
Abstract
In a number of contexts, and particularly in response to cellular stress, stimulation of the NF-kappaB (NF-κB) pathway promotes apoptosis. One mechanism underlying this pro-apoptotic activity is nucleolar sequestration of RelA, which is reported to cause cell death by repressing NF-κB-driven transcription. Here, we identify a novel and distinct nucleolar activity of RelA that induces apoptosis. We demonstrate, using a viral nucleolar localization signal (NoLS)–RelA fusion protein, that direct targeting of RelA to the nucleolus mediates apoptosis, independent of NF-κB transcriptional activity. We demonstrate a requirement for nucleophosmin (NPM, B23.1) in this apoptotic effect, and the apoptotic effect of stress-induced nucleolar RelA. We show by multiple approaches that nucleolar translocation of RelA is causally involved in the relocalization of NPM from the nucleolus to the cytoplasm and that RelA-induced cytoplasmic NPM mediates apoptosis by facilitating the mitochondrial accumulation of BAX. These data uncover a novel stress-response pathway and mechanism by which RelA promotes apoptosis, independent of its effects on NF-κB transcriptional activity. These findings are relevant to the design of novel anticancer agents that target RelA to this compartment.
Similar content being viewed by others


Main
The process of apoptosis is of fundamental importance to biological pathways ranging from embryogenesis to ageing and normal tissue homoeostasis to stress response. One group of proteins that have a pivotal role in regulating this process is the NF-kappaB (NF-κB) family of transcription factors.1, 2 The most common form of NF-κB is RelA(P65)/p50 heterodimers, which are generally retained inert in the cytoplasm by the inhibitor protein, I-kappaB (IκB).3 Following stimulation of the cell by a variety of agents, IκB is degraded, allowing NF-κB to translocate to the nucleus and bind to the promoter regions of its multiple target genes.3, 4
Although NF-κB is generally regarded as anti-apoptotic, it is now apparent that in particular contexts, and especially in response to cellular stress, NF-κB acts to promote apoptosis.2, 5, 6, 7 For instance, hypoxia, ultraviolet radiation-C (UV-C), serum withdrawal and DNA damaging agents require nuclear translocation of RelA/NF-κB complexes for their apoptotic effects.8 Stimulation of the NF-κB pathway is essential for p53-mediated cell death9 and expression of RelA alone promotes apoptosis in some cell types.10 However, despite evidence linking NF-κB to the promotion of apoptosis, the mechanisms underlying this effect have yet to be fully resolved.
One mechanism that has been suggested is the induction of repressive/inactive RelA–NF-κB complexes that mediate apoptosis by actively downregulating NF-κB-dependent, anti-apoptotic gene transcription.8, 11, 12 We have also suggested that nuclear translocation of NF-κB promotes apoptosis by downregulating NF-κB transcriptional activity. However, we described an alternative mechanism for this repression involving nucleolar sequestration of RelA.13, 14 We demonstrated that upon stimulation of the NF-κB pathway by specific stresses (including UV-C radiation, serum deprivation, cyclin-dependent kinase 4 inhibition and aspirin/related agents) the RelA component of NF-κB translocates from the cytoplasm to the nucleoplasm and then to the nucleolus.13, 15 We identified a signal at the N-terminus of RelA (aa 27–30) that was essential for nucleolar translocation of the protein and, using a dominant negative mutant deleted for this domain (green fluorescent protein (GFP)–RelAΔNoLS), demonstrated that nucleolar sequestration of RelA is causally involved in the repression of NF-κB-driven transcription and the induction of apoptosis.13 On the basis of these data, we proposed that, like the active repression model, nucleolar translocation of RelA mediates apoptosis purely through effects on NF-κB transcriptional activity.
In addition to its role in ribosome biogenesis, the nucleolus has an essential role in cell growth, stress response, transcriptional regulation and apoptotic cell death.16, 17 These multiple functions are achieved by over 4500 proteins that move rapidly and dynamically between this compartment, the nucleoplasm and cytoplasm, dependent on cell conditions.18 Previous studies have linked stress-induced disruption of nucleoli to the induction of apoptosis19 and a number of apoptotic regulators are known to localize to this compartment. Therefore, it is highly possible that, in addition to its effects on NF-κB transcriptional activity, nucleolar targeting of RelA mediates apoptosis through modulation of nucleolar pathways. In keeping with this suggestion, we have previously shown that nucleolar translocation of transfected GFP–RelA mediates apoptosis in rela null mouse embryo fibroblasts (MEFs), which do not require basal NF-κB activity to survive.13
Here, we demonstrate that targeting RelA to the nucleolus mediates apoptosis in the absence of repression of NF-κB-driven transcription. Furthermore, we demonstrate that this apoptosis, and that of stress-induced nucleolar RelA, occurs through relocalization of nucleophosmin (NPM, B23.1) from the nucleolus to the cytoplasm, which in turn facilitates the mitochondrial localization of BAX. These data reveal a novel mechanism by which stimulation of the NF-κB pathway induces cell death and a novel role for RelA, independent of its effects on NF-κB transcriptional activity.
Results
Targeting RelA to the nucleolus
Although RelA has a nucleolar localization signal (NoLS), this is only functional in response to specific stimuli.13 Therefore, to determine whether RelA mediates apoptosis through direct effects on nucleoli, we generated an NoLS–RelA fusion protein (GFP–NoLSRelA), to target RelA to this compartment in the absence of additional, complicating signals (Figure 1a). We utilized the NoLS from the HIV-1 Rev protein, as this has been shown to contain one of the most efficient signals for targeting foreign proteins to nucleoli and it is dominant to other NoLSs.20

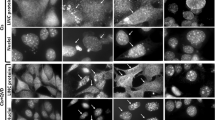
Targeting of RelA to the nucleolus. (a) Diagrammatic representation of the GFP-tagged proteins generated by the specified expression constructs. (b) The cellular distribution of GFP-tagged proteins was determined in live/adherent SW480 cells. Arrow indicates nucleolar presence of GFP–NoLSRelA as suggested by colocalization with dense bodies in phase images. Right: at 48h after transfection, the percentage of GFP-expressing cells showing GFP in the nucleolus was determined. At least 200 GFP-positive cells per sample were counted. Data are the mean± S.E. N=6. (c) GFP–NoLSRelA transfected SW480 cells were fixed. Left: cells were stained with DAPI. Arrow indicates colocalization of GFP bodies with areas devoid of DAPI (DNA) staining. Right: immunomicrographs showing colocalization of GFP nuclear bodies with UBF. (d) GFP–NoLSRelA transfected SW480 cells were either non-treated (NT) or treated with TNF (10 ng/ml) for 16 h. Live cell imaging demonstrates the localization of GFP–NoLSRelA as above. The percentage of transfected cells showing nucleolar GFP was determined as above (N=3). Scale bars throughout=10 μm
Analysis of the subcellular localization of GFP–NoLSRelA and controls (GFP–NoLS and GFP–RelA) revealed that, in keeping with previous data, GFP–NoLS localized predominantly to the nucleolus and GFP–RelA to the cytoplasm (Figure 1b). It also revealed that GFP–NoLSRelA was present in the cytoplasm, nucleoplasm and, in 20.2% of cells, in nuclear bodies (Figure 1b). These nuclear bodies colocalized with high-density nuclear bodies in brightfield images and in areas devoid of DAPI staining in fixed cells, which are both recognized to be nucleoli (Figures 1b and c). They also colocalized with the nucleolar protein, upstream-binding factor (UBF), confirming their nucleolar localization (Figure 1c).
Although these data indicated the RevNoLS targets RelA to the nucleolus, in a significant proportion of cells, the fusion protein remained cytoplasmic. As this was probably due to cytoplasmic retention of the RelA portion by IκBα, we postulated that treatment with tumor necrosis factor alpha (TNFα), which mediates the rapid degradation of IκBα, would increase the proportion of cells showing GFP–NoLSRelA in nucleoli. However, we found that while the fusion protein became predominantly nuclear in response to TNFα (confirming stimulation of the NF-κB pathway), fewer cells demonstrated nucleolar localization, compared with the non-TNFα-treated population (Figure 1d). We have previously demonstrated that RelA remains in the nucleoplasm in response to TNFα and we suggested that this is because the cytokine fails to induce the post-transcriptional modifications required for nucleolar targeting of the protein.21 However, these interesting data would suggest that RelA is actively excluded from the nucleolus in response to TNF, even when fused to the rev NoLS.
Nucleolar translocation of RelA and apoptosis
Having established that the RevNoLS can target RelA to the nucleolus, we next examined the effects on cellular apoptosis. As treatment with TNF did not increase the number of cells showing GFP–NoLSRelA in nucleoli, we reverted to expressing the proteins alone, in the absence of additional stimuli. Using annexin V apoptosis assays, we found that a significantly greater percentage of cells expressing GFP–NoLSRelA were apoptotic, compared with cells expressing GFP–NoLS or GFP–RelA (Figure 2a).

Targeting RelA to the nucleolus mediates apoptosis. (a) At 48h after transfection of SW480 cells with the specified vectors, the percentage of GFP-positive cells undergoing apoptosis was determined by annexin V-biotin/streptavidin Texas red apoptosis assay and fluorescence microscopy. At least 200 transfected cells were counted for each sample. Data are the mean±S.E. N=6. (b and c) dsRed-tagged versions of the vectors described in 1a were generated and transfected into SW480 cells. (b) Live cell imaging demonstrates the localization of the dsRed-tagged proteins. Arrow indicates dsRed–NoLSRelA in the nucleolus. (c) FITC–annexin V apoptosis assays and fluorescent microscopy were used to quantify the percentage of dsRed expressing cells undergoing apoptosis as in (a). N=3. (d and e) RKO cells were transfected with the specified vectors. (d) The cellular distribution of GFP-tagged proteins was determined in live/adherent cells. Arrow indicates GFP–NoLSRelA in nucleoli, identified in phase images. (e) Annexin V apoptosis assays were used to quantify the percentage of GFP-expressing cells undergoing apoptosis as in (a). N=3. (f) At 24 h after transfection of SW480 cells with the specified vectors, Mitotracker dye uptake was used to identify cells with active mitochondria. Fluorescent micrographs of fixed cells are shown. Arrows indicate lack of Mitotracker dye uptake in cells showing nucleolar localization of GFP–NoLSRelA (left) and dye uptake in cells showing cytoplasmic localization of the protein (right). (g) The percentage of GFP-expressing cells showing Mitotracker positivity was determined by fluorescent microscopy in at least 100 cells per sample. For GFP–NoLSRelA, quantification was restricted to cells with nucleolar localized GFP. The mean±S.E. is shown. N=3 (h) Anti-GFP immunoblot showing the expression levels of the specified proteins in whole cell lysates from transfected SW480 and RKO cells. Actin was used as a protein loading control. (i) The apoptotic effect of expressing GFP–NoLSRelA is not associated with repression of NF-κB-driven transcription. SW480 cells were transfected with the specified GFP-tagged vectors along with the 3 × κB ConA, NF-κB-dependent, luciferase reporter construct and a pCMVβ control plasmid. NF-κB activity was measured 48h after transfection and levels relative to β-galactosidase activity determined. The mean±S.E. is shown. N=3 Scale bars throughout=10 μm. The P-values shown in panels (a, c, e and g) are compared with GFP–NoLSRelA and are derived using a Student's t-test
Similar results were obtained using equivalent dsRed-tagged proteins in that the dsRed–NoLSRelA fusion protein was present in the nucleolus (Figure 2b) in 25% of transfected cells and expression of this protein was associated with increased apoptosis (Figure 2c). Similar results were also obtained using RKO cells in that GFP–NoLSRelA localized to the nucleolus (Figure 2d) and induced a significant increase in apoptosis (Figure 2e).
To further confirm the association between nucleolar RelA and apoptosis, we utilized Mitotracker assays in which apoptotic cells are identified by the demise of their mitochondrial activity and consequent inability to uptake a mitochondrial dye. Figure 2f clearly demonstrates that nucleolar localization of GFP–NoLSRelA is associated with loss of mitochondrial activity, while cytoplasmic localization of the same protein is not. Quantification indicated that significantly fewer cells expressing GFP–NoLSRelA in nucleoli were Mitotracker positive, compared with those expressing GFP–RelA or GFP–NoLS (Figure 2g). Western blot analysis confirmed that the differential effects of expressing the GFP-tagged constructs were not caused by differential expression of the proteins (Figure 2h), while NF-κB reporter assays ruled out the possibility that the apoptotic effect of expressing GFP–NoLSRelA was caused by decreased NF-κB-driven transcription (Figure 2i).
NPM is required for the pro-apoptotic effects of nucleolar targeting of RelA
The above data would suggest that nucleolar translocation of RelA is directly affecting a nucleolar pathway/protein(s) to mediate apoptosis. One protein of interest is NPM as this has a critical role in the regulation of cell growth and death, can interact with RelA and is critical for the apoptotic effects of UV-C, which causes apoptosis through nucleolar translocation of RelA.13, 22, 23, 24 Therefore, an siRNA approach was used to investigate the role of NPM in the apoptotic effects of nucleolar targeting of RelA.
We obtained efficient depletion of NPM on siRNA transfection and found that depleting NPM did not significantly affect expression levels (Figure 3a and data not shown) or localization (Figure 3b) of the GFP-tagged proteins. On examination of apoptosis, we found that depletion of NPM had a pro-apoptotic effect alone (data not shown). However, relative to this basal level of apoptosis, the apoptotic effects of expressing GFP–NoLSRelA, observed in control siRNA-transfected cells, were completely abrogated in cells transfected with NPM siRNA (Figure 3c).

NPM is required for the apoptotic effects of nucleolar targeting of RelA. (a–d) SW480 cells were depleted of NPM using siRNA (5′-UGAUGAAAAUGAGCACCAGUUTT-3′39) before transfection with the specified GFP-tagged plasmids. (a) Whole cell lysates were subjected to immunoblot analysis to determine the levels of the indicated proteins. (b) The localization of GFP-tagged proteins was determined in live/adherent cells. Scale bars=10 μm. (c) Annexin V-biotin/streptavidin Texas red apoptosis assays were used to determine the percentage of GFP-positive cells undergoing apoptosis. The data presented are the average fold increase in apoptosis compared with the equivalent siRNA, mock plasmid-transfected control (N=3, ±S.E.). A Student's t-test was performed and the P-value is indicated in the panel. (d) The requirement for NPM in the apoptotic effect of expressing GFP–NoLSRelA is not associated with modulation of NF-κB-driven transcription. The 3 × κB ConA, NF-κB-dependent, luciferase reporter construct and pCMVβ control plasmid were transfected along with the specified GFP expression vectors. Mean NF-κB activity, relative to β-galactosidase activity, is presented±S.E. N=3
Figure 2i demonstrates that expression of GFP–NoLSRelA is associated with increased NF-κB transcriptional activity. Therefore, it may be argued this increase in transcription mediates the apoptotic effects of the fusion protein, and not nucleolar translocation per se. However, we found that knockdown of NPM had no significant effect on GFP–NoLSRelA-mediated NF-κB-driven transcription, although this depletion abrogated apoptosis (Figure 3d). Taken together, these data suggest that the apoptotic effects of expressing GFP–NoLSRelA are independent of NF-κB transcriptional activity and are mediated through NPM.
NPM is required for the pro-apoptotic effects of stimulus-induced nucleolar translocation of RelA
Given that the above system for nucleolar targeting of RelA is experimental, we next examined the requirement for NPM in the pro-apoptotic effects associated with stress-induced nucleolar translocation of endogenous RelA. We used aspirin as a model NF-κB stimulus as this requires nucleolar translocation of RelA for its pro-apoptotic activity.13, 14, 15 Western blot analysis indicated NPM siRNA efficiently reduced expression of the protein in the presence and absence of aspirin, so we again used this approach (Figure 4a). First, we established that NPM knockdown does not inhibit aspirin effects on the NF-κB pathway (degradation of IκB, nuclear/nucleolar translocation of RelA and repression of NF-κB-driven transcription) (Figures 4b–d). In fact, NPM depletion appeared to enhance aspirin-mediated degradation of IκBα and nuclear/nucleolar translocation of RelA (Figures 4b and c). In contrast, annexin V apoptosis assays indicated that depletion of NPM blocked the apoptotic response to the agent (Figure 4e). These data were again confirmed using Mitotracker assays (Figure 4f). We found that aspirin induced a significant decrease in the number of cells showing active mitochondria (dye uptake) in cells transfected with control siRNA, but that this response was significantly abrogated in cells transfected with siRNA to NPM (Figures 4f and g). Taken together, these data indicate that NPM is required for the apoptotic effects of this model stimulus of the NF-κB pathway and that this requirement lies downstream of nucleolar translocation of RelA.

NPM is required downstream of nucleolar translocation of RelA in the apoptotic effects of a model pro-apoptotic stimulus of the NF-κB pathway. (a–g) SW480 cells were transfected with control or NPM siRNA then either non-stimulated (−) or stimulated (+) with aspirin ((a–e) 5 mM, 16 h and (f and g) 10 mM, 8 h)). (a) Anti-NPM immunoblot performed on whole cell lysates. (b) Anti-IκBα immunoblot performed on cytoplasmic extracts. (c) Immunomicrographs showing the localization of endogenous RelA. Inset: the percentage of cells in the total cell population showing nucleolar RelA was determined in at least 250 cells. The mean is shown. N=3. (d) SW480 cells were transfected with an NF-κB luciferase reporter construct (3 × κB ConA-Luc) and pCMVβ before aspirin exposure. NF-κB activity, relative to β-galactosidase activity, was calculated. The mean percentage of relative NF-κB activity, compared with basal levels (untreated controls) is shown (±S.E.). N=5. (e) Annexin V–FITC apoptosis assays and fluorescent microscopy were used to quantify the percentage of cells (at least 250 were examined) in the total cell population undergoing apoptosis. The mean±S.E. is shown (N=3). (f) Mitotracker dye uptake identified cells with active mitochondria. Fluorescent micrographs of fixed cells are shown. (g) The percentage of Mitotracker stained cells was determined by fluorescent microscopy in at least 250 cells per sample. The mean of three independent experiments±S.E. is shown. P-values in panels (e and g) were calculated using a Student's t-test. Scale bars throughout=10 μm
Nucleolar targeting of RelA causes nucleolar to cytoplasmic translocation of NPM
To begin to understand the pathway by which NPM mediates apoptosis in response to nucleolar RelA, we examined the cellular distribution of this protein. Using immunocytochemistry to detect endogenous NPM (Figure 5a), live cell imaging of GFP-tagged NPM (Figure 5b), and western blot analysis (Figure 5c), we found that aspirin mediates an increase in cytoplasmic levels of this protein. This cytoplasmic appearance coincided with a decrease in nuclear levels of the protein (Figures 5d and e). Furthermore, it was time dependent (Figures 5d–f), occurred in parallel with nucleolar translocation of RelA (Figures 5e and f) and preceded the induction of apoptosis.25 In contrast to effects on cytoplasmic and nuclear levels of NPM, aspirin had a minimal effect on total levels of the protein (Figure 4a).

Nucleolar translocation of RelA is associated with cytoplasmic presence of NPM. (a) Immunomicrographs showing the localization of endogenous NPM before (−) and after (+) aspirin stimulation (5 mM, 16 h). Dashed line indicates outline of nucleus, as determined by DAPI staining. (b and c) SW480 cells were transfected with GFP-NPM then left untreated (−) or stimulated with aspirin (+) as above. (b) The cellular localization of GFP-tagged NPM was determined in live cells using an Axiovert 100 inverted fluorescent microscope. (c) Immunoblot showing cytoplasmic (cyto) levels of endogenous and GFP-NPM. (d) Cells were transfected with GFP–NPM as above then exposed to aspirin (10 mM) for the specified times. Immunoblots show cytoplasmic (cyto) and nuclear (nuc) levels of the specified proteins. (e) SW480 cells were exposed to aspirin (10 mM) for the specified times. Top: immunoblot showing cytoplasmic and nuclear levels of endogenous NPM. Bottom: immunomicrographs demonstrating the cellular localization of RelA. The percentage of cells showing nucleolar RelA was determined in at least 200 cells per time point. N=4. (f) SW480 cells were transfected with GFP–RelA and dsRed-NPM then exposed to aspirin (10 mM) for the times specified. The cellular localization of the fluorescently tagged proteins was determined in live cells as above. Arrow indicates cytoplasmic NPM. Scale bars throughout=10 μm
Taken together, the above data suggest that aspirin causes the cellular redistribution of NPM. They also suggest the intriguing possibility that nucleolar translocation of RelA is responsible for this redistribution. To test this hypothesis, we initially utilized the NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), which we have shown blocks aspirin-induced degradation of IκBα and cytoplasmic to nuclear/nucleolar translocation of NF-κB/RelA.13 Here, we found that it also blocks aspirin-induced cytoplasmic accumulation of NPM (Figure 6a). To further confirm the importance of NF-κB in stress-mediated relocalization of NPM, RelA was depleted using siRNA. We obtained efficient knockdown of RelA on siRNA transfection and found that depleting RelA did not significantly affect the abundance of NPM (Figure 6b). However, we found that the nuclear-cytoplasmic relocalization of NPM, observed in response to aspirin in cells transfected with control siRNA, was completely abrogated in cells transfected with siRNA to RelA (Figures 6c and d).

NF-κB/RelA is required for stress-mediated nucleolar-cytoplasmic translocation of NPM. (a) SW480 cells were transfected with GFP-NPM then left untreated (−) or stimulated (+) with aspirin (5 mM, 16 h) in the presence (+) and absence (−) of the NF-κB inhibitor, PDTC (200 μM). The cellular localization of GFP–NPM was determined in live cells. (b) At 48 h after transfection with control or RelA (5′-GCUGAUGUGCACCGACAAG-3′),8 siRNA, SW480 cells were either left untreated (−), or treated (+) with aspirin (10 mM, 6 h). Immunoblots of whole cell (wc) lysates show siRNA depletion of RelA. (c) SW480 cells were transfected with control or RelA siRNA as in (a) then treated with aspirin (10 mM) for the indicated times. (c) Immunoblots demonstrate NPM levels in cytoplasmic (cyto) and nuclear (nuc) extracts. (d) Following siRNA depletion of RelA, cells were transfected with dsRed–NPM. Live cell images show NPM localization. Phase/fluorescent images are shown in the merged panel. Actin was used as a loading control. Scale bars throughout=10 μm
These data indicated that stimulation of the NF-κB pathway/RelA is required for relocalization of NPM to the cytoplasm. To determine whether nucleolar translocation of RelA is required, we utilized the nucleolar-deficient mutant, GFP–RelAΔNoLS. First, we established that the wild type (WT) and mutant proteins expressed comparably (Figure 7a) and confirmed that GFP–RelAWT translocates to the nucleolus in response to aspirin, while GFP–RelAΔNoLS remains nucleoplasmic, excluded from nucleoli (Figure 7b). In keeping with nucleolar RelA causing relocalization of NPM, we found that aspirin induced a time-dependent decrease in nuclear levels, and a parallel increase in cytoplasmic levels, of NPM in cells constitutively expressing GFP–RelAWT, but not in cells constitutively expressing GFP–RelAΔNoLS (Figure 7b). These data were confirmed using live cell imaging, which again indicated that expression of GFP–RelAΔNoLS blocked aspirin-induced cytoplasmic redistribution of NPM (Figure 7c).

Nucleolar translocation of RelA causes cytoplasmic translocation of NPM. (a–c) SW480 cells stably expressing GFP–RelAWT or ΔNoLS were utilized. (a) Cells were either left untreated (−) or treated (+) with aspirin (2.5 mM, 16 h). Anti-RelA immunoblots show levels of endogenous and GFP-tagged RelA in whole cell (wc) lysates. (b) Cells were stimulated with aspirin (10 mM) for the specified times. Immunomicrographs of fixed cells show the localization of GFP–RelA (top). Immunoblots show the cytoplasmic (cyto) and nuclear (nuc) levels of NPM. (c) Cells were transfected with dsRed–NPM then treated with aspirin (10 mM) for the specified times. Fluorescent images of live cells were captured. (d) Immunoblot showing cytoplasmic levels of the specified proteins in SW480 cells transfected with the indicated GFP-tagged plasmids. (e) Data from three independent experiments were analyzed by Image J and NPM intensities, relative to actin intensities, plotted as means±S.E. A Student's t-test was performed and the P-values, compared with GFP–NoLSRelA are indicated in the panel. Actin was used as a loading control throughout. Scale bars=10 μm
To further establish the relationship between nucleolar translocation of RelA and cytoplasmic redistribution of NPM, we utilized the RelA nucleolar targeting vector (GFP–NoLSRelA). We found that, although only a proportion of cells express the GFP-tagged proteins, transient transfection with GFP–NoLSRelA induced a significant increase in total cytoplasmic levels of NPM, compared with transfection with GFP–NoLS or GFP–RelA (Figures 7d and e). Anti-GFP western blot analysis indicated that this difference was not due to differences in expression levels of the fusion proteins (Figure 7d).
Nuclear export is required for the apoptotic effects of nucleolar targeting of RelA
Previous studies demonstrated NPM is exported from the nucleus via the RAN–exportin-1 (CRM1p) pathway.26 Therefore, to determine the relevance of the cytoplasmic relocalization of NPM with regard to the apoptotic effects of nucleolar RelA, we utilized the CRM1p specific inhibitor, leptomycin B (LMB). This inhibitor had no effect on aspirin-induced nucleolar accumulation of RelA (data not shown and Stark and Dunlop13). However, pretreatment with LMB blocked aspirin-mediated nucleolar–cytoplasmic translocation of NPM and apoptosis (Figures 8a and b). Similar results were obtained using the GFP–NoLSRelA construct. LMB blocked the cytoplasmic relocalization of NPM and apoptosis mediated by expression of this protein (Figures 8c and d). Taken together, these data provide a very strong indication that nuclear export of NPM is required for the apoptotic effects of nucleolar targeting of RelA.

Nuclear export of NPM is required for the apoptotic effects of nucleolar targeting of RelA. (a) SW480 cells were transfected with GFP–NPM then left untreated (−) or stimulated (+) with aspirin (5 mM, 16 h) in the presence (+) and absence (−) of the nuclear export inhibitor, leptomycin B (LepB, 20 nM). Fluorescent and phase contrast images of live, adherent cells were captured. (b) SW480 cells were exposed to aspirin and LepB as above. Annexin V apoptosis assays and fluorescent microscopy were used to quantify the percentage of cells undergoing apoptosis. At least 200 cells were counted per sample. The mean±S.E. is shown (N=3). A Student's t-test was performed and the P-value is indicated in the panel. (c and d) SW480 cells were transfected with GFP–NoLSRelA and 24 h after transfection cells were either left untreated (−) or treated (+) with LepB (20 nM, 16 h). (c) Immunoblot showing cytoplasmic levels of NPM. Actin acts as a loading control. (d) The percentage of GFP-expressing cells undergoing apoptosis was determined by annexin V–biotin/streptavidin Texas red apoptosis assays and fluorescent microscopy. At least 200 transfected cells were examined per sample. The mean of three independent experiments is shown±S.E. A Student's t-test was performed and the P-value indicated
NPM interacts with BAX following nucleolar translocation of RelA
One mechanism by which cytoplasmic NPM has been shown to influence apoptosis is by chaperoning the pro-apoptotic protein, BAX, to the mitochondria.24, 27 As a number of stimuli that induce nucleolar translocation of RelA are known to require BAX for their pro-apoptotic activity, we next examined this pathway to nucleolar RelA-mediated apoptosis.
Immunocytochemical analysis demonstrated that BAX accumulates in punctate cytoplasmic foci in response to aspirin (Figure 9a). These foci colocalized with the mitochondrial marker, heat shock protein 60 (HSP60), suggesting that BAX accumulates at the mitochondria after aspirin treatment (Figure 9b). To examine the relationship between aspirin-induced mitochondrial accumulation of BAX, NPM and RelA, we initially used time course studies. We found that the number of cells showing focal accumulations of BAX increased in a time-dependent manner after aspirin treatment (Figure 9c), which was concomitant with nucleolar translocation of RelA and cytoplasmic relocalization of NPM (Figures 5d–f). In keeping with a role for NPM in chaperoning BAX to the mitochondria, overexpression of NPM enhanced aspirin-mediated mitochondrial accumulation of BAX (compare transfected and non-transfected cells in Figure 9d). In addition, siRNA-mediated knockdown of NPM (confirmed by western blot analysis (Figure 4a)) abrogated this effect (Figure 9e), and aspirin-mediated permeabilization of the outer mitochondrial membrane (Figures 4f and g). Immunoprecipitation assays demonstrated that aspirin induces a specific interaction between NPM and BAX (Figure 9f). Furthermore, this interaction was blocked by RelA depletion, confirming a role for RelA in this process (Figure 9g).

RelA-induced cytoplasmic NPM facilitates the mitochondrial accumulation of BAX. SW480 cells were either untreated (−) or stimulated (+) with: (a, b, d–g) 5 mM, aspirin 16 h. (c) 10 mM aspirin for the times specified. (a–c) Immunomicrographs showing (a) bright, punctate BAX staining in response to the stimuli. (b) Localization of BAX and the mitochondrial protein, HSP60. Arrow indicates co-localization of BAX foci with the mitochondrial marker. (c) The percentage of cells showing bright BAX foci was determined in at least 200 cells per time point. N=2. (d) SW480 cells were transfected with dsRed-NPM before aspirin exposure. Immunomicrograph demonstrates the intensity and localization of BAX in transfected and non-transfected cells. White arrow indicates bright BAX foci in cell with cytoplasmic dsRed–NPM. Yellow arrow indicates cell with nucleolar dsRed-NPM showing diffuse BAX staining. (e) Depletion of NPM blocks mitochondrial accumulation of BAX. SW480 cells were depleted of NPM using siRNA then either non-treated (−) or exposed to aspirin (+) as in (a). Immunomicrographs demonstrate the localization/intensity of BAX. The percentage of cells showing bright, BAX foci (mitochondrial) was determined in the total cell population as in (b). The mean±S.E. is shown. N=3. (f) BAX interacts with NPM in response to aspirin. BAX was immunoprecipitated from whole cell lysates then precipitated proteins subjected to anti-NPM western blot analysis (WB). Immunoprecipitation with IgG controlled for non-specific binding. BAX and NPM levels in input samples are shown. NS, nonspecific protein. (g) RelA is required for the BAX–NPM interaction. Cells were transfected with control (Con) or RelA siRNA 48 h before aspirin treatment. Immunoprecipitations were performed on whole cell lysates as above. RelA levels in the inputs are shown. Scale bars throughout=10 μm
BAX is required for the apoptotic effects of nucleolar translocation of RelA
To determine whether BAX is absolutely required for the apoptotic effects of stimulus-induced nucleolar translocation of RelA, we utilized HCT116 colorectal cancer cells in which the BAX gene has been deleted through homologous recombination (HCT116BAX−/−)28 (Figure 10a). As has been observed for related non-steroidal anti-inflammatory drugs,28 the apoptotic effects of aspirin, observed in HCT116BAX+/+ cells, were greatly reduced in HCT116BAX−/− cells (Figure 10b).

BAX is required for nucleolar RelA-mediated apoptosis. (a and b) Wild type (HCT116+/+) and BAX deleted (HCT116−/−) HCT116 cells were either non-treated (−) or stimulated (+) with aspirin (5 mM, 16 h). (a) Immunoblot showing levels of BAX. (b) Annexin V apoptosis assays and fluorescent microscopy were used to quantify the percentage of cells (at least 200 per sample examined) undergoing apoptosis. The mean±S.E. is shown (N=4). (c–e) SW480 cells were depleted of BAX using siRNA before transfection with the specified GFP-tagged plasmids. (c) Whole cell lysates were subjected to immunoblot analysis to determine the levels of the indicated proteins. (d) The localization of GFP-tagged proteins was determined in live/adherent cells. Scale bars=10 μm. (e) Annexin V-biotin/streptavidin Texas red apoptosis assays were used to stain apoptotic cells. The percentage of GFP-positive cells undergoing apoptosis was determined by fluorescent microscopy in at least 200 transfected cells. The data presented are the mean±S.E. (N=2). (f) Model for pro-apoptotic effects of stress-mediated stimulation of the NF-κB pathway. Our model suggests that stress-induced nucleolar translocation of RelA mediates apoptosis by causing the nucleolar protein, NPM, to relocate to the cytoplasm, bind to BAX and chaperone BAX to the mitochondria. Nucleolar RelA-mediated repression of NF-κB-driven transcription may enhance this route to apoptosis by causing decreased expression of Bcl-xl, which inhibits BAX trafficking. See discussion for more details
To investigate the requirement for BAX in GFP–NoLSRelA-mediated apoptosis, an siRNA approach was used. Western blot analysis demonstrated efficient knockdown of BAX on siRNA transfection (Figure 10c) while live cell imaging indicated that depletion of this protein did not affect the expression or localization of GFP–NoLSRelA (Figure 10d). However, apoptosis assays indicated the apoptotic effects of expressing this protein were completely abrogated by depletion of BAX (Figure 10e).
Taken together, these data provide compelling evidence that nucleolar translocation of RelA causes the cytoplasmic relocalization of NPM and consequently, mitochondrial accumulation of BAX and apoptosis (Figure 10f).
Discussion
NF-κB is a critical regulator of the apoptotic process and can act as both a pro- and anti-apoptotic factor.1, 2 Although the anti-apoptotic function of NF-κB has been well characterized, the mechanisms that underlie its pro-apoptotic activity have yet to be fully elucidated. Previous studies indicate that in response to cell stress, NF-κB promotes apoptosis through the nucleolar accumulation of RelA.13, 29 Here, we demonstrate that RelA actively stimulates a nucleolar pathway to initiate apoptosis (Figure 10f). These data identify a novel, NF-κB regulated, pro-apoptotic pathway and a new mechanism by which RelA functions to induce apoptosis, independent of its effects on NF-κB transcriptional activity.
We initially used a viral NoLS–RelA fusion protein to show that targeting RelA to the nucleolus induces apoptosis (Figure 2). We then established that the nucleolar phospho-protein, NPM, is required (Figure 3). We also established that NPM is required for the apoptotic effects of a model stress stimulus of the NF-κB pathway and that this requirement lies downstream of nucleolar translocation of RelA (Figure 4). NPM has previously been shown to act as a co-activator of NF-κB-driven transcription.23 However, here we found that the role of NPM was independent of NF-κB transcriptional activity (Figures 2, 3 and 4). The most documented apoptotic pathway regulated by NPM is the p53 pathway.30 However, we have previously demonstrated that aspirin-mediated apoptosis does not require p53.31 Taken together, these data suggest that RelA directly modulates a p53 independent, NPM-dependent nucleolar pathway to mediate apoptosis.
Although shuttling of NPM from the nucleolus to the nucleoplasm and cytoplasm is well documented, the mechanisms that govern this relocalization are poorly understood.32, 33 On the basis of the following observations, we propose that stimulation of the NF-κB pathway/nucleolar translocation of RelA is one mechanism that drives the cytoplasmic redistribution of NPM. First, kinetic studies demonstrated a direct association between translocation of RelA to the nucleolus, a decrease in nuclear/nucleolar NPM and a corresponding increase in cytoplasmic levels of the protein (Figure 5). Second, blocking stimulation of the NF-κB pathway using a chemical inhibitor (PDTC) and RelA depletion, blocked the cellular re-distribution of NPM (Figure 6). Third, blocking nucleolar translocation of RelA, by expressing GFP–RelAΔNoLS, blocked cytoplasmic accumulation of NPM (Figure 7). Finally, and most convincingly, expression of GFP–NoLSRelA, which specifically targets RelA to the nucleolus, caused relocalization of NPM to the cytoplasm (Figure 7). In keeping with our proposal, UV-C radiation has previously been shown to induce both nucleolar translocation of RelA,13, 21 and cytoplasmic relocalization of NPM.24
It will now be extremely interesting to identify the pathway by which nucleolar RelA causes the re-distribution of NPM. RelA has previously been shown to bind to the nucleolar protein, LXXLL/leucine zipper containing ARF-binding protein (LZAP).34 This is of interest as LZAP also binds p14ARF,35 which retains NPM in the nucleolus.30 Therefore, one possibility is that RelA binding to LZAP in the nucleolus disrupts an LZAP, p14ARF NPM complex, thus releasing NPM from this compartment. In addition to protein binding, NPM localization is regulated by posttranslational modifications such as ubiquitinylation and sumoylation.22, 36 Therefore, another possible mechanism by which RelA may cause its relocalization is by facilitating modulation of its post-transcriptional modification state. In support of this suggestion, we found that aspirin and GFP–NoLSRelA expression induced modified (high molecular weight) forms of NPM in the cytoplasm (data not shown). Furthermore, RelA has previously been shown to directly bind NPM.23 These possible mechanisms of nucleolar RelA-mediated NPM relocalization are currently under investigation.
Our data suggest that on RelA-mediated relocalization to the cytoplasm, NPM initiates apoptosis by chaperoning the pro-apoptotic protein, BAX, to the outer mitochondrial membrane. This mechanism of NPM-mediated apoptosis has previously been shown in response to UV-C radiation and staurosporin in vitro and ischemic reperfusion in vivo.24, 27 However, ours is the first study to link this cytoplasmic apoptotic pathway to stimulation of NF-κB signaling (Figure 10). Lindenboim et al.33 have suggested that, contrary to NPM transporting BAX to the mitochondria, BAX is required for apoptotic stimuli-mediated nuclear to cytoplasmic relocalization of NPM. Here, we found that knockdown of BAX had no effect on nucleolar RelA-mediated cytoplasmic accumulation of NPM (data not shown), suggesting that in the pathway we have unraveled, BAX is not acting as an NPM chaperone but that NPM acts as a BAX chaperone.
Our data showing depletion of NPM blocks aspirin-mediated apoptosis, but not the effects of the agent on repression of NF-κB-driven transcription, may suggest that nucleolar RelA-mediated repression of NF-κB transcriptional activity is of no consequence. However, one of the anti-apoptotic proteins that is regulated by NF-κB is Bcl-xl, which acts by inhibiting BAX-mediated permeabilization of the outer mitochondrial membrane.37 Therefore, one may imagine a model whereby in response to stress agents, nucleolar translocation of RelA promotes BAX-mediated disruption of mitochondria both by causing a decrease in levels of Bcl-xl and by inducing cytoplasmic relocalization of NPM (Figure 10f).
In summary, use of a vector that targets RelA to the nucleolus has allowed us to systematically dissect a novel pathway by which RelA promotes apoptosis in response to cellular stress. Although we used aspirin as a model stress-inducing agent, it must be emphasized that data presented here, showing nucleolar RelA in non-stimulated cell populations (Figure 1b) and elsewhere,13, 21, 24 support the idea that this pathway is physiologically relevant and is general to stress response. These novel findings provide opportunities for additional research on RelA's function within the nucleolus. Furthermore, these findings have major ramifications for the design of new therapies that target RelA to this cellular compartment, which could be used in a variety of pathologies.
Materials and Methods
Cell culture, reagents
Cell culture and reagents have been described elsewhere.13, 21, 25
Plasmids
pEGFP–RelA, pEGFP–HIV-rev and the three enhancer CONA (3 × κB ConA-Luc) plasmids have been described elsewhere.13, 20, 38 pEGFP–NoLS and pEGFP–NoLSRelA were constructed by PCR subcloning the rev nucleolar targeting signal from pEGFP–HIV-rev in frame with the C-terminal of EGFP in either EGFP–C1 (Clonetech, Mountain View, CA, USA) or pEGFP–RelA. DsRed versions of these plasmids were generated in the same manner using DsRed–C1 and DsRed–RelA as backbone vectors. DsRed–NPM was supplied by P Roussel (University of Paris). GFP–NPM was generated by subcloning NPM from this vector into pEGFP–C1 (Clonetech), in fusion with GFP. The pCMV–β-galactosidase plasmid is commercially available (Promega, Southhampton, UK).
Transfections, RNA interference and reporter assays
Plasmid/siRNA transfections and NF-κB reporter assays were carried out as previously described.13, 21 siRNAs used were: NPM: 5′-UGAUGAAAAUGAGCACCAGUUTT-3′,39 BAX: 5′-AAGGUGCCGGAACUGAUCAGA-3′40 and RelA: 5′-GCUGAUGUGCACCGACAAG-3′.8 For studies involving both siRNA and plasmid transfection, cells were transfected with plasmid 24 h after transfection with siRNA.
Live cell imaging, immunoblotting, immunocytochemical staining, apoptosis assays and Mitotracker assays
Live cell imaging, immunoblotting, immunocytochemical staining and apoptosis assays were carried out as described previously.13, 21, 25 All were performed on the adherent cell population, apart from apoptosis assays, which included floating (dead) cells. The Mitotracker assay was carried out as per the manufacturer's instructions and uptake of the mitochondrial dye analyzed by fluorescent microscopy. The following primary antibodies were used for immunoblot analysis: rabbit anti-RelA(C-20), rabbit anti-GFP, rabbit anti-BAX (N20), mouse anti-B23 (NPM) (all from Santa Cruz, Santa Cruz, CA, USA), IκBα (gift from Professor R Hay (Dundee) and mouse anti-actin (Sigma, Dorset, UK). Immunocytochemistry was performed using antibodies against RelA (C20, Santa Cruz), NPM (Santa Cruz) UBF (Santa Cruz), BAX, (Santa Cruz) and Hsp60 (Sigma).
Fluorescence imaging and analysis
Images were captured using a Coolsnap HQ CCD camera (Photometrics Ltd, Tuscon, AZ, USA) Zeiss Axioplan II fluorescent microscope, 63 × Plan Neofluor objective, a 100 W Hg source (Carl Zeiss, Welwyn Garden City, UK) and Chroma 83 000 triple band pass filter set (Chroma Technology, Bellows Falls, UT, USA). Image capture and analysis were performed using scripts written for IPLab Spectrum 3.6 in house. For each panel, a constant exposure time was used. The percentage of cells showing annexin V binding, nucleolar RelA, uptake of Mitotracker dye or punctate BAX was determined in at least 200 cells from at least five random fields of view in three independent experiments, or as specified.
Immunoprecipitation
Immunoprecipitation assays were performed using 500 μg whole cell lysate as previously described.27 Rabbit polyclonal anti-BAX (Santa Cruz) was used to immunoprecipitate the appropriate protein. Rabbit IgG (pre-immune serum) was used as a control. Complexes were resolved by SDS polyacrylamide gel electrophoresis then analyzed by western blot analysis.
Abbreviations
- NF-κB:
-
NF-kappaB
- IκB:
-
I-kappaB
- NoLS:
-
nucleolar localization signal
- NPM:
-
nucleophosmin
- UV-C:
-
ultraviolet radiation-C
- GFP:
-
green fluorescent protein
- MEF:
-
mouse embryo fibroblast
- UBF:
-
upstream binding factor
- TNFα:
-
tumor necrosis factor alpha
- PDTC:
-
pyrrolidine dithiocarbamate
- Crm1P:
-
exportin-1
- LMB:
-
leptomycin B
- HSP60:
-
heat shock protein 60
- LZAP:
-
LXXLL/leucine zipper containing ARF-binding protein
References
Schmitz ML, Mattioli I, Buss H, Kracht M . NF-kappaB: a multifaceted transcription factor regulated at several levels. Chembiochem 2004; 5: 1348–1358.
Fan Y, Dutta J, Gupta N, Fan G, Gelinas C . Regulation of programmed cell death by NF-kappaB and its role in tumorigenesis and therapy. Adv Exp Med Biol 2008; 615: 223–250.
Thanos D, Maniatis T . NF-kappa B: a lesson in family values. Cell 1995; 80: 529–532.
Pahl HL . Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999; 18: 6853–6866.
Perkins ND, Gilmore TD . Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ 2006; 13: 759–772.
Radhakrishnan SK, Kamalakaran S . Pro-apoptotic role of NF-kappaB: implications for cancer therapy. Biochim Biophys Acta 2006; 1766: 53–62.
Strozyk E, Poppelmann B, Schwarz T, Kulms D . Differential effects of NF-kappaB on apoptosis induced by DNA-damaging agents: the type of DNA damage determines the final outcome. Oncogene 2006; 25: 6239–6251.
Campbell KJ, Rocha S, Perkins ND . Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol Cell 2004; 13: 853–865.
Ryan KM, Ernst MK, Rice NR, Vousden KH . Role of NF-kappaB in p53-mediated programmed cell death. Nature 2000; 404: 892–897.
Ricca A, Biroccio A, Trisciuoglio D, Cippitelli M, Zupi G, Del Bufalo D . relA over-expression reduces tumorigenicity and activates apoptosis in human cancer cells. Br J Cancer 2001; 85: 1914–1921.
Ho WC, Dickson KM, Barker PA . Nuclear factor-kappaB induced by doxorubicin is deficient in phosphorylation and acetylation and represses nuclear factor-kappaB-dependent transcription in cancer cells. Cancer Res 2005; 65: 4273–4281.
Tsuchiya Y, Asano T, Nakayama K, Kato Jr T, Karin M, Kamata H . Nuclear IKKbeta is an adaptor protein for IkappaBalpha ubiquitination and degradation in UV-induced NF-kappaB activation. Mol Cell 2010; 39: 570–582.
Stark LA, Dunlop MG . Nucleolar sequestration of RelA (p65) regulates NF-kappaB-driven transcription and apoptosis. Mol Cell Biol 2005; 25: 5985–6004.
Loveridge CJ, Macdonald AD, Thoms HC, Dunlop MG, Stark LA . The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the RelA(p65) subunit of NF-kappaB. Oncogene 2008; 27: 2648–2655.
Thoms HC, Dunlop MG, Stark LA . p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res 2007; 67: 1660–1669.
Mayer C, Grummt I . Cellular stress and nucleolar function. Cell Cycle 2005; 4: 1036–1038.
Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI . The multifunctional nucleolus. Nat Rev Mol Cell Biol 2007; 8: 574–585.
Trinkle-Mulcahy L, Lamond AI . Toward a high-resolution view of nuclear dynamics. Science 2007; 318: 1402–1407.
Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI . The nucleolus under stress. Mol Cell 2010; 40: 216–227.
Emmott E, Dove BK, Howell G, Chappell LA, Reed ML, Boyne JR et al. Viral nucleolar localisation signals determine dynamic trafficking within the nucleolus. Virology 2008; 380: 191–202.
Thoms HC, Loveridge CJ, Simpson J, Clipson A, Reinhardt K, Dunlop MG et al. Nucleolar targeting of RelA(p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res 2010; 70: 139–149.
Okuwaki M . The structure and functions of NPM1/nucleophosmin/B23, a multifunctional nucleolar acidic protein. J Biochem 2008; 143: 441–448.
Dhar SK, Lynn BC, Daosukho C, St Clair DK . Identification of nucleophosmin as an NF-{kappa}B Co-activator for the induction of the human SOD2 Gene. J Biol Chem 2004; 279: 28209–28219.
Thompson J, Finlayson K, Salvo-Chirnside E, MacDonald D, McCulloch J, Kerr L et al. Characterisation of the Bax-nucleophosmin interaction: the importance of the Bax C-terminus. Apoptosis 2008; 13: 394–403.
Stark LA, Din FVN, Zwacka RM, Dunlop MG . Aspirin-induced activation of the NF-κB signalling pathway: a novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J 2001; 15: 1273.
Wang W, Budhu A, Forgues M, Wang XW . Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat Cell Biol 2005; 7:823–830.
Kerr LE, Birse-Archbold JL, Short DM, McGregor AL, Heron I, Macdonald DC et al. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene 2007; 26: 2554–2562.
Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B . Role of BAX in the apoptotic response to anticancer agents. Science 2000; 290: 989–992.
Thoms HC, Dunlop MG, Stark LA . CDK4 inhibitors and apoptosis: a novel mechanism requiring nucleolar targeting of RelA. Cell Cycle 2007; 6: 1293–1297.
Lindstrom MS, Zhang Y . B23 and ARF: friends or foes? Cell Biochem Biophys 2006; 46: 79–90.
Din FV, Stark LA, Dunlop MG . Aspirin-induced nuclear translocation of NFkappaB and apoptosis in colorectal cancer is independent of p53 status and DNA mismatch repair proficiency. Br J Cancer 2005; 92: 1137–1143.
Hindley CE, Davidson AD, Matthews DA . Relationship between adenovirus DNA replication proteins and nucleolar proteins B23.1 and B23.2. J Gen Virol 2007; 88 (Part 12): 3244–3248.
Lindenboim L, Blacher E, Borner C, Stein R . Regulation of stress-induced nuclear protein redistribution: a new function of Bax and Bak uncoupled from Bcl-x(L). Cell Death Differ 2010; 17: 346–359.
Wang J, An H, Mayo MW, Baldwin AS, Yarbrough WG . LZAP, a putative tumor suppressor, selectively inhibits NF-kappaB. Cancer Cell 2007; 12: 239–251.
Wang J, He X, Luo Y, Yarbrough WG . A novel ARF-binding protein (LZAP) alters ARF regulation of HDM2. Biochem J 2006; 393 (Part 2): 489–501.
Tago K, Chiocca S, Sherr CJ . Sumoylation induced by the Arf tumor suppressor: a p53-independent function. Proc Natl Acad Sci USA 2005; 102: 7689–7694.
Giam M, Huang DC, Bouillet P . BH3-only proteins and their roles in programmed cell death. Oncogene 2008; 27 (Suppl 1): S128–S136.
Carlotti F, Chapman R, Dower SK, Qwarnstrom EE . Activation of nuclear factor kappaB in single living cells. Dependence of nuclear translocation and anti-apoptotic function on EGFPRELA concentration. J Biol Chem 1999; 274: 37941–37949.
Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG . Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol 2002; 4: 529–533.
Hastak K, Agarwal MK, Mukhtar H, Agarwal ML . Ablation of either p21 or Bax prevents p53-dependent apoptosis induced by green tea polyphenol epigallocatechin-3-gallate. FASEB J 2005; 19: 789–791.
Acknowledgements
We gratefully acknowledge P Roussel (University of Paris) for kindly providing the DsRed–NPM (B23) plasmid, RT Hay (University of Dundee) for providing the NF-κB reporter plasmids and IκBα antibody and E Quarnstrom (University of Sheffield) for kindly providing the GFP RelA expression vector. We are also very grateful to ND. Perkins (University of Newcastle) and N Hastie (MRC Human Genetics Unit) for their critical appraisal of the paper and P Perry, M Pearson and C Nicol (MRC HGU) for their help with microscopy and figure preparation. AW and JAH were funded by a University of Leeds Interdisciplinary award in Bionanoscience and a Yorkshire cancer Research Award. JAH is a Leverhulme Trust Research Fellow. The work was supported by grants from Cancer Research UK (C20658/A6656), AICR(10-0158) and the Melville Trust for the Cure and Care of Cancer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Deshmukh
Rights and permissions
About this article
Cite this article
Khandelwal, N., Simpson, J., Taylor, G. et al. Nucleolar NF-κB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death Differ 18, 1889–1903 (2011). https://doi.org/10.1038/cdd.2011.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.79
