
Abstract
Forkhead transcription factor family O (FoxO) maintains adult stem cell reserves by supporting their long-term proliferative potential. MicroRNAs (miRs) regulate neuronal stem/progenitor cell (NSPC) proliferation and differentiation during neural development by controlling the expression of a specific set of target genes. In the neurogenic subventricular zone, FoxO1 is specifically expressed in NSPCs and is no longer detected during the transition to neuroblast stage, forming an inverse correlation with miR-9 expression. The 3′-untranslated region of FoxO1 contains a conserved target sequence of miR-9 and FoxO1 expression is coordinated in concert with miR-9 during neuronal differentiation. Our study demonstrates that FoxO1 contributes to NSPC fate decision through its cooperation with the Notch signaling pathway.
Similar content being viewed by others


Main
Forkhead transcription factor family O (FoxO) is an evolutionarily conserved longevity factor found across species. FoxO’s most well-defined function is maintaining the adult stem cell compartment and supporting the long-term regenerative potential of tissues during organismal aging. In the nervous system, FoxO maintains adult neural stem cell reserves and life-long neurogenesis,1, 2 which may require functions beyond cell-cycle regulation and oxidative stress suppression. Given the importance of neural stem/progenitor cells (NSPCs) for the generation of new neurons, defining the mechanism of how FoxO contributes to the NSPC fate decision is important for the development of regenerative medicine.
MicroRNAs (miRs) are small noncoding RNAs that regulate gene expression by destabilizing or suppressing the translation of target mRNAs, mostly through the 3′-untranslated regions (3′-UTRs).3, 4 Several miRs have been found to participate in the neurogenic differentiation of NSPCs.5, 6, 7, 8 For example, let-7b inhibits NSPC proliferation and promotes neuronal differentiation by suppressing targets such as Hmga2, which promotes self-renewal.9 miR-124 promotes the neuronal differentiation of subventricular zone (SVZ) NSPCs by repressing Sox9 expression.10 In the developing mouse, brain-enriched miR-9 is expressed as early as embryonic day 10.5.11, 12, 13 miR-9 is a well-conserved miR; the mature sequence is 100% identical from flies to humans. It was shown that miR-9 promotes the neural differentiation of mouse embryonic stem cells by suppressing Sirt1 expression.14 Additionally, miR-9 was reported to accelerate neuronal differentiation by targeting Tlx in mouse NSPCs.15 By contrast, miR-9 promotes the proliferation and regulates the migration of human embryonic stem cell-derived NSPCs.16 Primary brain tumors highly express miR-917 and the inhibition of miR-9 in glioma-initiating stem cells leads to reduced neurosphere formation and increased cell differentiation.18 The double miR-9-2/miR-9-3-knockout mouse model had a complicated phenotype in that neural progenitor proliferation in the developing telencephalon was initially increased, but it was reduced at later stages because of the deregulation of multiple targets, including FoxG1, Tlx, Gsh2 and Meis2.19 In developing zebrafish, miR-9 suppresses the progenitor-promoting genes her6 and zic5 in the early progenitor state, leading to proliferation inhibition. However, at a later stage, miR-9 exerts an antagonistic neurogenesis-promoting action by controlling elavl3/HuC expression.20 Overall, the mechanisms of miR-9 action are context-dependent and remain incompletely understood.
Recent studies have begun to elucidate the interaction between miRs and FoxO. For example, miR-71 may promote longevity in C. elegans by functioning through the nervous system to facilitate the transcriptional activity of DAF-16/FoxO in the intestine.21 In addition, FoxO3-regulated miR-25 enhances NSPC proliferation and neuronal differentiation.22 In this study, we report that miR-9 regulates FoxO1 expression during neurogenesis from NSPCs and the endogenous expression of miR-9 and FoxO1 is inversely correlated in adult SVZ. Our study demonstrates that FoxO1 expression is downregulated in the early stage of neurogenesis and gain of function or loss of function of FoxO1 prevents or promotes NSPC differentiation, respectively. We propose that FoxO1-Notch pathway cooperation is an important mechanism for the inhibition of NSPC differentiation.
Results
Modulation of miR-9 expression alters NSPC differentiation
Although increasing evidence suggests that miR-9 favors neuronal differentiation over progenitor proliferation in vertebrates, the role of miR-9 in NSPC fate decision is context-dependent. To test this, we infected mouse primary NSPCs with miR-9-encoding retrovirus. Neuron-specific marker βIII-tubulin immunostaining demonstrated that the overexpression of miR-9 markedly promoted neuronal differentiation compared with control. By contrast, inhibiting miR-9 action by miR-9 sponge23 expression suppressed neuronal differentiation (Figure 1a). As a result of counting random fields of view, the percentage of βIII-tubulin-positive cells was increased ~2-fold by miR-9 overexpression and decreased by inhibition of miR-9 (Figure 1b). Consistent with this, exogenous miR-9 expression clearly increased the level of βIII-tubulin expression, whereas miR-9 sponge expression reduced the level of βIII-tubulin (Figures 1c and d). To support our observation, additional neural markers were also examined. As a result, the percentage of doublecortin (DCX)-positive cells (Supplementary Figures S1A and B) or glial fibrillary acidic protein (GFAP)-positive cells (Supplementary Figures S1C and D) were increased by exogenous miR-9 expression, albeit the latter was less significant. These results suggest that miR-9 enhances and is necessary for differentiation of NSPCs.

miR-9 promotes neuronal differentiation of adult NSPCs. (a) miR-9 sponge or miR-9-encoding virus-infected cells were induced to differentiate for 3 days, and they were immunostained with a βIII-tubulin-specific antibody (red). GFP-expressing retrovirus was used as a control. Nuclear DAPI (4',6-diamidino-2-phenylindole) staining is shown in blue. Scale bar=200 μm. (b) The quantification of βIII-tubulin-positive cell proportions in control, miR-9-overexpressing, miR-9 sponge-expressing NSPCs as in (a). (c) Western blot analysis of βIII-tubulin expression in control, miR-9 sponge or miR-9-encoding virus-infected neural stem cells. β-Actin was used as a loading control. (d) The mRNA expression of βIII-tubulin in control, miR-9-overexpressing, miR-9 sponge-expressing NSPCs, as measured by real-time RT-PCR analysis. The mRNA level of control cells was set to 1. *P<0.05. Representative results from multiple experiments are shown
FoxO1 and miR-9 expression is inversely correlated in the NSPC differentiation hierarchy
Although neurogenesis occurs in other regions of the mammalian brain,24 major neural stem cell niches are the SVZ of the lateral ventricles and subgranular zone of the dentate gyrus. Previously, it was reported that (1) miR-9 induces direct conversion of human adult fibroblasts into neurons,25 (2) miR-9 is highly expressed in neurogenic regions of the brain throughout development and adulthood26 and (3) the level of miR-9 in adult mouse neural stem cells is gradually increased during differentiation.15 It remained unclear, nonetheless, which cell types in the neural stem cell niche express miR-9. To better define the expression patterns of miR-9 in the neural stem cell niche, we performed RNA in situ hybridization (RISH) for miR-9 combined with immunohistochemistry (IHC) for lineage-specific neural markers. The results showed that miR-9 was highly expressed in DCX-positive neuroblasts (type A cells), but it was rarely detected in GFAP-positive type B cells and astrocytes in the SVZ (Figure 2a and Supplementary Figure S2A). In sharp contrast, we observed that FoxO1 was expressed in NSPCs (type B and C cells) and disappeared in type A neuroblasts (Figure 2b and Supplementary Figures S2B and C). This inverse correlation of expression between miR-9 and FoxO1 during adult neuronal differentiation raised a possibility that they could have an antagonistic effect on neurogenesis.

Expressional and functional interaction between FoxO1 and miR-9. (a) Cell-type-specific expression of miR-9 was determined by RISH coupled with IHC. Lower left inset shows schematic cellular composition of adult SVZ. GFAP is expressed in type B cells. Lower right inset is higher magnification of miR-9 and GFAP RISH-IHC with type A clusters demarcated. (b) Cell-type-specific expression of FoxO1 was determined on serial adjacent sections of adult SVZ by IHC. FoxO1 is expressed in GFAP-positive type B cells but absent in DCX-positive type A neuroblasts. GFAP or DCX staining visualizes type B or type A cells, respectively. Scale bars=50 μm (upper) and 200 μm (lower). (c) The overlap between predicted miR-9 downstream targets and genes whose expressions were affected by FoxO deficiency in NSPCs. The prediction of the miR-9 targets was performed by two different algorithms, miRanda and Targetscan. (d) The nucleotide pairing of miR-9 and FoxO1 3′-UTR. The 3′-UTR of FoxO1 mRNA harbors a putative miR-9 binding site at the 69–75 nt position, which is conserved in mammals. (e) Western blot analysis of FoxO1 and FoxO3 expression in control RNA or miR-9-encoding retrovirus-infected cells. α-Tubulin was used as a loading control. (f) miR-9 represses the luciferase reporter gene upstream of the FoxO1 3′-UTR. Luciferase reporter constructs under the control of wild-type (WT) or mutant (Mut) FoxO1 3′-UTR were transfected along with miR-9 RNA duplexes
To determine the functional interaction between FoxO and miRs, we performed a transcriptome analysis. As we reported earlier, the transcriptomic profile of FoxO-null NSPCs shows both up- and downregulation of gene sets.27 First, we used the Gene Set Enrichment Analysis (GSEA) algorithm to identify a particular miR that has a functional association with FoxO-mediated gene regulation. Interestingly, there was a high enrichment score for miR-9 target gene sets in the FoxO-null NSPC transcriptome (Supplementary Figure S3A). Next, we used two independent computational algorithms, TargetScan and miRanda, to identify miR-9 targetable genes. Overall, 24% of the entire FoxO-null NSPC transcriptome was predicted to be miR-9 targets, and 8% were common for both TargetScan and miRanda (Figure 2c). In accordance with the negatively correlated expression pattern of miR-9 and FoxO1 and overrepresented miR-9 signature in the FoxO-null NSPC transcriptome, we hypothesized that FoxO1 could be regulated by miR-9.
miR-9 and FoxO1 form a feedback regulatory circuit during NSPC differentiation
To determine whether miR-9 regulates FoxO1 expression during neurogenesis, we first analyzed the 3′-UTR of mouse FoxO1. Notably, the 69–75 position in the 3′-UTR of mouse FoxO1 was complementary to the miR-9 seed sequence. In addition, this putative miR-9 binding site was highly conserved among vertebrates, suggesting that miR-9-mediated FoxO1 regulation might be important across species (Figure 2d). To assess whether miR-9 regulates endogenous FoxO1 protein expression, we forced the expression of miR-9 in NSPCs using a retroviral vector. As a result, the overexpression of miR-9 clearly decreased the FoxO1 protein level in NSPCs (Figure 2e). To further confirm that miR-9 suppresses FoxO1 expression through the conserved 3′-UTR of FoxO1, we used a luciferase reporter that contains the mouse FoxO1 3′-UTR with either the miR-9 recognition sequence or mutated bases. Compared with the control, the reporter with mutated FoxO1 3′-UTR did not result in a decrease in the luciferase activity after miR-9 expression, indicating that miR-9 inhibits FoxO1 expression through binding to this 3′-UTR element (Figure 2f). In addition, we examined whether FoxO1 influences the expression of miR-9 in NSPCs. In FoxO1-null NSPCs, the expression of miR-9 was downregulated (Supplementary Figure S3B). In particular, the primary transcripts of miR-9-2 and miR-9-3 were significantly downregulated among three different precursors of miR-9 (Supplementary Figure S3C). Furthermore, miR-9-2 has a putative and conserved FoxO-binding element at the −117 bp region in the upstream of transcription start site (TSS). The expression of a constitutively active form of FoxO1 transactivated the miR-9-2 promoter (Supplementary Figure S3D). Taken together, FoxO1 and miR-9 form a feedback regulatory circuit during the NSPC differentiation.
FoxO1 expression is regulated during neuronal differentiation
Given that the level of miR-9 in neural stem cells is reportedly increased during neuronal differentiation15 and our own observation (Figure 3a), we examined the change in the FoxO1 protein level. Primary NSPCs were differentiated for the indicated times, and neuronal differentiation was determined by marker expression. The NSPC marker Nestin completely disappeared within 24 h after differentiation initiation. By contrast, βIII-tubulin expression was markedly increased within 3 days after the enforcement of differentiation. Importantly, the FoxO1 protein level was clearly downregulated in the early stage of neuronal differentiation, consistent with the prediction that the expression of FoxO1 is regulated by miR-9 (Figure 3b). Additionally, we examined phosphorylation status of FoxO1-Ser256 residue and found a concordant pattern with total FoxO1 expression. This suggests that downregulation of FoxO1 is unlikely to be mediated through increased inhibitory phosphorylation by AKT. Interestingly, FoxO1 was gradually increased at a later stage, suggesting that there is an alternative regulatory mechanism that overrides the action of miR-9 on FoxO1 and that FoxO1 expression is precisely controlled in multiple stages during differentiation. Taken together, our results indicate that FoxO1 may act as a molecular switch for NSPC differentiation and that FoxO1 may have additional roles in neural cell maturation and homeostasis. Indeed, it was reported that the genetic ablation of FoxO1 in mouse hypothalamic neurons affects food intake and energy expenditure.28, 29

FoxO1 expression patterns during neuronal differentiation. (a) Endogenous level of miR-9 was determined by quantitative real-time PCR (qRT-PCR) analysis at indicated time points during NSPC differentiation. Pr, proliferating NSPCs. (b) Western blot analysis of FoxO1 and neural markers during NSPC differentiation. Cells were harvested at the indicated time points. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as a loading control. (c–f) The coexpression of FoxO1 with neural markers was determined. FoxO1 staining is shown in green and neural markers, Nestin (c), GFAP (d), βIII-tubulin (e) and DCX (f), are shown in red. Nuclear DAPI (4',6-diamidino-2-phenylindole) staining is shown in blue. (d and e) Filled arrow heads point to cells that are positive for both FoxO1 and markers. (f) Empty arrowheads are for cells that are positive for DCX but do not express FoxO1. Scale bars=50 μm. (g) The percentage of each neural marker-positive cell of the FoxO1-positive cells
Next, we examined the neural markers of FoxO1-expressing cells to further characterize the relationship between FoxO1 expression and neuronal differentiation. We found that the majority of Nestin-positive proliferating NSPCs express FoxO1 (Figure 3c). In addition, ~80–90% of GFAP- (Figure 3d) or βIII-tubulin- (Figure 3e) positive astrocytes and neuronal cells also express FoxO1 upon differentiation. By contrast, cells coexpressing DCX and FoxO1 were rarely detected (Figures 3f and g). These results collectively suggest that the expression of FoxO1 is tightly regulated during neuronal differentiation.
Constitutively activated FoxO1 represses NSPC differentiation
To confirm whether the downregulation of FoxO1 in the initial step of NSPC differentiation is crucial, we expressed active FoxO1 by using adeno-FoxO1-ADA (Ade-FoxO1), non-phosphorylatable, constitutively nuclear form of FoxO1.30 In control adenovirus- (Ade-emp) infected cells, βIII-tubulin expression was increased during differentiation. However, constitutively activated FoxO1 significantly decreased neuronal differentiation as determined by βIII-tubulin and DCX levels (Figure 4a). Consistent with the immunoblotting result, activated mutant FoxO1-expressing NSPCs showed an ~50% reduction in the βIII-tubulin gene expression compared with control (Figure 4b).

Overexpression of FoxO1 inhibits neuronal differentiation. (a) Western blot analysis of βIII-tubulin and DCX expression in Ade-emp- or Ade-FoxO1-infected NSPCs. Cells were differentiated for the indicated time. The overexpression of FoxO1 was also confirmed. (b) Quantitative real-time PCR (qRT-PCR) of Nestin and βIII-tubulin mRNA in Ade-emp- or Ade-FoxO1-infected NSPCs. *P<0.05. (c–e) Immunofluorescence analysis of Nestin (c), GFAP (d) and βIII-tubulin (e) expression after expression of FoxO1-ADA was confirmed. Nuclear DAPI (4',6-diamidino-2-phenylindole) staining is shown in blue. Scale bars=50 μm (c) and 200 μm (d, e). (f) The quantification of Nestin-, GFAP- and βIII-tubulin-positive cell proportions in Ade-emp- or Ade-FoxO1-expressing NSPCs. NS, non-significant
To better characterize the inhibitory role of FoxO1 in NSPC differentiation, we examined the neural markers in NSPCs expressing FoxO1-ADA. Under a proliferating condition, a number of Nestin-positive NSPCs coexpressed FoxO1-ADA (Figure 4c). Exogenously expressed FoxO1 was also observed in GFAP-positive astrocytes upon differentiation (Figure 4d). However, the expression of FoxO1-ADA and βIII-tubulin were mutually exclusive (Figure 4e), suggesting that neuronal differentiation is suppressed with sustained FoxO1 activation. Regardless of exogenous FoxO1 expression, >60% of NSPCs expressed Nestin, which only had marginal effect on the distribution of different lineage marker-expressing cells under the proliferating condition. However, when cells were induced to differentiate, constitutively activated FoxO1 significantly reduced the proportion of GFAP or βIII-tubulin-positive cells (Figure 4f). Taken together, our data suggest that the downregulation of FoxO1 may be necessary for NSPC differentiation.
FoxO1 deficiency enhances the neuronal differentiation of NSPCs
The inhibitory role of FoxO1 in neuronal differentiation raised the possibility that reducing FoxO1 expression may enhance neurogenesis. To test this, primary FoxO1L/L NSPCs were infected with adeno-empty or adeno-Cre (Ade-Cre). Loss of FoxO1 expression by Cre-mediated recombination was confirmed by qPCR (Figure 5a) and immunoblotting (Figure 5b). Next, we examined the expression of neural markers (Figure 5c). As expected, proliferating NSPCs highly express Nestin, but they maintain βIII-tubulin and DCX at a low level. The mRNA levels of all of three neural markers were comparable between the control and FoxO1-deficient cells, indicating that the downregulation of FoxO1 was not sufficient to induce spontaneous neuronal differentiation. When NSPCs were differentiated, Nestin expression was significantly reduced and the levels of both βIII-tubulin and DCX were upregulated, and interestingly, FoxO1 deficiency led to higher expression levels of neuronal markers. This was confirmed by βIII-tubulin immunoblotting (Figure 5d). To better understand the inhibitory role of FoxO1 in NSPC differentiation, we performed immunostaining. Consistently, the number of βIII-tubulin-positive neurons was significantly increased (Figures 5e and f). To exclude the possibility of artifacts arising from adenoviral infection, we examined the expression level of neuronal markers in NSPCs derived from brain-specific FoxO-knockout mice (Nestin-Cre; FoxO1/3/4L/L). In this mouse model, the Nestin promoter drives Cre recombinase expression and FoxO expression is absent in neural stem cells. Both neuronal markers, DCX and βIII-tubulin, were upregulated by FoxO deficiency under differentiating conditions (Figure 5g). Collectively, these data suggest that downregulation of FoxO1 enhances differentiation of NSPCs into neurons.

Downregulation of FoxO1 enhances neuronal differentiation. FoxO1L/L primary NSPCs were infected with Ade-emp or Ade-Cre. (a) Quantitative real-time PCR (qRT-PCR) of FoxO1 mRNA. (b) Western blot analysis of FoxO1. (c) qRT-PCR of Nestin and neuronal marker (βIII-tubulin and DCX) mRNA. Cells were kept under either proliferation media or B-27-supplemented differentiation media before analysis. (d) Western blot analysis of βIII-tubulin expression in control Ade-emp- or Ade-Cre-infected and differentiated NSPCs. (e) Immunofluorescence analysis of GFAP (green) and βIII-tubulin (red) expression after the ablation of FoxO1 expression. Nuclear DAPI (4',6-diamidino-2-phenylindole) staining is in blue. Scale bars=200 μm. (f) The quantification of Nestin- (proliferating condition only), GFAP- and βIII-tubulin-positive cell proportions in control and FoxO1-deficient NSPCs. (g) qRT-PCR of DCX and βIII-tubulin mRNA in primary NSPCs which were prepared from Nestin-Cre+; FoxO1/3/4L/L (KO) and control littermate Nestin-Cre–; FoxO1/3/4L/L mice (WT) and they were induced to differentiate before analysis. *P<0.05, **P<0.01 and ***P<0.001
FoxO1 and Notch functionally interact during neuronal differentiation
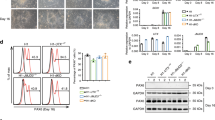
To evaluate the antagonistic mechanism of FoxO1 during neurogenesis, we compared the transcriptome of control and FoxO1-knockdown LN319 heterologous glioma cells. Notably, the expression levels of several genes involved in the Notch pathway were significantly changed (Figure 6a). The evaluation with the GSEA algorithm also suggested the functional association between FoxO1 and the Notch pathways (Figure 6b). The Notch signaling pathway has an important role in the cell fate decision of stem/progenitor cells, including embryonic stem cells,31 hematopoietic stem cells32, 33 and other tissue-specific stem cells.34, 35 It has been reported that the Notch pathway is essential for the maintenance of neural stem cells, and neuronal and glial differentiation was suppressed by expressing an active form of Notch1.36 Previously, the Notch ligand was shown to stimulate the neural specification of human ES cells,35 suggesting that the Notch pathway has a crucial role in neural system maintenance. To confirm whether the Notch pathway restricts the spread of NSPC differentiation, we used a γ-secretase inhibitor (DAPT) that blocks Notch signaling. Upon DAPT treatment, the cell morphology was clearly changed compared with control (Figure 6c). The action of DAPT was confirmed by the Hes1 expression level. Notably, DAPT treatment downregulated Nestin expression under proliferating conditions and highly upregulated the βIII-tubulin level under differentiating conditions (Figure 6d).

Functional cooperation between FoxO1 and the Notch pathway. (a) A heat map of the Notch pathway-related genes shows a strong correlation with FoxO1 expression. (b) GSEA profiles of the correlation between the FoxO1 expression and the Notch signaling pathway. The upper part plots the enrichment scores for each gene, and the lower part of the plot shows the value of the ranking metric moving down the list of ranked genes. (c) Morphologic changes of the NSPCs after Notch pathway inhibition. NSPCs were treated with vehicle or 5 μM of DAPT for the indicated time to inhibit the Notch pathway. Scale bar=200 μm. (d) Western blot analysis of Nestin and βIII-tubulin expression in vehicle-treated or DAPT-treated, differentiated NSPCs. Reduction in Hes1 protein expression was used to demonstrate the inhibition of the Notch signaling. (e) CSL-induced luciferase reporter activity was measured in (left) FoxO1L/L NSPCs infected with Ade-Cre or Ade-emp, or (right) Ade-emp- or Ade-FoxO1-infected NSPCs, with or without 5 μM DAPT treatment. (f) Quantitative real-time PCR (qRT-PCR) of Hes1 mRNA in (left) FoxO1L/L NSPCs infected with Ade-Cre or Ade-emp, or (right) Ade-emp or Ade-FoxO1 infected NSPCs. *P<0.05, **P<0.01 and ***P<0.001
Previously, FoxO1 was reported to interact directly with CSL, a transcription factor of Notch downstream genes, and this FoxO1-Notch cooperation regulates myogenic differentiation.37 Based on this, we measured the CSL-induced Notch reporter activity in the presence or absence of FoxO1. Consistent with the previous report, FoxO1 deficiency caused a marked reduction in the luciferase activity (Figure 6e). Similarly, the overexpression of FoxO1 caused a significant increase in the luciferase activity, and it was clearly decreased by DAPT treatment. To confirm the FoxO1-Notch cooperation, we analyzed the expression level of Hes1, one of the major target genes of the Notch pathway. Hes1 expression was significantly reduced by FoxO1 deficiency and upregulated by FoxO1 overexpression (Figure 6f). Next, we tested whether miR-9 modulates Notch signaling by suppressing FoxO1. Consistent with our prediction, enforced miR-9 expression significantly reduced CSL-induced reporter activity (Figure 7a). To determine whether miR-9-mediated Notch pathway regulation is FoxO1-dependent, we compared Notch reporter activity in both wild-type and FoxO1-knockout NSPCs, either mock or miR-9-encoding retrovirus infected. Notably, miR-9-mediated reduction of Notch reporter activity was abolished by FoxO1 deficiency (Figure 7b). These data suggest that FoxO1 is necessary for miR-9 regulation of Notch signaling. In addition, we performed a rescue experiment to confirm that FoxO1 is a functional target of miR-9 during neuronal differentiation. Overexpression of miR-9 enhanced βIII-tubulin and reduced Hes1 mRNA levels. This effect was, however, abolished by the expression of activated FoxO1 (Figure 7c).

miR-9 regulates Notch pathway through FoxO1. (a) CSL-induced luciferase reporter activity was measured in control or miR-9-overexpressed NSPCs. (b) Same experiment as in (a) was performed in wild-type (Ade-emp) or FoxO1-knockout (Ade-Cre) NSPCs. (c) Quantitative real-time PCR (qRT-PCR) analysis of βIII-tubulin and Hes1 mRNA in Ade-emp- or Ade-FoxO1-infected NSPCs, with or without miR-9 overexpression. (d) The FoxO1 occupancy of the Hes1 promoter in NSPCs was determined by chromatin immunoprecipitation (ChIP). (Upper) Predicted FoxO1 binding in the Hes1 promoter. TSS: transcription start site. (Lower) qPCR against two regions in the Hes1 promoter. (e) Western blot analysis of βIII-tubulin and Hes1 expression in Ade-empty- or Ade-Cre-infected FoxO1L/L NSPCs, with (D) or without (V) treatment of 5 μM DAPT. (f) qRT-PCR analysis of βIII-tubulin and Hes1 mRNA in Ade-empty- or Ade-Cre-infected FoxO1L/L NSPCs, with or without DAPT treatment. *P<0.05 and **P<0.01. (g) A graphical presentation of miR-9 and FoxO1 interaction on Notch target gene regulation
Based on the consensus binding sequence for CSL within the Hes1 promoter was already determined,38 we checked whether FoxO1 is bound to the CSL element in the Hes1 promoter using a chromatin immunoprecipitation (ChIP) assay (Figure 7d). We found that there was significant enrichment of the binding of FoxO1 in the CSL binding region of the Hes1 promoter (Region 1). In addition, we identified CSL-independent FoxO1 binding site within the Hes1 promoter (Region 2). This result raises a possibility that FoxO1 may regulate Hes1 expression and NSPC differentiation in Notch and CSL-independent manner. In conclusion, our data are consistent with the notion that FoxO1 cooperates with the Notch pathway and this functional interaction contributes to the inhibition of NSPC differentiation.
Discussion
Our results provide several pieces of evidence suggesting that FoxO1 has a suppressive role in the neuronal differentiation of NSPCs. As we reported previously, FoxOs regulate genes that are essential for neural stem cell homeostasis.27 In the absence of FoxO-dependent homeostatic processes, there was significant decrease in the NSC pool and accompanying neurogenesis in the adult mouse brain. In the present study, we posit that FoxO1 is an important switch for NSPC differentiation, and cooperation between FoxO1 and Notch pathway controls neural stem cell fate.
Several reports have demonstrated that FoxO1 is developmentally regulated and involved in the cell fate decision. For instance, FoxO1 is abundantly expressed in murine white and brown adipose tissues and a gain-of-function FoxO1 mutant inhibits the differentiation of preadipocytes.39 During B-cell development, early FoxO1 depletion caused a substantial block at the pro-B-cell stage, whereas deficiency of FoxO1 in late pro-B cells led to an arrest at the pre-B-cell stage.40 In addition, constitutively active FoxO1 expression prevented myoblast differentiation through the interaction with the Notch signaling pathway.37 Endothelial-specific FoxO1 knockout showed defective vasculature remodeling and lethality at E10.5, suggesting that endothelial FoxO1 is essential to cardiovascular development.41 Interestingly, FoxO3, another member of the FoxO family, was reported to regulate neuronal differentiation. In adult neural precursor cells, ASCL1, a proneuronal bHLH transcription factor, shares common targets with FoxO3 and FoxO3 could inhibit ASCL1-dependent transcription of neurogenic genes.42 The occupancy of FoxO3 and ASCL1 was especially enriched for Notch pathway target genes, such as Dll1 and Hes6, suggesting that there is crosstalk between Notch signaling and FoxO3. In this study, we demonstrated for the first time that FoxO1 regulates adult neural stem cell differentiation, cooperating with Notch. However, the role of FoxO1 in astroglial differentiation is still unclear. In our result, both loss and gain of function of FoxO1 reduced GFAP expressions. Our working hypothesis is that FoxO1 is necessary for maintaining undifferentiated state of NSPCs. Once differentiation initiates, FoxO1 is selectively silenced in neuroblasts and remains in astroglial cells. Consistent with this hypothesis, our results demonstrated that constitutive expression of hyperactivated FoxO1 reduced overall differentiation (both neuronal and glial). FoxO1-knockout NSPCs, on the other hand, have significantly increased neuronal commitment at the expense of glial differentiation. This suppressive effect of constitutively active FoxO1 in progenitor differentiation and lineage commitment was previously reported in adipogenesis.39
Although accumulating evidence has indicated that miR-9 favors neuronal differentiation over progenitor proliferation, it remains unclear how miR-9 induces neuronal differentiation, especially in mammalian adult neural system. We found that miR-9 had higher expression levels in type A cells compared with that in type B/C cells in adult SVZ, and FoxO1 was identified as a target of miR-9 during neurogenic differentiation. Although many target genes were already predicted and some of them were tested for miR-9-dependent expression, including TLX, REST, FoxG1 and Hes1,15, 43, 44, 45 few have been evaluated. Our study clearly demonstrated that miR-9 regulates FoxO1 in the neural stem cell fate decision. However, we cannot rule out the possibility that the coregulation of other miR-9 targets is necessary for eliciting the aberrant differentiation pattern observed in FoxO1-deficient NSPCs.
One interesting observation of this study is that the FoxO1 protein level was rapidly decreased in the early phase of in vitro NSPC differentiation, but it was restored after differentiation (Figure 3b). These data were consistent with in vivo observation that the protein level of FoxO1 is decreased during transition from type B/C to A cells in the SVZ (Figures 2a and b), but it is constitutively expressed in a subset of mature neurons.27 These data indicate that FoxO1 has a functional role in differentiated neurons, as well as in neural stem cells, and that the rapid downregulation of FoxO1 at the initiation of NSPC differentiation serves as a molecular switch. Also, our results suggest that miR-9-mediated FoxO1 regulation may depend on its threshold level and is then uncoupled over the course of differentiation or there may be an independent regulatory mechanism on FoxO1, which requires further investigation.
In this study, we proposed that the functional interaction between FoxO1 and Notch pathway is a mechanism for the suppression of neuronal differentiation. Interestingly, we also observed that there was an additive effect between FoxO1 deletion and DAPT-mediated Notch inhibition on βIII-tubulin and Hes1 expressions (Figures 7e and f). This raises the possibility that FoxO1 may regulate NSPC differentiation and Hes1 expression in a Notch-independent manner. Indeed, we found CSL-independent binding of FoxO1 in Hes1 promoter (Region 2; Figure 7d). We summarized our findings as a schematic model (Figure 7g).
Functional interaction between FoxO1 and Notch was reported in other tissue systems. It is required for Notch target gene induction to control the myogenic program.37 Additionally, FoxO1 and Notch coordinately regulate hepatic glucose metabolism,46 gluconeogenesis and maintenance of mouse embryonic stem cell self-renewal.47 We observed this interaction in mammalian adult SVZ neurogenesis. Our findings may be used to develop therapeutic strategies for acute brain injury or long-term degenerative conditions by regulating adult neurogenesis.
Materials and Methods
Neural stem cell culture and differentiation
Primary NSPCs were isolated from the brain of neonatal FoxO1L/L mice or wild-type mice and processed with Neural Tissue Dissociation Kits (Miltenyi Biotec, San Diego, CA, USA). NSPCs were maintained in a medium supplemented with 20 ng/ml EGF and bFGF. To induce the recombination and deletion of FoxO1, NSPCs were infected with Adeno-CMV-Cre or Adeno-empty virus (Ade-emp; Vector Biolab, Malvern, PA, USA). For differentiation, NSPCs were dissociated into single cells using TrypLE (Life Technologies, Carlsbad, CA, USA), and plated on polyornithine and fibronectin-coated plates in NSPC culture medium including B-27 Supplements (Life Technologies). Cells were harvested at indicated time points for further analysis.
Transfection and reporter assays
For Notch reporter plasmid transfection, NSPCs were dissociated and plated in NSPC culture medium, concurrent with the infection of Adeno-CMV-Cre (Ade-Cre for loss of function)/Adeno-FoxO1 (Ade-FoxO1 for gain of function) or Ade-emp. After 24 h incubation, the medium was replaced with NSPC culture medium including EGF and bFGF (for a proliferation condition) or B-27 supplement (for a differentiation condition). CSL-Notch reporter Plasmid DNA (kindly provided by Dr Jan Kitajewski) was transfected using Lipofectamine 3000 (Life Technologies) according to the manufacturer's instruction. After 48 h, cells were harvested and luciferase activity was measured. The relative luciferase activity was determined as the ratio of firefly to Renilla activity. For data presentation, the luciferase activity of pGL3 mock-negative control vector was set to 1. For miR-9-dependent FoxO1 mRNA level, DNA fragment encoding the mouse FoxO1 3′-UTR was subcloned into a pGL4 luciferase vector (Promega, Madison, WI, USA).
ISH-IHC
The miR-9 probes were 3′-end labeled with digoxigenin (DIG)-ddUTP with terminal transferase using the DIG-3′-End Labeling Kit (Roche, Indianapolis, IN, USA). Paraffin-embedded sections were hydrated and hybridized at 65 °C overnight and washed. After blocking, sections were labeled with anti-DIG antibody (Roche) at 4 °C overnight and washed, stained with BM purple (Roche). Sections were processed for immune detection of neural markers using Vector Elite Kit (Vector Laboratories, Burlingame, CA, USA) and developed with DAB substrate.
Quantitative real-time RT-PCR
Total RNA was isolated from primary NSPCs using RNA Purification Kit (Thermo Scientific, Waltham, MA, USA). Five hundred nanograms of total RNA was treated with RNase-free DNase (Sigma-Aldrich, St. Louis, MO, USA) for 15 min. After inactivation of DNase with EDTA and heating, RNA was reverse transcribed using First Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer's instructions. Quantitative RT-PCR was performed on cDNA samples using the Power SYBR Green Master mix and was performed the qPCR on the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The relative mRNA level was presented as values of 2^(Ct(β-actin)−Ct(gene of interest)). The sequences of the forward and reverse primers are as follows: Nestin, 5′-GGAAGAAGTTCCCAGGCTTC-3′ and 5′-ATTAGGCAAGGGGGAAGAGA-3′; Hes1, 5′-CCAGCCAGTGTCAACACGA-3′ and 5′-AATGCCGGGAGCTATCTTTCT-3′; Tubb3 (βIII-tubulin), 5′-TAGACCCCAGCGGCAACTAT-3′ and 5′-GTTCCAGGTTCCAAGTCCACC-3′; Dcx, 5′-CATTTTGACGAACGAGACAAAGC-3′ and 5′-TGGAAGTCCATTCATCCGTGA-3′; FoxO1, 5′-TTCAATTCGCCACAATCTGTCC-3′ and 5′-GGGTGATTTTCCGCTCTTGC-3′. For quantification of miRNA miScript Kit and primers (Qiagen, Valencia, CA, USA) were used.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as previously described.48 In brief, five millions of NSPCs were crosslinked with 1% formaldehyde followed by quenching with 125 mM glycine. The cells were lysed in lysis buffer containing 1% SDS, 10 mM EDTA and 50 mM Tris (pH 8.1), and then sonicated 15 times for 30s. The sheared DNA was diluted in dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl (pH 8.1) and 167 mM NaCl), precleared and incubated with anti-rabbit IgG and anti-FoxO1 IgG overnight at 4 °C and collected with protein G gamma plus sepharose beads. Precipitates were washed sequentially and DNA was purified. TRANSFAC algorithm was used to predict the FoxO1 binding site on regulatory region of target genes (up to 2 kb upstream of TSS) with minimal false-positive stringency. Enrichment was calculated based on Ct values of qPCR and plotted as fraction of total input. For negative control, we used Hes1 promoter region, which was not predicted as FoxO1 binding element and the sequences of primers are as follows: control, 5′-GCCTGGCCACAAAAGAAATA-3′ and 5′-CGGAGGCTACAACGTCAATC-3′; Hes1 Region 1, 5′- CCTCCCATTGGCTGAAAGT-3′ and 5′-CAGCTCCAGATCCTGTGTGA-3′; Hes1 Region 2, 5′-TAGAGAGAGTGGCGGAGGAA-3′ and 5′-TTCGGAGCAAGGTGTCTTTT-3′.
Protein preparation and immunoblot analysis
For immunoblotting, cells were disrupted directly with Laemmli buffer, followed by sonication and heat denaturation. Immunoblot analyses were performed with anti-βIII-tubulin, (Abcam, Cambridge, MA, USA), anti-FoxO1, anti-GAPDH (Cell Signaling, Danvers, MA, USA), anti-Nestin (EMD Millipore, Billerica, MA, USA), anti-α-tubulin (DHSB, Iowa City, IA, USA). The signals were detected with Supersignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL, USA).
Retrovirus production and purification
In total, 1.5 × 107 293T cells were seeded in 150 mm tissue culture dishes. After 24 h, the medium was replaced and cells were transfected with 22.5 μg of MDH1-PGK-GFP-miR-9 (Addgene, Cambridge, MA, USA; no. 25036) or pBabe-puro-miR-9 sponge (Addgene; no. 25040) along with 22.5 μg of pCL-Ecotrophic retrovirus packaging vector using polyethylenimine (Polysciences, Warrington, PA, USA; no. 23966-2). The medium containing retrovirus particles were collected at 48 and 72 h after transfection. To precipitate retrovirus particles, polyethylene glycol solution was added to four volumes of the medium and incubated at 4 °C overnight, followed by centrifugation at 3 000 r.p.m. for 1 h. Viral particles were resuspended in the media and titered before the infection of NSPCs.
Statistical analysis
The unpaired two-tailed Student’s t-test was used for experiments comparing two sets of data unless noted. Otherwise, one-way analysis of variance was conducted with Tukey's HSD as a post-test for significant differences as noted.
Abbreviations
- FoxO:
-
Forkhead transcription factor O
- miRs:
-
microRNAs
- NSPC:
-
neural stem/progenitor cells
- SVZ:
-
subventricular zone
- DCX:
-
doublecortin
- GFAP:
-
glial fibrillary acidic protein
- UTR:
-
untranslated regions
References
Ro SH, Liu D, Yeo H, Paik JH . FoxOs in neural stem cell fate decision. Arch Biochem Biophys 2013; 534: 55–63.
Rafalski VA, Brunet A . Energy metabolism in adult neural stem cell fate. Prog Neurobiol 2011; 93: 182–203.
Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R . Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 2006; 20: 515–524.
Ambros V . microRNAs: tiny regulators with great potential. Cell 2001; 107: 823–826.
Stefani G, Slack FJ . Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 2008; 9: 219–230.
Gao FB . Context-dependent functions of specific microRNAs in neuronal development. Neural Dev 2010; 5: 25.
Liu C, Zhao X . MicroRNAs in adult and embryonic neurogenesis. Neuromolecular Med 2009; 11: 141–152.
Kawahara H, Imai T, Okano H . MicroRNAs in neural stem cells and neurogenesis. Front Neurosci 2012; 6: 30.
Nishino J, Kim I, Chada K, Morrison SJ . Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell 2008; 135: 227–239.
Cheng LC, Pastrana E, Tavazoie M, Doetsch F . miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci 2009; 12: 399–408.
De Pietri, Tonelli D, Pulvers JN, Haffner C, Murchison EP, Hannon GJ, Huttner WB . miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 2008; 135: 3911–3921.
Smirnova L, Gräfe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG et al. Regulation of miRNA expression during neural cell specification. Eur J Neurosci 2005; 21: 1469–1477.
Krichevsky AM, Sonntag KC, Isacson O, Kosik KS . Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 2006; 24: 857–864.
Saunders LR, Sharma AD, Tawney J, Nakagawa M, Okita K, Yamanaka S et al. miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging (Albany, NY) 2010; 2: 415–431.
Zhao C, Sun G, Li S, Shi Y . A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat Struct Mol Biol 2009; 16: 365–371.
Delaloy C, Liu L, Lee JA, Su H, Shen F, Yang GY et al. MicroRNA-9 coordinates proliferation and migration of human embryonic stem cell-derived neural progenitors. Cell Stem Cell 2010; 6: 323–335.
Nass D, Rosenwald S, Meiri E, Gilad S, Tabibian-Keissar H, Schlosberg A et al. MiR-92b and miR-9/9* are specifically expressed in brain primary tumors and can be used to differentiate primary from metastatic brain tumors. Brain Pathol 2009; 19: 375–383.
Schraivogel D, Weinmann L, Beier D, Tabatabai G, Eichner A, Zhu JY et al. CAMTA1 is a novel tumour suppressor regulated by miR-9/9* in glioblastoma stem cells. EMBO J 2011; 30: 4309–4322.
Shibata M, Nakao H, Kiyonari H, Abe T, Aizawa S . MicroRNA-9 regulates neurogenesis in mouse telencephalon by targeting multiple transcription factors. J Neurosci 2011; 31: 3407–3422.
Coolen M, Thieffry D, Drivenes O, Becker TS, Bally-Cuif L . miR-9 controls the timing of neurogenesis through the direct inhibition of antagonistic factors. Dev Cell 2012; 22: 1052–1064.
Boulias K, Horvitz HR . The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab 2012; 15: 439–450.
Brett JO, Renault VM, Rafalski VA, Webb AE, Brunet A . The microRNA cluster miR-106b~25 regulates adult neural stem/progenitor cell proliferation and neuronal differentiation. Aging (Albany, NY) 2011; 3: 108–124.
Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 2010; 12: 247–256.
Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J et al. Neurogenesis in the striatum of the adult human brain. Cell 2014; 156: 1072–1083.
Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011; 476: 228–231.
Deo M, Yu JY, Chung KH, Tippens M, Turner DL . Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev Dyn 2006; 235: 2538–2548.
Paik JH, Ding Z, Narurkar R, Ramkissoon S, Muller F, Kamoun WS et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 2009; 5: 540–553.
Ren H, Orozco IJ, Su Y, Suyama S, Gutiérrez-Juárez R, Horvath TL et al. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 2012; 149: 1314–1326.
Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC et al. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci 2006; 9: 901–906.
Qiang L, Banks AS, Accili D . Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem 2010; 285: 27396–27401.
Kobayashi T, Mizuno H, Imayoshi I, Furusawa C, Shirahige K, Kageyama R . The cyclic gene Hes1 contributes to diverse differentiation responses of embryonic stem cells. Genes Dev 2009; 23: 1870–1875.
Mercher T, Cornejo MG, Sears C, Kindler T, Moore SA, Maillard I et al. Notch signaling specifies megakaryocyte development from hematopoietic stem cells. Cell Stem Cell 2008; 3: 314–326.
Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity 1999; 11: 299–308.
Blanpain C, Lowry WE, Pasolli HA, Fuchs E . Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev 2006; 20: 3022–3035.
Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR et al. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 2008; 3: 429–441.
Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS et al. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev 2002; 16: 846–858.
Kitamura T, Kitamura YI, Funahashi Y, Shawber CJ, Castrillon DH, Kollipara R et al. A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J Clin Invest 2007; 117: 2477–2485.
Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M . Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res 1994; 22: 965–971.
Nakae J, Kitamura T, Kitamura Y, Biggs WH 3rd, Arden KC, Accili D et al. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell 2003; 4: 119–129.
Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, Castrillon DH et al. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol 2008; 9: 1388–1398.
Sengupta A, Chakraborty S, Paik J, Yutzey KE, Evans-Anderson HJ . FoxO1 is required in endothelial but not myocardial cell lineages during cardiovascular development. Dev Dyn 2012; 241: 803–813.
Webb AE, Pollina EA, Vierbuchen T, Urbán N, Ucar D, Leeman DS et al. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep 2013; 4: 477–491.
Wu J, Xie X . Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol 2006; 7: R85.
Bredenkamp N, Seoighe C, Illing N . Comparative evolutionary analysis of the FoxG1 transcription factor from diverse vertebrates identifies conserved recognition sites for microRNA regulation. Dev Genes Evol 2007; 217: 227–233.
Bonev B, Stanley P, Papalopulu N . MicroRNA-9 Modulates Hes1 ultradian oscillations by forming a double-negative feedback loop. Cell Rep 2012; 2: 10–18.
Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J et al. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat Med 2011; 17: 961–967.
Jeon JH, Suh HN, Kim MO, Ryu JM, Han HJ . Glucosamine-induced OGT activation mediates glucose production through cleaved Notch1 and FoxO1, which coordinately contributed to the regulation of maintenance of self-renewal in mouse embryonic stem cells. Stem Cells Dev 2014; 23: 2067–2079.
Yeo H, Lyssiotis CA, Zhang Y, Ying H, Asara JM, Cantley LC et al. FoxO3 coordinates metabolic pathways to maintain redox balance in neural stem cells. EMBO J 2013; 32: 2589–2602.
Acknowledgements
We thank Ms Rujuta Narukar for performing IHC-RISH. This work was supported by the Ellison Medical Foundation (AG-NS-0646-10 to JP), and the Sidney Kimmel foundation (SKF-092 to JP) and National Institutes of Health Grant AG048284 (to JP).
Author contributions
D-YK, IH, FLM and J-HP performed experiments; D-YK, IH and J-HP designed experiments, analyzed data and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by Y Shi
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Kim, DY., Hwang, I., Muller, F. et al. Functional regulation of FoxO1 in neural stem cell differentiation. Cell Death Differ 22, 2034–2045 (2015). https://doi.org/10.1038/cdd.2015.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.123
This article is cited by
-
FoxO1 Controls Redox Regulation and Cellular Physiology of BV-2 Microglial Cells
Inflammation (2023)
-
Elucidating the Possible Role of FoxO in Depression
Neurochemical Research (2021)
-
Translating neural stem cells to neurons in the mammalian brain
Cell Death & Differentiation (2019)
-
Transcription factor Ptf1a in development, diseases and reprogramming
Cellular and Molecular Life Sciences (2019)
-
Cross-talk among HMGA1 and FoxO1 in control of nuclear insulin signaling
Scientific Reports (2018)
