. Real-time polymerase chain reaction analysis of MDM2 and CDK4 expression using total RNA from core-needle biopsies is useful for diagnosing adipocytic tumors. BMC Cancer 2014; 14: 468.
Louis-Brennetot C, Coindre JM, Ferreira C, Perot G, Terrier P, Aurias A . The CDKN2A/CDKN2B/CDK4/CCND1 pathway is pivotal in well-differentiated and dedifferentiated liposarcoma oncogenesis: an analysis of 104 tumors. Genes Chromosomes Cancer 2011; 50: 896–907.
Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 2012; 13: 1133–1140.
Lo Furno D, Graziano AC, Caggia S, Perrotta RE, Tarico MS, Giuffrida R et al. Decrease of apoptosis markers during adipogenic differentiation of mesenchymal stem cells from human adipose tissue. Apoptosis 2013; 18: 578–588.
Tang YA, Lin RK, Tsai YT, Hsu HS, Yang YC, Chen CY et al. MDM2 overexpression deregulates the transcriptional control of RB/E2F leading to DNA methyltransferase 3A overexpression in lung cancer. Clin Cancer Res 2012; 18: 4325–4333.
Chen L, Li Z, Zwolinska AK, Smith MA, Cross B, Koomen J et al. MDM2 recruitment of lysine methyltransferases regulates p53 transcriptional output. EMBO J 2010; 29: 2538–2552.
Mungamuri SK, Benson EK, Wang S, Gu W, Lee SW, Aaronson SA . p53-mediated heterochromatin reorganization regulates its cell fate decisions. Nature Struct Mol Biol 2012; 19: 478–484 S471.
Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012; 483: 598–602.
Singer S, Socci ND, Ambrosini G, Sambol E, Decarolis P, Wu Y et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res 2007; 67: 6626–6636.
Li B, Tsao SW, Chan KW, Ludwig DL, Novosyadlyy R, Li YY et al. Id1-induced IGF-II and its autocrine/endocrine promotion of esophageal cancer progression and chemoresistance—implications for IGF-II and IGF-IR-targeted therapy. Clin Cancer Res 2014; 20: 2651–2662.
Cristancho AG, Lazar MA . Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol 2011; 12: 722–734.
Iguchi H, Urashima Y, Inagaki Y, Ikeda Y, Okamura M, Tanaka T et al. SOX6 suppresses cyclin D1 promoter activity by interacting with beta-catenin and histone deacetylase 1, and its down-regulation induces pancreatic beta-cell proliferation. J Biol Chem 2007; 282: 19052–19061.
Goldstein I, Yizhak K, Madar S, Goldfinger N, Ruppin E, Rotter V . p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer Metab 2013; 1: 9.
Lee DF, Su J, Ang YS, Carvajal-Vergara X, Mulero-Navarro S, Pereira CF et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell Stem Cell 2012; 11: 179–194.
Marxer M, Ma HT, Man WY, Poon RY . p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of Aurora kinases. Oncogene 2014; 33: 3550–3560.
He W, Zhang MG, Wang XJ, Zhong S, Shao Y, Zhu Y et al. AURKA suppression induces DU145 apoptosis and sensitizes DU145 to docetaxel treatment. Am J Transl Res 2013; 5: 359–367.
Acknowledgements
This study was funded by a National Institutes of Health PO1 grant (CA087497) to CC-C and AJL.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Puzio-Kuter, A., Laddha, S., Castillo-Martin, M. et al. Involvement of tumor suppressors PTEN and p53 in the formation of multiple subtypes of liposarcoma. Cell Death Differ 22, 1785–1791 (2015). https://doi.org/10.1038/cdd.2015.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.27
This article is cited by
-
Molecular signatures of tumor progression in myxoid liposarcoma identified by N-glycan mass spectrometry imaging
Laboratory Investigation (2020)
-
Immune checkpoint inhibitors in sarcomas: in quest of predictive biomarkers
Laboratory Investigation (2018)
-
Activity of trabectedin and the PARP inhibitor rucaparib in soft-tissue sarcomas
Journal of Hematology & Oncology (2017)
-
Differential regulated microRNA by wild type and mutant p53 in induced pluripotent stem cells
Cell Death & Disease (2016)
-
The roles and therapeutic potential of cyclin-dependent kinases (CDKs) in sarcoma
Cancer and Metastasis Reviews (2016)

{kind=link}