
Abstract
Myogenic differentiation requires the coordination between permanent cell cycle withdrawal, mediated by members of the cyclin-dependent kinase inhibitor (CKI) family, and activation of a cascade of myogenic transcription factors, particularly MYOGENIN (MYOG). Recently, it has been reported that the Protein aRginine Methyl Transferase PRMT5 modulates the early phase of induction of MYOG expression. Here, we show that the histone- and PRMT5-associated protein COPR5 (cooperator of PRMT5) is required for myogenic differentiation. C2C12 cells, in which COPR5 had been silenced, could not irreversibly exit the cell cycle and differentiate into muscle cells. This phenotype might be explained by the finding that, in cells in which COPR5 was downregulated, p21 and MYOG induction was strongly reduced and PRMT5 recruitment to the promoters of these genes was also altered. Moreover, we suggest that COPR5 interaction with the Runt-related transcription factor 1 (RUNX1)–core binding factor-β (CBFβ) complex contributes to targeting the COPR5–PRMT5 complex to these promoters. Finally, we present evidence that COPR5 depletion delayed the in vivo regeneration of cardiotoxin-injured mouse skeletal muscles. Altogether, these data extend the role of COPR5 from an adaptor protein required for nuclear functions of PRMT5 to an essential coordinator of myogenic differentiation.
Similar content being viewed by others


Main
Coordination between permanent cell cycle exit and initiation of terminal differentiation is critical during tissue development, a process that requires a controlled balance between cell proliferation, apoptosis and differentiation. The regulated expression of the cyclin-dependent kinase inhibitors (CKIs) p21Cip1/Waf1 (p21), p27Kip1 (p27) and p57Kip2 (p57) has a key role in promoting and maintaining cell cycle arrest in quiescent cells and during differentiation of many cell types.1 Importantly, only quiescent cells, and not terminally differentiating cells, retain the ability to reverse the proliferation block induced by p21. Moreover, quiescent cells are less likely to differentiate than proliferating cells, indicating that they employ active mechanisms to prevent the adoption of a non-dividing status (i.e., differentiation) associated with permanent cell cycle withdrawal.2 Skeletal muscle formation is characterized by the fusion of myoblasts into myotubes and increased synthesis of muscle contractile proteins. Differentiating myoblasts strongly accumulate CKIs3, 4, 5, 6 and the differentiation programme is initiated by the ordered expression of several bHLH transcription factors of the myogenic regulatory factor family.7 Among them, MYOD1, which is expressed in proliferating, undifferentiated committed myoblasts, and MYOGENIN (MYOG), which is expressed early during the differentiation programme, are essential for inducing muscle cell differentiation.7 In the C2C12 myoblast cell line, MYOD1-dependent differentiation involves induction of MYOG while cells are still proliferating, followed by p21-dependent exit from the cell cycle.3, 5 Coupling the onset of differentiation with cell cycle withdrawal involves the direct activation by MYOD1 of genes involved in the regulation of the cell cycle, such as p21, RB and CYCLIN D3 (CCND3).3, 4
A growing body of evidence suggests that members of the Protein aRginine Methyl Transferases (PRMTs) family participate actively in the regulation of cell proliferation. Indeed, they modulate chromatin dynamics, transcriptional regulation, RNA metabolism, signal transduction and cell cycle checkpoint controls through methylation of histone and non-histone proteins.8, 9 Particularly, PRMT5 is essential for cell proliferation and PRMT5 deficiency triggers cell cycle arrest in G1.10 Conversely, stable expression of PRMT5 stimulates cell proliferation and transforms NIH3T3 cells, which can then grow in an anchorage-independent manner.11 Moreover, PRMT5 is an important cytosolic factor, as evidenced by a recent study that implicates PRMT5 in the maintenance of ES cell pluripotency.12 However, PRMT5 associates also with nuclear complexes to mediate mainly transcriptional repression,13, 14, 15, 16, 17 although a recent work has reported that PRMT5 facilitates the activation of the MYOG promoter during MYOD-induced muscle differentiation.18
We previously reported that the adaptor protein COPR5 (cooperator of PRMT5) strongly binds to PRMT5 and histone H4, and that its silencing in proliferating U2OS cells reduces PRMT5 recruitment to the promoter of CCNE1, a key regulator of proliferation.19 We now investigated whether COPR5 could play a role in muscle cell differentiation by modulating the recruitment of PRMT5 to the promoter of genes involved in the coordination between cell cycle exit, such as p21, and differentiation, for instance, MYOG. We show that C2C12 cells in which COPR5 is downregulated exit the cell cycle in a state which is not permissive for differentiation due to imbalanced CKI and MYOG expression. COPR5 silencing impaired the recruitment of PRMT5 to the p21 and MYOG promoters early during differentiation. Finally, we observe that in vivo downregulation of COPR5 delayed skeletal muscle regeneration in mice following cardiotoxin (CTX)-induced injury. Collectively, these data suggest that COPR5 is a coordinator of PRMT5 activity targeted to a subset of key differentiation genes.
Results
COPR5 silencing interferes with myogenic differentiation
To investigate the role of the chromatin- and PRMT5-associated protein COPR5 during myogenic differentiation, C2C12 myoblasts, which can be induced to differentiate into muscle cells ex vivo,20 were transduced with retroviral vectors that express COPR5 shRNA (shCOPR5) or as control Luciferase shRNA (shLUC). As expected, upon shift to differentiation medium (DM), control shLUC cells stopped proliferating, aligned and fused to form multinuclear myotubes (Figure 1a, upper panels). In contrast, shCOPR5 cells failed to form differentiated myotubes (Figure 1a, lower panels). COPR5 downregulation in shCOPR5 cells could be confirmed by quantitative RT-PCR (RT-qPCR) analysis (Figure 1b), but not by western blotting because the used antibody did not react with the mouse protein. Similar results were obtained with two other COPR5 shRNAs, as well as with an shRNA directed against PRMT5 (Supplementary Figures S1A and S1B). The failure of shCOPR5 cells to differentiate was also confirmed by RT-PCR and immunofluorescence studies that showed the very low expression of the myogenic marker Myosin (MHC) in comparison with those detected in shLUC cells soon after shifting to DM (Figure 1b, Supplementary Figures S1A and S1C). Similar results were obtained by silencing COPR5 expression in primary human myoblasts (Figures 1c–e). Importantly, although we observed that ectopic COPR5 expression did not promote obviously by itself the differentiation phenotype in C2C12 cells (Supplementary Figure S1D), myotube formation was restored in shCOPR5 C2C12 cells upon transduction with viral particles encoding human COPR5, the expression of which is not affected by the mouse anti-COPR5 shRNAs (Figure 1f). Collectively, these data indicate that COPR5 is required for myogenic differentiation of C2C12 cells.

COPR5 downregulation with shRNA interferes with myogenic differentiation. (a) Phase-contrast micrographs of proliferating (pro), confluent (day 0; D0) and differentiating (D1 and D3) C2C12 cells that express control (shLUC) or COPR5 (shCOPR5) shRNAs. (b) Expression analysis of COPR5 and myosin heavy chain (MHC) was performed by RT-qPCR. Results were normalized to S26 RNA and values are expressed as the fold change compared with control cells. Values are the means±SD of three independent experiments. (c) Phase-contrast micrographs of proliferating (pro) and differentiating (D1 and D3) human skeletal myoblasts transduced either with LUC- or COPR5-shRNA encoding viral particles. (d) Expression of MHC mRNA in primary human skeletal myoblasts transduced as in a and induced to differentiate (is shown at D3). The effect of COPR5 downregulation on MHC1 expression was monitored by RT-qPCR and results were normalized to RPLP0 expression; analysis was performed as in b. (e) Protein extracts from LUC or COPR5 shRNA-transduced human primary myoblasts were analysed by western blotting using an anti-COPR5 antibody. (f) Human HA-tagged COPR5 was expressed in mouse C2C12 cells in which endogenous COPR5 was silenced. The phenotype in differentiating conditions is presented and visualized by the formation of myotubes (left panels) at D3 after induction of differentiation. Detection of HA–COPR5 by western blotting confirmed its resistance to the effects of shRNAs specific for mouse COPR5 (right panels)
C2C12 cells in which COPR5 has been downregulated fail to irreversibly exit the cell cycle
Differentiating myoblasts must arrest in the G1 phase of the cell cycle to terminally differentiate.21 Thus, to address whether the differentiation defect observed in shCOPR5 C2C12 cells could be due to a failure to irreversibly exit the cell cycle, we compared growth curves, cell cycle profiles and reversibility of cell cycle arrest in control shLUC and shCOPR5 C2C12 cells. In growth medium (GM), both cell populations behaved similarly (Figures 2a and b). Similarly, no differences were observed when cells were cultured in DM (Figures 2a and b), or in the presence of methylcellulose (+MeC) to promote G1 or G0 arrest (Supplementary Figure S2). Conversely, when cells were first growth arrested in DM and then switched back to GM (DM → GM), a significant fraction of shCOPR5 cells started to proliferate again and entered S phase, whereas most of control shLUC cells remained arrested, as indicated by the growth curves and FACS analysis of BrdU-labelled cells (Figures 2c and d). These results show that shCOPR5 cells can exit the cell cycle when transferred to DM, but not in a permanent way, suggesting that COPR5 controls indirectly or directly a subset of genes required for establishing the G1 arrest compatible with induction of myogenic conversion.

COPR5-silenced cells exit the cell cycle in a refractory state for differentiation. (a) Proliferation curves of selected C2C12 myoblast populations transduced with LUC or COPR5 shRNA and maintained in growth medium (GM) or differentiation medium (DM) until day 6. Values represent the means±SD of two independent experiments made in triplicates. (b) BrdU incorporation in proliferating (GM) or differentiating (DM) shLUC or shCOPR5 C2C12 cells analysed by FACS. The percentage of cells in the different phases of the cell cycle is indicated. (c) Proliferation curves as in a of selected C2C12 populations cultivated in DM for 4 days and then switched to GM. (d) The same experiment as in b but with cell culture conditions described in c is presented
The expression of a subset of myogenic inducers and cell cycle regulators is altered in C2C12 cells, in which COPR5 has been downregulated
To understand the mechanisms underlying the defects observed in shCOPR5 C2C12 cells induced to differentiate, we first examined the expression of the cell cycle regulators involved in the tight control of the permanent G1 arrest required for differentiation.21 As expected, quantification of the protein levels of the CKIs p21, p27 and p57 as well as of RB and CCND3 showed a significant increase in control shLUC cells after switching to DM (Figure 3a). Conversely, in shCOPR5 cells, expression of p21 and p27 was significantly lower, whereas the level of RB, CCND3 and p57 was only slightly affected (Figure 3a). Similar analysis of the main myogenic inducers MYOD1 and MYOG showed that both protein expression levels increased significantly during differentiation of shLUC cells (Figure 3b). Conversely, in shCOPR5 cells, MYOG induction was strongly reduced, while MYOD1 level was largely unaffected (Figure 3b). Moreover, the mRNA level of both the myogenic and cell cycle-regulated genes affected by COPR5 knockdown showed that it followed the protein variation level (Figure 3c). Interestingly, expression of PRMT4 and PRMT5, the two PRMTs involved in C2C12 differentiation,22 was not altered by COPR5 downregulation (Figure 3c). By contrast, mRNA level of p8, which encodes an HMG protein involved in the mechanisms that restrict myogenic differentiation to the G1 phase,23 was reduced, while those of HES1 and HES6, which are involved in the control of cell cycle exit and myogenic differentiation, respectively,24, 25 remained unaltered (Supplementary Figure S3A). Finally, COPR5 expression in shLUC cells increased during muscle differentiation, reinforcing the hypothesis of a role of COPR5 in this process.

The expression of some cell cycle regulators and myogenic inducers are altered in COPR5-depleted cells. (a) Protein extracts from LUC or COPR5 shRNA-transduced C2C12 cells were recovered at different time points (pro, D0, D1 and D3 of differentiation) and analysed by western blotting using antibodies against different cell cycle regulators involved in myogenic differentiation (upper panel). Quantification of the western blot is shown in lower panel. (b) The same analysis as in a was performed to analyse the protein level of different myogenic markers or inducers, as indicated. (c) The expression profile of different cell cycle regulators was assessed by RT-qPCR with RNA from shLUC and shCOPR5 C2C12 cells at different differentiation time points, as indicated. Results were normalized to S26 RNA and values are expressed as the fold change compared with control cells (means±SD of three independent experiments)
Altogether, these results indicate that the differentiation defect observed in shCOPR5 cells is associated with reduced expression of p21, p27 and p8 (but not of RB, CCND3 and p57) and of MYOG (but not MYOD1), thus suggesting that only a subset of the differentiation programme is modulated by COPR5.
Early recruitment of PRMT5 to the p21 and MYOG promoters requires COPR5, which can associate with the RUNX1 complex
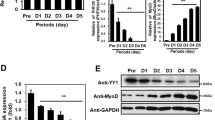
We next investigated how COPR5 controls the expression of p21 and MYOG. As COPR5 is a histone- and PRMT5-associated protein,19 we first tested whether it was present in the chromatin environment of the genes encoding these factors. To address this question, we performed chromatin immunoprecipitations (ChIPs) using an anti-HA antibody on C2C12 cells that had been stably transfected with a plasmid expressing HA-tagged COPR5. Consistent with the effect of COPR5 silencing on p21, MYOG and p8 expression, transient recruitment of HA–COPR5 to the p21, MYOG and p8, but not to the p57 and MYOD1, promoters was detected at day 1 (D1) after switching to DM (Figure 4a and Supplementary Figure S3B). Interestingly, reCHIP experiment using the anti-HA and anti-PRMT5 antibodies as first and second antibody, respectively, showed on the p21 promoter, but not the MYOG promoter, that COPR5 and PRMT5 were detected as part of a same complex (Supplementary Figure S4). As PRMT5 regulates the early phase of MYOG activation,18 we then assessed whether COPR5 downregulation could hinder the recruitment of PRMT5 to the MYOG, but also p21 and p8 promoters. ChIPs using differentiating shLUC and shCOPR5 C2C12 cells showed the presence of PRMT5 at the p21 as well as MYOG and p8 promoters in shLUC cells at D1 after the switch to DM, whereas the recruitment of PRMT5 to these promoters was strongly reduced in shCOPR5 cells (Figure 4b and Supplementary Figure S3C). Consequently to this observation we analysed whether the detection level of two PRMT5-mediated histone methylation marks (H3R8me2s and H4R3me2s) was affected as well. Strikingly, we failed to detect the presence of the H3R8me2s mark on both the MYOG and p21 promoters on chromatin from C2C12 cells infected with a CTL shRNA (LUC), while the presence of the H4R3me2s mark was detected (Supplementary Figure S5). Interestingly, the detection of the latter mark decreased at D1 and D3 on the MYOG promoter only in COPR5 shRNA-treated cells, probably consequently to the decreased recruitment of PRMT5 observed at D1 on this promoter. However, this mark could not be considered as a full readout of PRMT5 activity (see D0 with the MYOG promoter and results with the p21 promoter). A very slight increase, if any, in the detection of the H3R8me2s mark was observed in COPR5 shRNA-treated cells at D3. We next tested whether COPR5 silencing could affect the pool of PRMT5 associated with chromatin during differentiation. A fractionation assay showed that a small amount of PRMT5 was associated with the chromatin-enriched fractions (P3) in control shLUC cells during differentiation. Conversely, the PRMT5 level in the P3 fraction of shCOPR5 cells was strongly reduced, while it remained unchanged in the soluble cytosolic (S1) and nuclear (S2) fractions (Figure 4c). This effect is specific for PRMT5 as no alteration in the cellular distribution of another PRMT family member, PRMT1, was observed. Moreover, those of the chromatin remodeler BRG1 and MEKK1, used as controls of the S1 and P3 fractions, respectively, did not vary in shLUC and shCOPR5 cells (Figure 4c). Altogether, these results indicate that COPR5 downregulation affects the activation of genes that play a key role during muscle cell differentiation by hindering the recruitment of PRMT5 to their promoters. We then asked whether COPR5 or the COPR5–PRMT5 complex could bind to specific factors present on these promoters. As both the p21 and MYOG promoters are characterized by the presence of adjacent Runt-related transcription factor 1 (RUNX1) and MYOD1 functional DNA binding sites26 and because MYOD1 is known to interact with PRMT5,22 we assessed whether the COPR5–PRMT5 complex could interact with RUNX1 and its partner protein core binding factor-β (CBFβ), which cooperate with MYOD1 in regulating target genes in myoblasts.26 GST pull-down experiments using GST–COPR5 (GC) and protein extracts from U2OS cells transfected with HA–RUNX1 or Flag–CBFβ confirmed that COPR5 interacts with PRMT5, as expected,19 as well as with CBFβ and RUNX1 (Figure 4d). Moreover, when using a GST–COPR5 deletion mutant (ΔC4), which does not interact with PRMT5,19 only the interaction with CBFβ was observed (Figure 4d). The capacity of COPR5 to associate with CBFβ was confirmed by ChIP experiments (Figure 4e), suggesting that COPR5 could be targeted via the RUNX1 complex to promoters. Conversely, no RUNX1 DNA binding site was found in the p27 promoter, although COPR5 binds to this promoter and recruits PRMT5 at a later stage of differentiation (data not shown), suggesting that alternative mechanisms are involved in COPR5 regulation of p27 transcriptional activity.

Early recruitment of PRMT5 to the MYOG and p21 promoters requires COPR5, which binds to the RUNX1 complex. (a) Chromatin immunoprecipitation was performed using C2C12 cells that express HA–COPR5 and were grown in different culture conditions, as indicated. The recruitment of COPR5 to the p21, p57, MYOG and MYOD1 promoters was analysed. Relative values are expressed as the percentage of immunoprecipitated chromatin relative to the input and represent the means±SD of three independent experiments. (b) Chromatin immunoprecipitation was performed using shLUC and shCOPR5 C2C12 cells in different culture conditions, as indicated. The recruitment of PRMT5 was performed on the p21 and MYOG gene promoters. Results were analysed as in a. (c) Western blot analysis using anti-PRMT1 and -PRMT5 antibodies revealed a specific alteration in the repartition of PRMT5, compared with PRMT1, in a fractionation assay of shCOPR5 C2C12 cells performed at D0 and D1. S1: cytoplasmic soluble, S2: nuclear soluble and P3: chromatin fraction. The anti-MEKK1 and -BRG1 antibodies were used as control of the S1 cytosolic and P3 chromatin fractions, respectively. (d) GST pull-down assay was performed after incubation of full-length GST–COPR5 (GC) or a mutant GST–COPR5 in which the C-terminal part had been deleted (ΔC4) with protein extracts from U2OS cells that had been transfected with HA-tagged RUNX1 or Flag-tagged CBFβ. (−) Corresponds to extracts of non-transfected cells. Anti-HA, -Flag and -PRMT5 antibodies were used. (e) Immunoprecipitation of COPR5 was validated in C2C12 cells that express HA–COPR5 and that had been transfected with Flag-CBFβ. (−) Corresponds to extracts from non-transfected cells. Inp: input (10% of total extract); IP: immunoprecipitation with anti-Flag antibodies
Collectively, these results indicate that COPR5 regulates recruitment of PRMT5 to the promoters of p21 and MYOG, a subset of genes that play key roles during muscle cell differentiation.
COPR5 downregulation delays in vivo muscle regeneration
To provide evidence of the role of COPR5 in myogenic differentiation in vivo, the two tibialis anterior muscles of mice were damaged by injection of CTX to reactivate satellite cells, which are considered the adult stem cells responsible for post-natal growth, regeneration and repair of skeletal muscle. Two days later, COPR5 or control LUC shRNA (contralateral muscle) plasmid DNA was electroporated and then mice were killed at different time points to follow muscle regeneration. At D2 after electroporation, muscle tissue sections showed increased cellularity attributable to both proliferation of satellite cells and recruitment of inflammatory cells to damaged fibres (Figure 5a; haematoxylin–eosin staining). At this stage no obvious difference could be detected between control shLUC and shCOPR5 muscles. In contrast, at D5, high level of necrosis was only observed in muscles in which COPR5 was silenced in comparison with control muscles (Figure 5a). At D7.5, regeneration had largely taken place in control muscles and many newly regenerated fibres were present with central nuclei, a known hallmark of recent muscle regeneration (Figure 5a). Such fibres were detected also in muscles in which COPR5 was downregulated, but to a lesser extent than in controls. In accordance with results in C2C12 cells, the mRNA levels of MYOG and p21 were reduced significantly in COPR5-depleted muscles compared with control but transiently (Figure 5b), probably due to a milder efficiency of the knockdown in vivo in the muscle compared with an in vitro-infected cell population. Consistently, immunohistochemical analysis using an anti-MYOG antibody on muscle sections performed at D5 showed less myofibres stained compared with control (Supplementary Figure 6). To assess whether this phenotype was due to a reduction in the pool of satellite cells activated following CTX-induced muscle injury, muscles were injected with retroviral particles expressing LUC or COPR5 shRNAs that could only transduce activated and proliferating cells. An enriched satellite cell/isolated myoblast preparation was recovered from the tibialis anterior muscle at D3 after injury and the effect of COPR5 downregulation on satellite cells was monitored by quantifying the number of CD34-positive (a satellite cell marker) cells that normally remains steady during homeostasis and injury-induced regeneration.27 The number of CD34-positive cells in COPR5 shRNA-transduced muscles was lower than in control muscles, indicating that the pool of satellite cells to generate committed myogenic progenitors was reduced following downregulation of COPR5. In accordance with the delayed regeneration observed in CTX-treated muscles, these findings strongly suggest that COPR5 plays a role during muscle regeneration, probably at the level of the satellite cell amplification and/or differentiation programme.

COPR5 silencing slows down muscle regeneration in vivo. (a) Haematoxylin–eosin (HE) staining of paraffin-embedded muscle sections from tibialis anterior (TA) muscles of mice in which muscle necrosis was induced by cardiotoxin (CTX) injection, followed 2 days later by electroporation of LUC or COPR5 shRNA-encoding plasmids. Bars: 200 μm. (b) mRNA level of COPR5, p21 and MYOG genes was analysed by RT-qPCR after RNA extraction at D5 from muscle that had been treated as in a. Results were normalized and expressed as in Figure 3. Error bars of the means correspond to two independent experiments, except at D5. (c) Cells from muscles were injured and infected with ecotropic Moloney-based retroviral particles encoding either LUC or COPR5 shRNAs, recovered at D3 and analysed by flow cytometry to quantify the CD34+ population (i.e., satellite cells). x-axis: CD34; y-axis: SSC (side scatter)
Discussion
Here we show that the PRMT5 adaptor COPR5 plays a role in myogenic differentiation of cultured C2C12 cells and modulated the transcriptional regulation of genes that are important for the control of cell cycle exit (p27 and p21) and muscle differentiation (MYOG). Increased levels of p27 and p21 have been correlated with the irreversible growth arrest required for differentiation.3, 4 Indeed, the level of p27 expression is critical for initiating growth arrest in differentiating myoblasts and in maintaining this arrest in terminally arrested mature myotubes, while in the presence of differentiation signals, p21 and p57 enhance cell survival.28 C2C12 cells, in which COPR5 was downregulated, could exit the cell cycle when switched to DM (Figure 2), suggesting that the residual levels of the CKIs p21 and p27, and or a compensatory increase of p57 expression, are sufficient to trigger cell survival and this initial growth arrest. Consistent with this, while p21 and p27 expression was strongly reduced, p57 expression was slightly increased in differentiated COPR5-silenced cells (Figure 3). A similar MYOD1-mediated upregulation of p57 was previously reported in cells lacking p21.29 Noteworthy, commitment to terminal differentiation is confined to the G1 phase of the cell cycle in differentiating myoblasts.21 Strikingly, we also observed a decreased mRNA level of the G1-induced p8 gene, which encodes an HMG protein involved in the mechanism to restrict myogenic differentiation to the G1 phase.23 These findings together with our previous report that the COPR5–PRMT5 complex represses the CCNE1 gene in U2OS cells13 suggest that this complex is involved in coordinating CKI and p8 activation, while repressing CCNE1 expression as required for a stable cell cycle exit competent for differentiation. Consistent with this, we found that HA–COPR5 is recruited to the p21, MYOG and p8 promoters (Figure 4a, Supplementary Figure S3). Moreover, the reduction of PRMT5 recruitment to these promoters in C2C12 cells, in which COPR5 had been silenced (Figure 4b, Supplementary Figure S3), positions COPR5 centrally within the transcriptional regulatory mechanism that coordinates cell cycle exit and terminal muscle differentiation, and future studies will aim at identifying additional components and the upstream signalling that control this novel network. Our reCHIP data suggest that the accessibility of PRMT5 within COPR5-containing complex is different on the p21 and MYOG promoters and might reflect a difference in the BRG1-mediated chromatin remodelling regulation at these promoters. In accordance with this, previous work supports that BRG1 is required for the induction of all muscle-specific gene expression by MyoD, thus including MYOG, whereas induction of the cell cycle regulators, p21, Rb, and cyclin D3 occurred independently of SWI/SNF function.30 Our data also showed that COPR5–PRMT5 complex can interact with the transcription factor RUNX1–CBFβ complex, suggesting how COPR5 could target PRMT5 activity to a subset of genes, including p21 and MYOG. Both the p21 and MYOG promoters are characterized by the presence of adjacent RUNX1 and MYOD1 functional DNA binding sites. Interestingly, the interaction between the RUNX1 complex and MYOD1 participates in regulating the balance between proliferation and differentiation.26 Moreover, the methylation of RUNX1 by PRMT1 regulates its interaction with SIN3A and modulates RUNX1-inducible gene expression.31 Furthermore, crystal studies indicate that another GRG motif resembling those methylated by PRMT5 in other proteins is important for DNA binding.32 Although we failed to methylate RUNX1 by COPR5–PRMT5 in vitro (data not shown), we do not exclude the possibility that such methylation could exist in a chromatin-dependent context to dissociate RUNX1 complex from MYOD1 and/or from DNA, allowing promoter activation.
Finally, consistent with a key role of COPR5 in myogenic conversion, its downregulation in CTX-injured muscles in vivo delayed muscle regeneration by reducing the pool of CD34-positive satellite cells (Figure 5). The presence of residual COPR5 indicated the in vivo depletion of COPR5 was milder than those obtained in vitro. Interestingly, muscle regeneration is also impaired in p21−/− mice,33 thus stressing again the importance of COPR5 as a regulator of the expression of p21 and reinforcing the notion that PRMT5 is essential for myogenesis. Accordingly, beyond its impact on histone methylation, PRMT5 was shown recently to methylate Ash2L, a factor associated with PAX in a complex that directly controls entry into the myogenic programme of satellite cell-derived myoblasts.34, 35 Whether COPR5 downregulates directly the expression of satellite cell-driver genes such as PAXs or whether the phenotype due to COPR5 silencing is an indirect consequence of the dysfunction of other myogenic regulators remains to be investigated.
This study clearly positions the COPR5–PRMT5 complex as a potential sensor and integrator of differentiation signals and a novel coordinator of cell cycle exit and differentiation. Noteworthy, the coupling of these two events is often perturbed during tumourigenesis and several types of tumours, including rhabdomyosarcoma, are characterized by an undifferentiated phenotype. Moreover, PRMT5 expression is deregulated and/or mislocalized in several types of cancer.36, 37 Therefore, it would be interesting to test whether alteration of COPR5 could contribute to the abnormal differentiation of tumours of mesenchymal origin.
Materials and Methods
Cell culture conditions
The C2C12 cell line was cultured in Dulbecco's modified Eagle medium supplemented with 15% fetal calf serum using standard conditions. To induce myogenic differentiation, GM was replaced with DM containing 2% serum (FetalClone III PerBio, Brebières, France). Primary human skeletal muscle myoblasts were cultured similar to C2C12 cells in the presence of Ultroser G (Gibco, St Aubin, France). Generation of retroviral particles and infection have already been described.19
RNA isolation, cDNA synthesis and RT-qPCR amplification
RNA isolation, reverse transcription and qPCR were performed as described.38
ChIP
ChIP was carried out essentially as described13 using anti-H4R3me2s (Abcam, Cambridge, UK), PRMT5 (Euromedex, Mundosheim, France) and anti-HA (Sigma-Aldrich, St Quentin Fallavier, France) antibodies. H3R8me2s antibody was a gift from S Sif.
The sequences of the oligonucleotides used for shRNA, PCR and CHIP experiments are listed in Supplementary Information.
Western blot analysis
Proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes (Whatman, Maidstone, Kent, UK), and probed with anti-PRMT1, PRMT5, BRG1, MYOG, MYOD1, RB, p21 or p27 antibodies (Santa Cruz, Heidelberg, Germany), as indicated or an anti-HA antibody (12CA5, Sigma-Aldrich). Membranes were then incubated with the appropriate HRP-conjugated secondary antibodies. Immunoreactive bands were detected by chemiluminescence. Quantification was performed using AIDA software (Dalian Software, Dalian, China).
Flow cytometry
For cell cycle analysis, experiments and analysis were performed as described.38 Briefly, cells were incubated with 300 μM BrdU (Sigma-Aldrich) for 2 h, fixed in 70% ethanol solution and permeabilized with 0.2% Triton X-100 for 10 min. Then, cells were treated with 0.2 N HCl before incubation with a mouse anti-BrdU antibody (1 : 30 diluted in PBS with 0.2% Tween 20 and 1% BSA; BD Biosciences, Le Pont de Claix, France) at room temperature for 1 h followed by 1-h incubation with an FITC-conjugated secondary antibody (1 : 300; BD Biosciences). DNA was then counterstained with 7-amino-actinomycin D (1 : 50; Sigma-Aldrich) in the presence of RNase overnight. Cell cycle profiles were analysed by flow cytometry (FACScan; BD Biosciences) using the CellQuest software (BD Biosciences). For CD34 expression, 106 cells were labelled with a biotinylated anti-CD34 antibody (marker) in 100 μl PBS with 0.3% BSA on ice for 45 min, then washed and incubated with a streptavidin-Texas Red-conjugated secondary antibody for 45 min.
Cardiotoxin muscle injury
Tibialis anterior of mice (type of mice) were injected with 10 μM CTX (Calbiochem, Nottingham, UK) and processed 2 days later for electroporation, as described.39 At the indicated time points, mice were killed, tibialis anterior muscles harvested and fixed in 4% formaldehyde overnight before to be paraffin-embedded for further analysis. For enriched satellite cell/myoblast preparations, isolation was performed as previously described,22 after muscles were treated as above with CTX and co-injected with ecotropic Moloney-retroviral particles that encoded shLUC or shCOPR5.
Mice and animal care
Animal experiments were approved by and performed in accordance with the guidelines of the Ethics Committee of the Languedoc-Roussillon region (France).
Abbreviations
- CKI:
-
cyclin-dependent kinase inhibitor
- MYOG :
-
MYOGENIN
- PRMT5 :
-
protein arginine methyl transferase 5
- COPR5 :
-
cooperator of PRMT5
- RUNX1 :
-
Runt-related transcription factor 1
- CBFβ:
-
core binding factor-β
References
Sherr CJ, Roberts JM . Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995; 9: 1149–1163.
Sang L, Coller HA, Roberts JM . Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science 2008; 321: 1095–1100.
Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ et al. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 1995; 267: 1018–1021.
Guo K, Wang J, Andres V, Smith RC, Walsh K . MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol 1995; 15: 3823–3829.
Andres V, Walsh K . Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol 1996; 132: 657–666.
Zhang P, Wong C, Liu D, Finegold M, Harper JW, Elledge SJ . p21(CIP1) and p57(KIP2) control muscle differentiation at the myogenin step. Genes Dev 1999; 13: 213–224.
Puri PL, Avantaggiati ML, Balsano C, Sang N, Graessmann A, Giordano A et al. p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J 1997; 16: 369–383.
Bedford MT, Richard S . Arginine methylation an emerging regulator of protein function. Mol Cell 2005; 18: 263–272.
Litt M, Qiu Y, Huang S . Histone arginine methylations: their roles in chromatin dynamics and transcriptional regulation. Biosci Rep 2009; 29: 131–141.
Scoumanne A, Zhang J, Chen X . PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res 2009; 37: 4965–4976.
Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S . Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004; 24: 9630–9645.
Tee WW, Pardo M, Theunissen TW, Yu L, Choudhary JS, Hajkova P et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev 2011; 24: 2772–2777.
Fabbrizio E, El Messaoudi S, Polanowska J, Paul C, Cook JR, Lee JH et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep 2002; 3: 641–645.
Kwak YT, Guo J, Prajapati S, Park KJ, Surabhi RM, Miller B et al. Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Mol Cell 2003; 11: 1055–1066.
Pal S, Yun R, Datta A, Lacomis L, Erdjument-Bromage H, Kumar J et al. mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol Cell Biol 2003; 23: 7475–7487.
Hou Z, Peng H, Ayyanathan K, Yan KP, Langer EM, Longmore GD et al. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol 2008; 28: 3198–3207.
Ancelin K, Lange UC, Hajkova P, Schneider R, Bannister AJ, Kouzarides T et al. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol 2006; 8: 623–630.
Dacwag CS, Ohkawa Y, Pal S, Sif S, Imbalzano AN . The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol Cell Biol 2007; 27: 384–394.
Lacroix M, Messaoudi SE, Rodier G, Le Cam A, Sardet C, Fabbrizio E . The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep 2008; 9: 452–458.
Blau HM, Pavlath GK, Hardeman EC, Chiu CP, Silberstein L, Webster SG et al. Plasticity of the differentiated state. Science 1985; 230: 758–766.
Clegg CH, Linkhart TA, Olwin BB, Hauschka SD . Growth factor control of skeletal muscle differentiation: commitment to terminal differentiation occurs in G1 phase and is repressed by fibroblast growth factor. J Cell Biol 1987; 105: 949–956.
Dacwag CS, Bedford MT, Sif S, Imbalzano AN . Distinct protein arginine methyltransferases promote ATP-dependent chromatin remodeling function at different stages of skeletal muscle differentiation. Mol Cell Biol 2009; 29: 1909–1921.
Sambasivan R, Cheedipudi S, Pasupuleti N, Saleh A, Pavlath GK, Dhawan J . The small chromatin-binding protein p8 coordinates the association of anti-proliferative and pro-myogenic proteins at the myogenin promoter. J Cell Sci 2009; 122: 3481–3491.
Kobayashi T, Kageyama R . Hes1 regulates embryonic stem cell differentiation by suppressing Notch signaling. Genes Cells 2009; 15: 689–698.
Cossins J, Vernon AE, Zhang Y, Philpott A, Jones PH . Hes6 regulates myogenic differentiation. Development 2002; 129: 2195–2207.
Philipot O, Joliot V, Ait-Mohamed O, Pellentz C, Robin P, Fritsch L et al. The core binding factor CBF negatively regulates skeletal muscle terminal differentiation. PLoS One 2010; 5: e9425.
Ieronimakis N, Balasundaram G, Rainey S, Srirangam K, Yablonka-Reuveni Z, Reyes M . Absence of CD34 on murine skeletal muscle satellite cells marks a reversible state of activation during acute injury. PLoS One 2010; 5: e10920.
Myers TK, Andreuzza SE, Franklin DS . p18INK4c and p27KIP1 are required for cell cycle arrest of differentiated myotubes. Exp Cell Res 2004; 300: 365–378.
Figliola R, Maione R . MyoD induces the expression of p57Kip2 in cells lacking p21Cip1/Waf1: overlapping and distinct functions of the two cdk inhibitors. J Cell Physiol 2004; 200: 468–475.
Roy K, de la Serna IL, Imbalzano AN . The myogenic basic helix-loop-helix family of transcription factors shows similar requirements for SWI/SNF chromatin remodeling enzymes during muscle differentiation in culture. J Biol Chem 2002; 277: 33818–33824.
Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S et al. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev 2008; 22: 640–653.
Bartfeld D, Shimon L, Couture GC, Rabinovich D, Frolow F, Levanon D et al. DNA recognition by the RUNX1 transcription factor is mediated by an allosteric transition in the RUNT domain and by DNA bending. Structure 2002; 10: 1395–1407.
Hawke TJ, Meeson AP, Jiang N, Graham S, Hutcheson K, DiMaio JM et al. p21 is essential for normal myogenic progenitor cell function in regenerating skeletal muscle. Am J Physiol Cell Physiol 2003; 285: C1019–C1027.
Butler JS, Zurita-Lopez CI, Clarke SG, Bedford MT, Dent SY . Protein arginine methyltransferase 1 (PRMT1) methylates Ash2L, A shared component of mammalian histone H3K4 methyltransferase complexes. J Biol Chem 2011; 286: 12234–12244.
McKinnell IW, Ishibashi J, Le Grand F, Punch VG, Addicks GC, Greenblatt JF et al. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol 2008; 10: 77–84.
Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin Cancer Res 2005; 11: 473–482.
Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S . Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J 2007; 26: 3558–3569.
El Messaoudi S, Fabbrizio E, Rodriguez C, Chuchana P, Fauquier L, Cheng D et al. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc Natl Acad Sci USA 2006; 103: 13351–13356.
Schertzer JD, Gehrig SM, Ryall JG, Lynch GS . Modulation of insulin-like growth factor (IGF)-I and IGF-binding protein interactions enhances skeletal muscle regeneration and ameliorates the dystrophic pathology in mdx mice. Am J Pathol 2007; 171: 1180–1188.
Acknowledgements
We thank G Carnac for primary human skeletal muscle myoblasts; S Ait-Sit-Ali for RUNX1 constructs; our animal facility staff, the RHEM facility (Réseau d’Histologie Expérimentale de Montpellier) and all members of the CS laboratory for helpful discussions. This work was realized with institutional supports of the French CNRS and grants from the FRM, ANR (CS,équipe labélisée 2007) and ARC (EF).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by R De Maria
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Paul, C., Sardet, C. & Fabbrizio, E. The histone- and PRMT5-associated protein COPR5 is required for myogenic differentiation. Cell Death Differ 19, 900–908 (2012). https://doi.org/10.1038/cdd.2011.193
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.193
Keywords
This article is cited by
-
NF1 microdeletion syndrome: case report of two new patients
Italian Journal of Pediatrics (2019)
-
Emerging genotype–phenotype relationships in patients with large NF1 deletions
Human Genetics (2017)
-
The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond
Cellular and Molecular Life Sciences (2015)
-
miR-24 affects hair follicle morphogenesis targeting Tcf-3
Cell Death & Disease (2013)
