
Abstract
Human cytomegalovirus (HCMV) is the most frequent viral cause of congenital defects and HCMV infection in immunocompromised patients may trigger devastating disease. Cytotoxic lymphocytes control HCMV by releasing granzymes towards virus-infected cells. In mice, granzyme M (GrM) has a physiological role in controlling murine CMV infection. However, the underlying mechanism remains poorly understood. In this study, we showed that human GrM was expressed by HCMV-specific CD8+ T cells both in latently infected healthy individuals and in transplant patients during primary HCMV infection. We identified host cell heterogeneous nuclear ribonucleoprotein K (hnRNP K) as a physiological GrM substrate. GrM most efficiently cleaved hnRNP K in the presence of RNA at multiple sites, thereby likely destroying hnRNP K function. Host cell hnRNP K was essential for HCMV replication not only by promoting viability of HCMV-infected cells but predominantly by regulating viral immediate-early 2 (IE2) protein levels. Furthermore, hnRNP K interacted with IE2 mRNA. Finally, GrM decreased IE2 protein expression in HCMV-infected cells. Our data suggest that targeting of hnRNP K by GrM contributes to the mechanism by which cytotoxic lymphocytes inhibit HCMV replication. This is the first evidence that cytotoxic lymphocytes target host cell proteins to control HCMV infections.
Similar content being viewed by others


Main
Human cytomegalovirus (HCMV) is a widespread β-herpesvirus that infects the majority of the population.1, 2 It is the most frequent viral cause of congenital defects.1, 2 Primary HCMV infection induces a lifelong latent infection in cells of the myeloid lineage with asymptomatic episodes of viral replication, which is controlled by a vigorous immune response. In the absence of an adequate immune response, however, HCMV can cause invasive disease with end-organ failure, morbidity, and mortality, for example, after allogeneic stem cell or solid organ transplantation, or in HIV-infected patients.1, 2 Treatment of HCMV infection is an urgent clinical problem. Vaccines against HCMV are not available and treatment by antiviral drugs has not definitively proven efficient to improve overall survival due to toxicity of antiviral agents and continual appearance of drug-resistant viruses.2 Cellular anti-HCMV immunotherapy is now emerging as a promising alternative approach.3 Understanding the mechanism by which our immune system counteracts HCMV infections could provide new possibilities for the development of novel (immuno)therapeutic antiviral strategies.
HCMV is the largest and most complex member of the human herpesvirus family. Activation of the HCMV major immediate-early promotor (MIEP) is central in controlling the lytic phase and essential for HCMV reactivation. During productive infection, the HCMV genome is expressed in a temporally coordinated cascade of transcriptional events that leads to the expression of immediate-early (IE), early, and late viral proteins.4
HCMV infection is controlled by cytotoxic lymphocytes, that is, antigen-specific CD8+ and CD4+ αβT cells, γδT cells, and NK cells.2, 5, 6 Cytotoxic lymphocytes produce interferon-γ to block HCMV replication, but most importantly release cytotoxic granules towards infected host cells.7, 8, 9, 10 These granules contain the pore-forming protein perforin and a family of structurally homologous serine proteases called granzymes. While perforin facilitates entry of granzymes into infected cells, granzymes are believed to be the death executors during the antiviral immune response. In humans, five granzymes exist (GrA, GrB, GrH, GrK, and GrM) that display distinct proteolytic substrate specificities.11 Although all five human granzymes are able to induce cell death, evidence is emerging that granzymes also use noncytotoxic strategies to control virus replication.12, 13, 14, 15, 16, 17
In mice, GrM has been shown to be important for murine cytomegalovirus (MCMV) clearance.18 We have recently discovered that human GrM can efficiently inhibit HCMV replication in vitro in the absence of host cell death.17 GrM efficiently cleaves HCMV phosphoprotein 71 (pp71) and completely abolishes its function to transactivate the MIEP,17 which is indispensable for effective HCMV replication.19 In this study, we addressed the possibility that GrM targets host cell proteins that HCMV hijacks for its own replication. We demonstrate that human GrM cleaves host cell protein heterogeneous nuclear ribonucleoprotein K (hnRNP K) that is essential for IE2 protein translation and HCMV replication. This may provide a novel mechanism by which cytotoxic lymphocytes mediate direct anti-HCMV activity.
Results
GrM is expressed in HCMV-specific CD8+ T cells
GrM knockout mice are more susceptible to MCMV infections, indicating a role of GrM in CMV clearance at least in mice.18 If GrM has a major role in controlling HCMV infections in humans, one would expect that GrM is present in HCMV-specific cytotoxic lymphocytes. Therefore, we analyzed GrM expression in CD8+ T cells of healthy human HCMV-seropositive individuals as well as in seronegative renal transplant recipients receiving a kidney from an HCMV-seropositive donor, followed by primary HCMV infections. First, pp65- and IE1-tetramer-specific CD8+ T cells – comprising both CD27− effector and CD27+CD45RA− memory phenotypes – of a healthy HCMV-seropositive individual were analyzed (Figure 1a). Both pp65- and IE1-tetramer-specific CD8+ T cells expressed increased levels of GrM as compared with naive CD8+ T cells (Figure 1b). A similar increase of GrM levels was observed in pp65-specific CD8+ T cells from a second HCMV-seropositive (latently infected) healthy individual (Supplementary Figure 1). Increased GrM protein levels were not restricted to HCMV-specific CD8+ T cells, as EBV- and influenza-specific CD8+ T cells also contained higher GrM protein levels (Supplementary Figure 1). Second, GrM protein expression was analyzed longitudinally in HCMV-specific CD8+ T cells from an HCMV-seronegative renal transplant recipient of an HCMV-seropositive donor, who experienced a primary HCMV infection (Figure 1c). IE1-specific effector CD8+ T cells appeared around the peak of the viral load and expressed increased levels of GrM as compared with naive CD8+ T cells. After cessation of the viral load, there was an increase in the percentage of CD8+CD27− T cells within the IE1-specific CD8+ T-cell compartment, which also coincided with an increase in GrM expression in these cells as compared with the naive CD8+ T-cell pool. The percentage of circulating GrM-expressing IE1-specific CD8+CD27− T cells reached a peak after 1 year post-transplantation and these cells were maintained for at least 5 years. Maintenance of CD8+CD27− T cells, known as vigilant resting effector cells, is characteristic for latent HCMV infections.6, 20 Identical results were obtained when pp65-specific CD8+ T cells were analyzed longitudinally in a second renal transplant patient (Supplementary Figure 2). Collectively, these data indicate for the first time that HCMV-reactive CD8+ T cells not only express GrM but also show elevated levels of GrM as compared with naive CD8+ T cells, both in HCMV-seropositive healthy individuals and in the peripheral blood of seronegative renal transplant recipients in response to primary HCMV infection. Furthermore, GrM-positive HCMV-reactive CD8+ T cells are maintained in the blood during latency. This supports the concept of an active antiviral role of GrM in HCMV infection and HCMV reactivation in humans.

GrM is expressed in HCMV-specific CD8+ T cells. (a) FACS plots show the expression of CD8- and HCMV-specific pp65 and IE1 tetramers on CD3+ T cells (upper row) or the expression of CD27 and CD45RA (bottom row) on total CD3+CD8+ T cells (left) or HCMV-specific CD3+CD8+ T cells (middle, right). (b) FACS histogram visualizes the relative intracellular GrM protein levels in naive CD3+CD8+ T cells (filled gray) and HCMV-specific CD3+CD8+ T cells (lines). (c) PBMCs were isolated from a renal transplant patient pre- and post-transplantation. Graph depicts kinetically the absolute numbers of IE1-specific CD3+CD8+ cells (line) and HCMV viral load (filled gray). FACS plots show the expression of CD8 and IE1 tetramers on CD3+ T cells (upper row), and CD27 and CD45RA on total or IE1-specific CD3+CD8+ T cells (middle rows). Histograms (bottom row) visualize the relative intracellular GrM protein levels in naive CD3+CD8+ T cells (filled gray) and IE1-specific CD3+CD8+ T cells (line)
GrM targets host cell protein hnRNP K
To investigate the mechanism by which GrM inhibits HCMV replication, we addressed the possibility that GrM targets host cell proteins that are required for efficient HCMV replication. Recently, we have employed multiple proteomic approaches to identify potential macromolecular cellular substrates of GrM.21 Both gel-based (2D-DIGE) (Figure 2a) and complementary positional (cofradic) proteomics have identified hnRNP K as a potential GrM substrate.21 We focused on hnRNP K because it has been shown to have a role in herpes simplex virus-1 (HSV-1) and hepatitis B virus replication.22, 23 To validate hnRNP K cleavage by GrM, cell lysates were incubated with increasing concentrations of GrM or catalytically inactive control GrM-SA. Cleavage of hnRNP K by GrM was apparent with a decrease of full-length hnRNP K (∼62 kDa) and the appearance of a clear ∼45 kDa cleavage product and several smaller cleavage fragments, using an antibody directed against the mid-region of hnRNP K (Figure 2b). GrM also cleaved purified recombinant His-hnRNP K with the appearance of the ∼45 kDa fragment (Figure 2c), indicating that GrM directly cleaves hnRNP K. Using complementary positional proteomics, we identified at least three GrM cleavage sites in hnRNP K, that is, Leu125, Leu133, and Met359 (Figure 2d).21 We mutated Leu125, Leu133, Met359, or combinations into Ala and we evaluated these mutants in cellular lysates for GrM cleavage using immunoblotting with an anti-His antibody to detect the hnRNP K N terminus (Figure 2e). Cleavage of wild-type hnRNP K resulted in a decrease of full-length hnRNP K and the appearance of a ∼20 kDa N-terminal cleavage fragment. The hnRNP KL133A single mutant, hnRNP KL125A/L133A double mutant, and hnRNP KL125A/L133A/M359A triple mutant showed no ∼20 kDa cleavage fragment, but did show an N-terminal ∼45 kDa cleavage fragment upon GrM treatment. This indicates that Leu133 is an important GrM cleavage site in hnRNP K. However, GrM was still able to cleave all hnRNP K mutants, indicating that other GrM cleavage sites exist in hnRNP K other than Leu125, Leu133, and Met359. First, GrM cleavage of hnRNP KL133A, hnRNP KL125A/L133A, and hnRNP KL125A/L133A/M359A mutants showed a cleavage fragment that migrated 1–2 kDa below the ∼20 kDa band (Figure 2e), indicating at least one additional GrM cleavage site N-terminal of Leu125 (Figure 2d). Second, the hnRNP KL125A/L133A/M359A triple mutant still showed accumulation of the N-terminal ∼45 kDa cleavage fragment, indicating at least one additional GrM cleavage site in close proximity to Met359 (Figure 2e). Thus, hnRNP K proteolysis by GrM is complex in that GrM cleaves hnRNP K after at least five independent sites that are clustered in two apparent proteolysis-sensitive hot spots (Figure 2d). Next, human foreskin fibroblasts (HFFs) were incubated with purified GrM or GrM-SA in the presence or absence of perforin-analog streptolysin O (SLO). Whereas hnRNP K remained unaffected in all control settings, it was cleaved when cells were incubated with both GrM and SLO with the concomitant appearance of at least the ∼45 kDa hnRNP K cleavage fragment (Figure 2f). GrM induced minor cell death in HFFs (Figure 2g), which was not affected by the pan-caspase inhibitor zVAD-fmk and virtually no pro-caspase 3 activation was observed (Figures 2g and h). GrB did trigger efficient cell death in HFFs with pro-caspase-3 activation (Figures 2g and h). HnRNP K cleavage fragments were also observed when living HFFs were challenged with living lymphokine-activated killer (LAK) cells (Figure 2i). No decrease in full-length hnRNP K was observed because of the presence of endogenous hnRNP K in LAK cells that were present in analyzed cellular lysates. Taken together, these data indicate that hnRNP K is directly cleaved by GrM and targeted during cytotoxic lymphocyte-mediated attack.

GrM cleaves host cell protein hnRNP K. (a) HeLa cell lysates were incubated with GrM (1 μM) or GrM-SA (1 μM) for 1 h at 37°C and then subjected to 2D-DIGE. A representative protein spot that is identified by mass spectrometry as a cleavage fragment of hnRNP K is depicted. (b) Jurkat lysates (5 μg) were incubated with increasing concentrations of GrM or GrM-SA (300 nM) for 4 h at 37°C and immunoblotted for hnRNP K. (c) Purified His-hnRNP K (1.5 μg) was incubated with increasing concentrations of GrM or GrM-SA (300 nM) for 16 h at 37°C and subjected to SDS-PAGE. Proteins were stained with InstantBlue. (d) Schematic overview of GrM cleavage sites within hnRNP K (NLS, nuclear localization signal; KH, K homology domain; KI, K interactive region; KNS, K nuclear shuttling signal; RGG, arginine–glycine–glycine). (e) HeLa cells were transfected with His-tagged hnRNP K wild-type (wt) or mutant expression plasmids and lysed 2 days post-transfection. Expression of recombinant proteins was determined by subjecting untreated lysates to immunoblotting using anti-6 × His antibody (bottom panel). Lysates were incubated with 300 nM GrM or left untreated for 4 h at 37°C and immunoblotted using anti-6 × His antibody (upper panel) (NS, nonspecific). (f) HFFs were incubated with GrM (2.8 μM) or GrM-SA (2.8 μM) in the presence or absence of SLO (2 μg/ml) for 16 h at 37°C. Lysates were immunoblotted for hnRNP K. (g) HFFs were incubated with GrM (1 μM), GrM-SA (1 μM), GrB (100 nM), or left untreated in the presence or absence of SLO (1 μg/ml) with or without zVAD-fmk (100 μM) for 16 h at 37°C. Cell viability was assessed by staining cells with annexin-V-FLUOS (fluorescein) and PI followed by flow cytometry analysis. Annexin-V and PI double-negative cells were considered to be viable. Bars represent the mean±S.D. of three independent experiments. (h) HFFs were incubated with GrM (1 μM), GrM-SA (1 μM), GrB (100 nM), or left untreated in the presence or absence of SLO (1 μg/ml) with or without zVAD-fmk (100 μM) for 16 h at 37°C. Cells were lysed and immunoblotted using antibodies against active caspase-3 (D175). (i) HFFs were challenged with increasing effector:target (E:T) ratios of LAK cells for 4 h at 37°C. Lysates were subjected to immunoblotting using hnRNP K antibodies. Data depicted are representative of at least two independent experiments
RNA promotes GrM-mediated cleavage of hnRNP K
Cleavage of hnRNP K by GrM was more efficient in cell lysates (Figure 2b) as compared with recombinant purified hnRNP K (Figure 2c), suggesting that cellular factors contribute to this proteolysis. Because hnRNP K is an RNA-interacting protein,24, 25 the effects of RNA on GrM-mediated hnRNP K cleavage were examined. Cell lysates were preincubated with or without RNase, followed by GrM treatment. Immunoblot analysis showed that hnRNP K was less efficiently cleaved by GrM when lysates were preincubated with RNase, whereas RNase had no effect on the cleavage of GrM substrate α-tubulin (Figure 3a). RNase did not affect the proteolytic activity of GrM itself as determined by the small synthetic chromogenic substrate AAPL-pNA (data not shown). Consistent with this finding, purified His-hnRNP K was more efficiently cleaved by GrM when preincubated with RNA (Figure 3b). These data suggest that GrM cleaves hnRNP K more efficiently in its functional RNA-bound conformation.

GrM cleaves hnRNP K more efficiently in the presence of RNA. (a) Jurkat cell lysates were pretreated with RNase (100 μg/ml) or left untreated for 30 min at 37°C, followed by incubations with increasing concentrations of GrM or GrM-SA (300 nM) for 4 h at 37°C and immunoblotted for hnRNP K or α-tubulin (upper panel). Band intensities were quantified and depicted in graphs (lower panel). (b) Purified His-hnRNP K (1.5 μg) was pretreated with RNA (100 ng) or left untreated, followed by incubations with increasing concentrations of GrM or GrM-SA (300 nM) for 16 h at 37°C. Samples were subjected to SDS-PAGE and gels were stained with InstantBlue (upper panel). Band intensities were quantified and depicted in graphs (lower panel). Data depicted are representative of at least two independent experiments
HnRNP K is required for efficient HCMV replication
The role of hnRNP K during HCMV replication is unknown. To investigate this, we downregulated hnRNP K protein levels in HFFs (Figure 4a), infected these cells with HCMV, and viral replication was kinetically determined by quantifying HCMV viral load in supernatants. The viral load was 10- to 17-fold decreased in hnRNP K-deficient HFFs as compared with control HFFs (Figure 4b), indicating that hnRNP K is required for efficient HCMV replication. This dramatic decrease in viral load in hnRNP K-deficient HFFs was accompanied with a slow decrease in cell viability (up to two- to threefold) (Figure 4c). Quite remarkable is the observation that cell viability is only decreased in HCMV-infected cells but not in uninfected hnRNP K-deficient cells, suggesting that hnRNP K is important in coping with cell survival after stress induced by HCMV infection. As there is a linear correlation between the number of cells and viral load (Figure 4d), the observed two- to threefold decrease in cell viability does not explain the major decrease in viral load (10- to 17-fold) when hnRNP K protein levels are downregulated. These data indicate that hnRNP K is required for efficient HCMV replication, predominantly in a host cell death-independent manner.

HnRNP K is essential for HCMV replication. (a) HFFs were transduced with hnRNP K-specific or control shRNA and lysed at different time points. Lysates were subjected to immunoblotting using antibodies against hnRNP K or α-tubulin (loading control). (b) HnRNP K-deficient and control HFFs were infected with HCMV and viral load in supernatants at different time points was assessed by quantitative PCR. (c) Cell viability of uninfected or HCMV-infected transduced HFFs was determined by WST-1 assay and relative cell viability of uninfected control shRNA-treated cells at 0 h.p.i. was set to 1. (d) HFFs were seeded at different cell densities and infected with HCMV at a multiplicity of 1.0 plaque-forming unit per cell. Viral load in supernatants at 96 h.p.i. was assessed by quantitative PCR and a linear regression curve was plotted. Data depicted represent the mean±S.D. of triplicates
HnRNP K regulates IE2 translation
The temporally controlled expression of HCMV proteins during viral replication allows us to determine at which stage of the HCMV viral life cycle hnRNP K is involved. Lysates of HCMV-infected hnRNP K-deficient and control HFFs were immunoblotted for several HCMV proteins. Interestingly, the 86 kDa IE2 but not the 72 kDa IE1 protein level was dramatically reduced in HCMV-infected HFFs when hnRNP K was downregulated (Figure 5a). Furthermore, a slight if any reduction in early HCMV proteins pp65 and pp71, as well as a dramatic reduction of the late HCMV protein pp28, were observed in hnRNP K-deficient HFFs. However, these latter effects likely are the functional consequence of decreased IE2 protein levels. These data indicate that hnRNP K has a dominant role during the initial IE phase of HCMV replication by regulating IE2 but not IE1 protein expression.

HnRNP K regulates IE2 protein expression. (a) HFFs were transduced with hnRNP K-specific or control shRNA, infected with HCMV, and lysed at different time points. Lysates were subjected to immunoblotting using antibodies against IE1/2, pp71, pp65, pp28, hnRNP K, and α-tubulin. (b) HeLa cells were co-transfected with pGL3-SV40 (50 ng), pRL-CMV (10 ng), complemented with pCGN-71 vector (1 μg) or pSG5-His-hnRNP K, and pcDNA3.1+ vector (1 μg total DNA). Cells were lysed at 48 h post-transfection, and luciferase activity was assessed. Relative luciferase activity is represented as fold CMV promoter (MIEP) induction. Lysates used in the luciferase reporter assay were subjected to immunoblotting using an anti-pp71 or anti-6 × His antibody. (c) HCMV-infected hnRNP K-deficient and control HFFs were lysed at 8 h.p.i. and mRNA was isolated and subjected to RT-PCR and quantitative PCR using IE2 and GAPDH specific primers and probes. Relative IE2 mRNA levels were normalized for GAPDH mRNA levels and the mean from control cells was set at 1. (d) HCMV-infected transduced HFFs were lysed at different time points and immunoblotted for the C terminus of IE2 or α-tubulin. (e) HCMV-infected hnRNP K-deficient and control HFFs were treated with MG132 (250 nM) or left untreated at 48 h.p.i. and lysed at 72 h.p.i. Lysates were subjected to immunoblotting using antibodies against IE2, hnRNP K, α-tubulin, and ubiquitin. Data depicted are representative of triplicate experiments. (f) HCMV-infected HFFs were lysed at 72 h.p.i. and incubated with His-hnRNP K or His-β-tubulin as a negative control for 16 h at 4°C. Immunoprecipitation of His-tagged proteins was followed by isolation of mRNA, RT-PCR, and quantitative PCR using specific primers and probes against UL123 (IE1), UL122 (IE2), UL54, and GAPDH. The mean mRNA levels from His-β-tubulin control samples for each mRNA were set to 1 and all samples were normalized for GAPDH mRNA levels. Bars represent the mean±S.D. of triplicates. (Inset) Samples after immunoprecipitation were subjected to immunoblotting using anti-6 × His antibody
In view of the importance of IE2 as an absolutely indispensable protein in HCMV replication,26, 27 we further examined the mechanism by which hnRNP K regulates IE2 protein levels. First, we investigated the transcriptional activity of hnRNP K towards the MIEP, which regulates IE2 gene (UL122) expression. In contrast to control pp71, no transcriptional activity of hnRNP K towards the MIEP was detected in a luciferase MIEP reporter assay (Figure 5b). In line with this observation, IE2 mRNA levels were similar between hnRNP K-deficient and control HFFs after HCMV infection at 8 (Figure 5c), 24, and 72 h.p.i. (data not shown). These data indicate that hnRNP K does not regulate IE2 protein levels via transcription of the UL122 gene or promoting IE2 mRNA stability. Second, besides the 86 kDa IE2 protein, there are two alternative (unspliced) IE2 variants of 60 and 40 kDa, each regulated by its own promoter.28, 29 Protein levels of all three IE2 protein variants were decreased when hnRNP K was downregulated (Figure 5d), further suggesting that hnRNP K regulates IE2 protein levels at the post-transcriptional level. Third, we investigated whether hnRNP K is required for IE2 protein stability by using the chemical compound MG132, a proteasome inhibitor that does not affect IE gene expression.30 MG132 failed to restore IE2 protein levels in HCMV-infected hnRNP K-deficient HFFs (Figure 5e), indicating that hnRNP K does not protect IE2 protein from proteasomal degradation. Finally, as hnRNP K is an RNA-binding protein, we addressed the possibility that hnRNP K interacts with IE2 mRNA. Purified His-tagged hnRNP K or unrelated control His-β-tubulin was incubated with lysates of HCMV-infected HFFs followed by pull-down of His-tagged proteins (inset of Figure 5f). Interestingly, hnRNP K pull-down resulted in a clear enrichment of IE2 mRNA, whereas no enrichment was observed for viral mRNAs IE1 and UL54 and host cell mRNA glyceraldehyde 3-phosphate dehydrogenase (GAPDH), or when His-β-tubulin was used (Figure 5f). These data suggest that hnRNP K specifically binds to IE2 mRNA, pointing to a role of hnRNP K during IE2 translation.
GrM reduces IE2 protein expression during HCMV infection
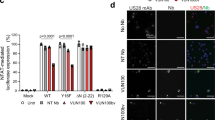
The diverse functions of hnRNP K require both nuclear and cytoplasmic localization and bidirectional shuttling. Viruses can enrich hnRNP K in a specific subcellular compartment in favor of the replication of the virus.22, 31 In uninfected HFFs, hnRNP K was predominantly localized in the cytoplasm (Figure 6a, upper panels). After infection with HCMV, HFFs showed typical rounded morphology and hnRNP K predominantly remained cytoplasmic (Figure 6a, bottom panels). Thus, during HCMV infection hnRNP K is located at the site where IE2 mRNA translation occurs. Previously, we demonstrated that GrM can inhibit HCMV replication independent of host cell death.17 Our findings that GrM cleaved hnRNP K (Figure 2), that hnRNP K interacted with IE2 mRNA (Figure 5g), and that hnRNP K downregulation dramatically reduced IE2 protein expression during HCMV infection (Figure 5a) prompted us to investigate whether GrM-treatment of HCMV-infected fibroblasts affects IE2 protein expression. Indeed, immunoblot analysis showed a clear decrease in IE2 protein levels when HCMV-infected fibroblasts were treated with GrM/SLO as compared with control GrM-SA/SLO treatment (Figure 6b). The decrease in IE2 protein levels correlated with decreased viral loads (Figure 6c) and little cell death (Figure 6d) in GrM-treated HCMV-infected fibroblasts. These data indicate that GrM reduces IE2 protein expression to inhibit HCMV replication.

HnRNP K localization and GrM-mediated decrease of IE2 protein expression. (a) Immunofluoresence images of uninfected (upper panel) and HCMV-infected (lower panel) HFFs at 72 h.p.i. Nuclei are stained blue and hnRNP K is visualized in green. Bars, 50 μm. (b) HCMV-infected fibroblasts were treated with GrM-SA (500 nM) or GrM (500 nM) in the presence of SLO (1 μg/ml) at 2 h.p.i. and cells were lysed at indicated time points. Lysates were subjected to immunoblotting using antibodies against IE2 or β-tubulin. Data depicted are representative of at least two independent experiments performed in triplicates. (c) HCMV-infected fibroblasts were incubated with GrM (500 nM) or GrM-SA (500 nM) in the presence of SLO (1 μg/ml) at 2 h.p.i. HCMV viral load in supernatants at 72 h.p.i. was assessed by quantitative PCR. Bars represent the mean±S.D. of triplicates. (d) HCMV-infected fibroblasts were incubated with GrM (500 nM) or GrM-SA (500 nM) in the presence of SLO (1 μg/ml) at 2 h.p.i. Relative cell viability at 24 h.p.i. was assessed by WST-1 assay. Bars represent the mean±S.D. of triplicates
Discussion
The granule exocytosis pathway is a crucial mechanism that cytotoxic lymphocytes use to control virus infections. The physiological importance of GrM as an antiviral mediator has been demonstrated in GrM-deficient mice, which are more susceptible to MCMV infections.18 However, both murine GrM and MCMV have been largely diverged from their human counterparts,1, 21 emphasizing that caution is essential when extrapolating data from mice to humans. In this study, we demonstrated for the first time that human GrM is expressed in HCMV-specific CD8+ T cells after primary infection and during latency (Figure 1). HCMV-specific CD8+CD27− T cells, the so-called vigilant resting effector T cells, are proposed to act in response to reactivation and to maintain HCMV latency.6, 20 Interestingly, accumulation of these vigilant resting effector cells after primary HCMV infection coincided with increased GrM protein expression in these HCMV-specific CD8+ T cells (Figure 1). This supports a model in which GrM actively mediates an antiviral function in HCMV infections in vivo. In this study, we demonstrated that GrM cleaved hnRNP K and that hnRNP K was required for efficient HCMV replication by promoting cell viability in virus-infected cells and more importantly by regulating IE2 protein expression (Figures 2, 3, 4, 5). This provides the first evidence that GrM targets a host cell protein that is hijacked by HCMV and that is critical for its replication. Apart from HCMV, GrM protein levels were also elevated in EBV- and influenza-specific CD8+ T cells (Supplementary Figure 1), pointing to an antiviral role of GrM in a broad range of viral infections.
A previous study has shown that hnRNP K is important for the egress of HSV-1.22 In HCMV infection, however, hnRNP K appears to have a different role. Knockdown of hnRNP K not only limited HCMV replication but also decreased cell viability in HCMV-infected but not in uninfected fibroblasts (Figure 4). The latter finding suggests that hnRNP K is required for cell survival only following HCMV infection. However, host cell death only partly explains the dramatic reduction in HCMV replication in hnRNP K-deficient fibroblasts. More importantly, decreased viral load was accompanied with markedly reduced IE2 but not IE1 protein levels in hnRNP K-deficient HCMV-infected fibroblasts (Figure 5). IE2 is absolutely indispensable for HCMV replication.26, 27 Therefore, we explored how hnRNP K regulates IE2 protein levels. As a multifunctional protein, hnRNP K is involved in many biological processes, including transcription, mRNA splicing, translation, and protein signaling.24 The decrease of IE2 protein levels in hnRNP K-deficient fibroblasts is most likely the result of decreased IE2 mRNA translation, because hnRNP K did not affect transcription of the IE2-encoding gene nor IE2 mRNA and protein stability, but rather bound IE2 mRNA (Figure 5). This is compatible with the previous observation that hnRNP K binds to and thereby regulates translation of the mRNA of cytoskeleton genes and myelin basic protein.32, 33
Binding of hnRNP K to RNA is mediated by three KH domains within hnRNP K that act synergistically to provide both high-affinity binding and specificity (Figure 2).24, 25 Hence, loss of only a single KH domain dramatically reduces the RNA binding capacity of hnRNP K.25 GrM cleaved hnRNP K at least at five independent sites, including Leu125, Leu133, and Met359, that are located in two apparent proteolysis-sensitive hot spots, thereby dissecting the functional KH domains (Figure 2). This strongly suggests that GrM abrogates the ability of hnRNP K to bind to IE2 mRNA, resulting in reduced IE2 protein expression and reduced HCMV replication. It should be mentioned that hnRNP K was more efficiently cleaved by GrM in the presence of RNA (Figure 3). This suggests that the conformational change of hnRNP K after binding to RNA allows for efficient proteolysis by GrM or that RNA facilitates the interaction between hnRNP K and GrM. In this model, the functional interaction between hnRNP K protein and IE2 mRNA promotes the antiviral activity of GrM to cleave hnRNP K.
Previously, we have demonstrated that GrM cleaves and inactivates the HCMV tegument protein pp71,17 which is essential for IE protein expression and effective HCMV replication.19 In this study, we demonstrated that GrM also targets a host cell substrate (hnRNP K) that has a major role in HCMV replication and is necessary for efficient IE2 protein expression (Figures 5 and 6). Apparently, cytotoxic lymphocytes and GrM target the initial IE machinery that HCMV requires for both lytic replication and reactivation from latency. As viruses highly depend on the transcription/translation machinery of the host cell, it could be possible that other hnRNPs also have a role in viral infections and are also cleaved by GrM. Indeed, several proteomic screens have identified hnRNP A1, A2/B1, C1/C2, E1, M, and U as potential GrM substrates.21, 34 Interestingly, many members of the hnRNP protein family have also been identified as potential substrates of other granzymes than GrM.35, 36, 37, 38 Further studies are required to validate these findings in the context of an antiviral immune response.
It remains an intriguing question why GrM targets viral pp7117 and host cell hnRNP K to attack the initial IE events of HCMV replication rather than just killing HCMV-infected cells. Consistent with our findings, IE1- and pp65-specific CD4+ T cells and CD8+ T cells can recognize and kill IE1 and pp65 peptide-loaded target cells, but fail to induce cell death in HCMV-infected target cells after recognition and degranulation, whereas HCMV replication is efficiently inhibited39, 40 (Dr. MR Wills, personal communication (Department of Medicine, University of Cambridge, Cambridge, UK)). This apparently noncytotoxic mechanism to block HCMV replication might be an important pathway to prevent HCMV reactivation from latency and to preserve latently-infected CD34+ hematopoietic progenitor cells. This model is compatible with the recent finding that antigen-specific CD8+ T cells recognize HSV-1 latently infected ganglion cells, release GrB towards these cells, and inhibit HSV-1 reactivation without inducing cell death.13
Materials and Methods
Subjects
Two healthy HCMV-seropositive volunteers (HLA-B7+, HLA-B8+, and/or HLA-A2+) and two HCMV-seronegative renal transplant recipients who received a kidney from an HCMV-seropositive donor (HLA-B8+ and HLA-A2+) were included in this study. Both patients received immunosuppressive therapy (consisting of CD25mAb induction therapy, followed by prednisolone, mycophenolate mofetil, and tacrolimus), experienced primary HCMV infection, and had no acute cellular rejection episodes. Patients gave written informed consent, and the study was approved by the medical ethics committee of the Academic Medical Center (Amsterdam, The Netherlands). Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood and subsequently cryopreserved until flow cytometry analysis. HCMV viral load was determined in EDTA-treated blood by quantitative PCR.
Flow cytometry stainings
PBMCs were washed in phosphate-buffered saline (PBS) containing 0.01% NaN3 and 0.5% bovine serum albumin. PBMCs (2 × 106) were incubated with an appropriate concentration of APC-conjugated tetrameric complexes for 30 min at 4°C. For the analysis of surface marker expression, cells were incubated with fluorescence-labeled monoclonal antibodies for 30 min at 4°C at concentrations according to the manufacturer’s instructions. After tetramer and surface marker stainings, cells were fixed (FACS Lysing Solution; BD Biosciences, Franklin Lakes, NJ, USA) and permeabilized (FACS Permeabilizing Solution 2; BD Biosciences), followed by incubation with Alexa Fluor 488-conjugated anti-GrM (1.2 μg/ml) for 30 min at 4°C to stain intracellular GrM. Live/Dead Fixable Red Dead Cell Stain Kit (Life Technologies, Grand Island, NY, USA) was used to exclude dead cells from all analysis. Cells were measured on a LSR-Fortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star Inc., Ashland, OR, USA). APC-conjugated tetrameric complexes were obtained from Sanquin (Amsterdam, The Netherlands): HLA-B0702 tetramer loaded with the HCMV pp65-derived TPRVTGGGAM peptide, HLA-B0801 tetramer loaded with the HCMV IE1-derived QIKVRVDMV peptide, HLA-B0801 tetramer loaded with the HCMV IE-1-derived ELRRKMMYM peptide, HLA-A0201 tetramer loaded with the HCMV pp65-derived NLVPMVATV peptide, HLA-B0801 tetramer loaded with the EBV EBNA3A-derived FLRGRAYGL peptide, HLA-B0801 tetramer loaded with the EBV BZLF1-derived RAKFKQLL peptide, and HLA-A0201 tetramer loaded with the influenza matrix protein (FLU MP)-derived GILGFVFTL peptide. The following antibodies were obtained from commercial sources: CD3-V500 and CD8-V450 (BD Biosciences); CD45RA eFluor 605NC (eBioscience Inc., San Diego, CA, USA); and CD27 APC-Alexa Fluor 750 (Life Technologies). Alexa Fluor 488-conjugated anti-GrM (clone 4B2G4) was generated as described previously.41
Cell culture and cell-free protein lysates
Cells were cultured in a 5% CO2 atmosphere at 37°C. HFFs, HEK293T, and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). Jurkat cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum, sodium bicarbonate (Life Technologies), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). Cell-free protein lysates were generated by washing cells three times in PBS and subsequent lysis by three cycles of freeze-thawing in PBS. Samples were centrifuged at 18 000 × g for 10 min at 4°C and protein concentration was determined by the method of Bradford (Bio-Rad, Hercules, CA, USA).
Antibodies and reagents
HCMV-purified virus, strain AD169, was purchased from Advanced Biotechnologies Inc. (Columbia, MD, USA). Primary antibodies to the following proteins were obtained from commercial sources: cleaved caspase-3 (rabbit polycolonal, D175; Cell Signaling, Boston, MA, USA), hnRNP K (rabbit polyclonal, ab18195; Abcam, Cambridge, MA, USA), IE1/2 (mouse monoclonal; Argene, Verniolle, France), IE2 (mouse monoclonal, 5A8.2; Merck Millipore, Billerica, MA, USA), α-tubulin (mouse monoclonal, B-5-1-2; Sigma-Aldrich, St. Louis, MO, USA), β-tubulin (mouse monoclonal, TUB 2.1; Sigma-Aldrich), pp28 (mouse monoclonal, 5C3; Santa Cruz Biotechnology, Santa Cruz, CA, USA), pp65 (mouse monoclonal, 1-L-11; Santa Cruz Biotechnology), pp71 (goat polyclonal, vC-20; Santa Cruz Biotechnology), HRP-conjugated 6xHis (mouse monoclonal; BD Biosciences), and HRP-conjugated anti-ubiquitin (mouse monoclonal; Enzo Life Sciences, Farmingdale, NY, USA). Mouse monoclonal antibodies against pp71 (2H10-9 and 10G11) were kindly provided by Dr. T Shenk (Princeton University, Princeton, NJ, USA). Secondary HRP-conjugated goat anti-mouse and goat anti-rabbit antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). HRP-conjugated rabbit anti-goat was obtained from Dako (Glostrup, Denmark). Alexa Fluor 488-conjugated goat anti-rabbit and DAPI were purchased from Life Technologies and Roche (Basel, Switzerland), respectively. Immunoblotted proteins were detected using the Enhanced Chemiluminescence detection system (GE Healthcare, Piscataway, NJ, USA) and ChemiDoc XRS+ (Bio-Rad). InstantBlue stain was purchased from Expedeon (Harston, Cambridge, UK) and MG132 was obtained from Sigma-Aldrich. Pan-caspase inhibtor zVAD-fmk was obtained from Enzo Life Sciences. RNase A was purchased from Roche and dissolved in granzyme activity buffer (20 mM Tris (pH 7.4), 150 mM NaCl). Total RNA was isolated using Trizol according to the manufacturers’ protocol (Life Technologies).
HnRNP K mutagenesis
The hnRNP K mutants (L125A, L133A, M359A, L125A/L133A, and L125A/L133A/M359A) were generated using the QuikChange Site-Directed Mutagenesis (Agilent Technologies, Santa Clara, CA, USA) using the pSG5-His-hnRNP K expression plasmid (a kind gift from Dr. A Ostareck-Lederer, Martin Luther University Halle-Wittenberg, Halle, Germany) as template. DNA sequences of all constructs were verified by sequencing.
Purified recombinant proteins
The cDNA encoding mature human GrM was amplified and cloned into yeast expression vector pPIC9 (Life Technologies). Using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies), the Ser195 residue in the catalytic center of GrM was replaced by an Ala residue, resulting in the catalytically inactive GrM variant (GrM-SA) that we have used as a control in this study. GrM and GrM-SA proteins were expressed and purified as described previously.34 Briefly, Pichia pastoris GS115 cells were transformed with the pPIC9-granzyme expression plasmids and granzymes were expressed for 72 h in conditioned media, according to the manufacturers’ protocol (Life Technologies). GrM and GrM-SA were purified to homogeneity using an SP-Sepharose column (GE Healthcare) with 1 M NaCl for elution. GrM protein fractions were dialyzed against 50 mM Tris (pH 7.4) and 150 mM NaCl, and stored at −80°C. Recombinant GrM and GrM-SA proteins were pure (>98%) as determined by SDS-PAGE and active as measured by hydrolysis of synthetic chromogenic leucine substrates Suc-AAPL-pNA (Bachem, Bubendorf, Switzerland) and Suc-KVPL-pNA (GL Biochem, Shanghai, China), and known macromolecular substrates (data not shown).
BL21-CodonPlus competent Escherichia coli cells (Agilent Technologies) were transformed with the pET16-b-His-hnRNP K expression plasmid (a kind gift from Dr. A Ostareck-Lederer, Martin Luther University Halle-Wittenberg) or pQE-80L-His-β-tubulin and cultured in LB medium. At an OD600 between 0.4 and 1.0, cells were stimulated with 0.4 mM IPTG and incubated for 3 h at 37°C at 225 r.p.m. Cells were harvested and lysed by sonification. N-terminal His-tagged hnRNP K and β-tubulin were purified to homogeneity using Talon metal ion chelate affinity chromatography (Clontech, Mountain View, CA, USA) with 125 mM imidazole in PBS for elution and stored at 4°C.
2D-DIGE
2D-DIGE was performed as described previously.21 Briefly, cell-free protein extracts of HeLa cells (100 μg) were incubated with 1 μM GrM or GrM-SA for 1 h at 37°C. Samples were precipitated, solubilized, labeled, rehydrated into immobilized pH gradient strips, and isoelectrically focused. Strips were reduced and overlaid on a 12% SDS-PAGE gel. Images were acquired on a Typhoon 9410 scanner (GE Healthcare). Each condition was performed at least five times and a dye swab was included to exclude preferentially labeled proteins from the analysis. Relative quantification of matched gel features was performed using the Decyder DIA and BVA software (GE Healthcare). For inter-gel analyses, the internal standard method was used.
GrM cleavage assay in living HFFs
HFFs (2 × 104) were seeded in 96-well plates, washed twice with serum-free DMEM, and incubated with the perforin-analog SLO (Aalto Bio Reagents, Dublin, Ireland) and purified granzyme in a final volume of 30 μl for 30 min at 37°C. Then, supernatant was removed and cells were cultured in 100 μl DMEM containing 10% fetal calf serum for 16 h. For analyzing caspase activation and cell death, HFFs were treated with zVAD-fmk (100 μM) or vehicle only (DMSO) for 30 min at 37°C before granzyme treatment, and zVAD-fmk (100 μM) or DMSO was added after granzyme treatment. Cells were washed two times with PBS and directly lysed in reducing sample buffer (50 μl) before immunoblotting. To measure GrM-mediated cell death, cells were trypsinized, harvested, and washed twice with PBS. Cells were pelleted and stained with annexin-V-FLUOS (Roche) and PI in 100 μl annexin-V binding buffer (140 mM NaCl, 4 mM KCl, 0.75 mM MgCl2, 1.5 mM CaCl2, and 10 mM Hepes (pH 7.4)) for 15 min at room temperature, after which 500 μl annexin-V binding buffer was added. Fluorescence was measured by FACS on a FACS Calibur (BD Biosciences) and data were analyzed with CellQuest Pro Software (BD Biosciences).
shRNA-mediated knockdown of hnRNP K protein expression
Lentiviral particles were produced using third-generation lentivirus vectors. HEK293T cells were transfected with pCMV-VSV-G, pMDLg-RRE, pRSV-REV (kind gifts from Dr. EJHJ Wiertz, University Medical Center, Utrecht, The Netherlands) and hnRNP K shRNA plasmid (sc-38282-SH; Santa Cruz Biotechnology) or control shRNA plasmid (sc-108060; Santa Cruz Biotechnology) using polyethylenimine (PEI; Polysciences Inc., Warrington, PA, USA) in a 3 : 1 ratio with total DNA. Supernatant with viral particles was harvested at 48 and 72 h.p.i., filtered (0.45 μm), and stored at −80°C. HFFs were pretreated with polybrene (10 μg/ml) for 20 min and transduced four times for 2 h.
HCMV infection model
HFFs (2 × 104) were seeded in 96-well plates and infected with purified HCMV at a multiplicity of 1.0 plaque-forming unit per cell for 2 h at 37°C. Cells were washed three times and cultured in 100 μl medium. For analysis of protein levels, cells were washed two times with PBS and directly lysed in reducing sample buffer before immunoblotting. To measure viral load by quantitative PCR, supernatant was collected at the indicated time points and HCMV was inactivated for 30 min at 60°C. Cell viability was measured using a WST-1 assay (Roche) according to the manufacturer’s protocol.
mRNA isolation, reverse transcriptase PCR, and quantitative PCR
For direct mRNA isolation, cells were lysed (100 mM Tris (pH 7.5), 500 mM LiCl, 10 mM EDTA, 5 mM EDTA, 1% SDS) and treated with proteinase K for 1 h at 56°C. For mRNA isolation after hnRNP K immunoprecipitation, cells were lysed (100 mM Tris (pH 7.5), 500 mM LiCl, 10 mM EDTA, 5 mM EDTA, 1% Triton X-100, supplemented with a protease cocktail inhibitor from Roche) and incubated with purified His-tagged hnRNP K or His-tagged β-tubulin overnight at 4°C. His-tagged proteins were immunoprecipitated using Talon metal ion chelate affinity chromatography (Clontech) and beads were washed five times, followed by proteinase K treatment for 1 h at 56°C. To isolate mRNA, samples were incubated with Oligo(dT) Dynabeads (Life Technologies) for 10 min and beads were washed twice with 10 mM Tris (pH 7.5), 150 mM LiCl, 1 mM EDTA, 0.1% SDS, and twice with 10 mM Tris (pH 7.5), 150 mM LiCl, and 1 mM EDTA. The mRNA was eluted in Milli-Q water by incubating 2 min at 80°C and used as a template for reverse transcriptase PCR (RT-PCR) with SuperScript III First-Strand Synthesis System for RT-PCR (Life Technologies). Amplification of cDNA was performed using quantitative PCR (LightCycler 480 II; Roche). Primers and probes against UL123 (IE1), UL122 (IE2), and UL54 are listed in Table 1. Taqman gene expression assay for GAPDH was purchased from Life Technologies.
To measure viral replication, supernatants from HCMV-infected HFFs or samples for the standard curve were diluted 50-fold in water and amplification of HCMV DNA was performed using quantitative PCR (ABI Prism 7900 HT; Applied Biosystem). Primers and probes were located in the HCMV DNA polymerase gene UL54 (Table 1). The viral load of supernatants was determined by plotting Cq-values on the standard curve.
Immunofluoresence
HFFs were grown in white 96-well flat-bottom plates. Cells were infected with purified HCMV at a multiplicity of 2.0 plaque-forming units per cell for 2 h at 37°C. Cells were washed three times and cultured in 100 μl medium. At different time points, cells were fixed in 100 μl 4% paraformaldehyde for 30 min at 37°C, washed three times with PBS, and stored in PBS at 4°C. For protein detection, fixed cells were incubated with anti-hnRNP K for 1 h, followed by Alexa Fluor 488-conjugated secondary antibody for 1 h. DAPI was used for nuclear counterstaining and fluorescence was analyzed using a Leica DMI4000 B (Leica Microsystems, Wetzlar, Germany).
Dual luciferase reporter assay
HeLa cells were seeded in 12-well plates and transfected with 50 ng of pGL3-SV40 (Life Technologies), 10 ng of pRL-CMV (Life Technologies), complemented with pCGN-71, pSG5-His-hnRNP K, or pcDNA3.1+ to a total of 1 μg DNA. The pp71 protein activates the CMV promoter (MIEP) but does not activate the SV40 promotor and is used as a positive control.42 SV40-firefly was used as a correction for transfection efficiency to allow adequate comparison between samples. After 48 h, cells were washed twice with PBS, freeze-thawed once at −80°C, and lysed with 200 μl passive lysis buffer (Promega, Madison, WI, USA). Supernatant was collected and used for immunoblot analysis with an anti-6 × His or anti-pp71 antibody and to determine both Renilla and firefly luciferase activities using the Dual-GLO Luciferase Assay System (Promega) in a Veritas Microplate Luminometer (Turner Biosystems, Sunnyvale, CA, USA) according to the manufacturer’s protocol (Promega). Both luciferase activities were normalized for background values. Then, changes in CMV-Renilla luciferase activities relative to SV40-firefly luciferase activities (i.e., Renilla luciferase/firefly luciferase ratio) were determined. The mean Renilla luciferase activity from control (pcDNA 3.1+)-transfected cells was set at 1.
Abbreviations
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- Gr:
-
granzyme
- HCMV:
-
human cytomegalovirus
- hnRNP K:
-
heterogeneous nuclear ribonucleoprotein K
- HFF:
-
human foreskin fibroblast
- HSV-1:
-
herpes simplex virus-1
- IE1:
-
immediate-early 1
- IE2:
-
immediate-early 2
- LAK:
-
lymphokine-activated killer
- MCMV:
-
murine cytomegalovirus
- MIEP:
-
major immediate-early promotor
- PBMCs:
-
peripheral blood mononuclear cells
- PBS:
-
phosphate-buffered saline
- PI:
-
propidium iodide
- pp71:
-
phosphoprotein 71
- SLO:
-
streptolysin O
References
Mocarski ES, Shenk T, Pass RF . Cytomegaloviruses. In: Knipe PM, Howley DE, Griffin DE, Lamb RA, Martin MA, Roizman B, et al. (eds) Fields Virology. 5th edn., Vol. 2. Lippincott Williams & Wilkins: Philadelphia, PA, 2007 pp, 2701–2772.
Crough T, Khanna R . Immunobiology of human cytomegalovirus: from bench to bedside. Clin Microbiol Rev 2009; 22: 76–98.
Moss P, Rickinson A . Cellular immunotherapy for viral infection after HSC transplantation. Nat Rev Immunol 2005; 5: 9–20.
Wathen MW, Stinski MF . Temporal pat terns of human cytomegalovirus transcription: mapping the viral RNAs synthesized at immediate early, early, and late times after infection. J Virol 1982; 41: 462–477.
Gamadia LE, Remmerswaal EB, Weel JF, Bemelman F, van Lier RA, Ten Berge IJ . Primary immune responses to human CMV: a critical role for IFN-gamma-producing CD4+ T cells in protection against CMV disease. Blood 2003; 101: 2686–2692.
van Leeuwen EM, de Bree GJ, ten Berge IJ, van Lier RA . Human virus-specific CD8+ T cells: diversity specialists. Immunol Rev 2006; 211: 225–235.
Barry M, Bleackley RC . Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol 2002; 2: 401–409.
Chowdhury D, Lieberman J . Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol 2008; 26: 389–420.
Bovenschen N, Kummer JA . Orphan granzymes find a home. Immunol Rev 2010; 235: 117–127.
Trapani JA, Smyth MJ . Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol 2002; 2: 735–747.
Mahrus S, Craik CS . Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell-mediated lysis of target cells. Chem Biol 2005; 12: 567–577.
van Domselaar R, Bovenschen N . Cell death-independent functions of granzymes: hit viruses where it hurts. Rev Med Virol 2011; 21: 301–314.
Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL . Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008; 322: 268–271.
Romero V, Fellows E, Jenne DE, Andrade F . Cleavage of La protein by granzyme H induces cytoplasmic translocation and interferes with La-mediated HCV-IRES translational activity. Cell Death Differ 2009; 16: 340–348.
Zhong C, Li C, Wang X, Toyoda T, Gao G, Fan Z . Granzyme K inhibits replication of influenza virus through cleaving the nuclear transport complex importin alpha1/beta dimer of infected host cells. Cell Death Differ 2011; 19: 882–890.
Tang H, Li C, Wang L, Zhang H, Fan Z . Granzyme H of cytotoxic lymphocytes is required for clearance of the hepatitis B virus through cleavage of the hepatitis B virus X protein. J Immunol 2011; 19: 882–890.
van Domselaar R, Philippen LE, Quadir R, Wiertz EJ, Kummer JA, Bovenschen N . Noncytotoxic inhibition of cytomegalovirus replication through NK cell protease granzyme M-mediated cleavage of viral phosphoprotein 71. J Immunol 2010; 185: 7605–7613.
Pao LI, Sumaria N, Kelly JM, van Dommelen S, Cretney E, Wallace ME et al. Functional analysis of granzyme M and its role in immunity to infection. J Immunol 2005; 175: 3235–3243.
Bresnahan WA, Shenk TE . UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc Natl Acad Sci USA 2000; 97: 14506–14511.
van Leeuwen EM, ten Berge IJ, van Lier RA . Induction and maintenance of CD8+ T cells specific for persistent viruses. Adv Exp Med Biol 2007; 590: 121–137.
de Poot SA, Westgeest M, Hostetter DR, Van Damme P, Plasman K, Demeyer K et al. Human and mouse granzyme M display divergent and species-specific substrate specificities. Biochem J 2011; 437: 431–442.
Schmidt T, Striebinger H, Haas J, Bailer SM . The heterogeneous nuclear ribonucleoprotein K is important for Herpes simplex virus-1 propagation. FEBS Lett 2010; 584: 4361–4365.
Ng LF, Chan M, Chan SH, Cheng PC, Leung EH, Chen WN et al. Host heterogeneous ribonucleoprotein K (hnRNP K) as a potential target to suppress hepatitis B virus replication. PLoS Med 2005; 2: e163.
Bomsztyk K, Denisenko O, Ostrowski J . hnRNP K: one protein multiple processes. BioEssays 2004; 26: 629–638.
Paziewska A, Wyrwicz LS, Bujnicki JM, Bomsztyk K, Ostrowski J . Cooperative binding of the hnRNP K three KH domains to mRNA targets. FEBS Lett 2004; 577: 134–140.
Heider JA, Bresnahan WA, Shenk TE . Construction of a rationally designed human cytomegalovirus variant encoding a temperature-sensitive immediate-early 2 protein. Proc Natl Acad Sci USA 2002; 99: 3141–3146.
White EA, Clark CL, Sanchez V, Spector DH . Small internal deletions in the human cytomegalovirus IE2 gene result in nonviable recombinant viruses with differential defects in viral gene expression. J Virol 2004; 78: 1817–1830.
Stenberg RM, Depto AS, Fortney J, Nelson JA . Regulated expression of early and late RNAs and proteins from the human cytomegalovirus immediate-early gene region. J Virol 1989; 63: 2699–2708.
White EA, Del Rosario CJ, Sanders RL, Spector DH . The IE2 60-kilodalton and 40-kilodalton proteins are dispensable for human cytomegalovirus replication but are required for efficient delayed early and late gene expression and production of infectious virus. J Virol 2007; 81: 2573–2583.
Kaspari M, Tavalai N, Stamminger T, Zimmermann A, Schilf R, Bogner E . Proteasome inhibitor MG132 blocks viral DNA replication and assembly of human cytomegalovirus. FEBS Lett 2008; 582: 666–672.
Lin JY, Li ML, Huang PN, Chien KY, Horng JT, Shih SR . Heterogeneous nuclear ribonuclear protein K interacts with the enterovirus 71 5′ untranslated region and participates in virus replication. J Gen Virol 2008; 89: 2540–2549.
Liu Y, Szaro BG . hnRNP K post-transcriptionally co-regulates multiple cytoskeletal genes needed for axonogenesis. Development 2011; 138: 3079–3090.
Laursen LS, Chan CW, Ffrench-Constant C . Translation of myelin basic protein mRNA in oligodendrocytes is regulated by integrin activation and hnRNP-K. J Cell Biol 2011; 192: 797–811.
Bovenschen N, de Koning PJ, Quadir R, Broekhuizen R, Damen JM, Froelich CJ et al. NK cell protease granzyme M targets alpha-tubulin and disorganizes the microtubule network. J Immunol 2008; 180: 8184–8191.
Bovenschen N, Quadir R, van den Berg AL, Brenkman AB, Vandenberghe I, Devreese B et al. Granzyme K displays highly restricted substrate specificity that only partially overlaps with granzyme A. J Biol Chem 2009; 284: 3504–3512.
Bredemeyer AJ, Lewis RM, Malone JP, Davis AE, Gross J, Townsend RR et al. A proteomic approach for the discovery of protease substrates. Proc Natl Acad Sci USA 2004; 101: 11785–11790.
Van Damme P, Maurer-Stroh S, Hao H, Colaert N, Timmerman E, Eisenhaber F et al. The substrate specificity profile of human granzyme A. Biol Chem 2010; 391: 983–997.
Van Damme P, Staes A, Bronsoms S, Helsens K, Colaert N, Timmerman E et al. Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat Methods 2010; 7: 512–515.
Delmas S, Brousset P, Clement D, Le Roy E, Davignon JL . Anti-IE1 CD4+ T-cell clones kill peptide-pulsed, but not human cytomegalovirus-infected, target cells. J Gen Virol 2007; 88: 2441–2449.
Besold K, Wills M, Plachter B . Immune evasion proteins gpUS2 and gpUS11 of human cytomegalovirus incompletely protect infected cells from CD8 T cell recognition. Virology 2009; 391: 5–19.
de Koning PJ, Tesselaar K, Bovenschen N, Colak S, Quadir R, Volman TJ et al. The cytotoxic protease granzyme M is expressed by lymphocytes of both the innate and adaptive immune system. Mol Immunol 2010; 47: 903–911.
Shen W, Westgard E, Huang L, Ward MD, Osborn JL, Chau NH et al. Nuclear trafficking of the human cytomegalovirus pp71 (ppUL82) tegument protein. Virology 2008; 376: 42–52.
Acknowledgements
We thank Dr. RA van Lier (Sanquin, Amsterdam, The Netherlands) for critical reading of the manuscript. This work was supported by The Netherlands Organization for Scientific Research (Grant 916.66.044) and the Dutch Cancer Society (UU-2009-4302) (to NB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by JM Hardwick
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
van Domselaar, R., de Poot, S., Remmerswaal, E. et al. Granzyme M targets host cell hnRNP K that is essential for human cytomegalovirus replication. Cell Death Differ 20, 419–429 (2013). https://doi.org/10.1038/cdd.2012.132
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.132
Keywords
This article is cited by
-
Perforin and granzymes: function, dysfunction and human pathology
Nature Reviews Immunology (2015)
-
NK cell intrinsic regulation of MIP-1α by granzyme M
Cell Death & Disease (2014)
-
Granzyme M: behind enemy lines
Cell Death & Differentiation (2014)
-
Caspase-3 cleaves hnRNP K in erythroid differentiation
Cell Death & Disease (2013)

{kind=link}
{kind=link}