
Abstract
Normal stem cells reside in functional niches critical for self-renewal and maintenance. Neural and hematopoietic stem cell niches, in particular, are characterized by restricted availability of oxygen and the resulting regulation by hypoxia-inducible factors (HIFs). Glioblastoma multiforme (GBM) is the most common malignant brain tumor and also contains high degrees of hypoxia. Heterogeneity within the neoplastic compartment has been well characterized in GBM and may be derived from genetic and epigenetic sources that co-evolve during malignant progression. Recent experimental evidence has supported the importance of hypoxia in glioma stem cell (GSC) niches. We hypothesized that HIFs require epigenetic-modifying proteins to promote tumor malignancy in GBM. Here we demonstrate that in GBM the histone methyltransferase mixed-lineage leukemia 1 (MLL1) is induced by hypoxia and enhances hypoxic responses. Loss of MLL1 reduces the expression of HIF transcripts and HIF2α protein. Targeting MLL1 by RNA interference inhibited the expression of HIF2α and target genes, including vascular endothelial growth factor (VEGF). GSCs expressed higher levels of MLL1 than matched non-stem tumor cells and depletion of MLL1 reduced GSC self-renewal, growth, and tumorigenicity. These studies have uncovered a novel mechanism mediating tumor hypoxic responses linking microenvironmental regulation of epigenetic-modifying proteins to cellular heterogeneity and provide rationale for the design of more sophisticated clinical approaches targeting epigenetic regulation.
Similar content being viewed by others


Main
Glioblastoma multiforme (GBM) is one of the most lethal adult malignancies with current therapy offering only palliation.1 Recent elegant genetically engineered models2 and systematic genomic analyses3, 4 have provided new insights into GBM pathogenesis. Characterization of GBM molecular subtypes has further refined our understanding of the intertumoral heterogeneous nature of the disease.5 Additional complexity in tumor biology is derived from intratumoral heterogeneity within the neoplastic compartment. Cellular heterogeneity is driven by two complementary forces: stochastic genetic mutations and epigenetic hierarchies.6 At the apex of the tumor hierarchy are cancer stem cells (CSCs, also known as tumor-initiating cells or tumor-propagating cells) that are functionally defined by their capacities for self-renewal and ability to propagate tumors.6 Although some cancers may not follow the CSC model, GBMs have been the subject of numerous studies supporting the presence of glioma stem cells (GSCs).7, 8, 9, 10 GSCs remain controversial owing to the unresolved nature of the cell(s) of origin, immunophenotypes (enrichment markers), and frequency within tumors. Rigorous functional studies have permitted the identification and characterization of GSCs8, 11 and provided evidence that GSCs are resistant to conventional therapy.7, 12, 13 GSCs are enriched in regions around tumor vessels and necrosis,14 the latter associated with restricted oxygen/hypoxia. GSC maintenance requires both hypoxia-inducible factor-1α (HIF1α) and HIF2α, with HIF2α being preferentially expressed in the GSC compartment.10, 15, 16, 17 However, the molecular mechanisms mediating the effects of hypoxia and HIF2α to promote a stem-like state are poorly characterized, and the regulation of HIF2α expression in GSCs is unknown. Microenvironmental conditions such as hypoxia and acidic stress actively promote the expression of GSC markers and functional characteristics,15, 16, 17, 18 suggesting that the GSC phenotype is plastic and can be modulated by the microenvironment, reminiscent of induced pluripotency.19, 20 Indeed, molecular regulators of induced pluripotent stem cells, such as Sox2 and c-Myc, are expressed by GSCs and regulated by the microenvironment.21, 22 As normal stem cells also exhibit characteristic chromatin patterns,23 we interrogated GSC chromatin regulation in response to hypoxia.
The polycomb genes (e.g. Bmi1) that repress transcription have received substantial attention in glioma and GSCs,24, 25 but the trithorax group that activates transcription has been less well studied and may represent important molecular regulators of cancer phenotypes. One member of the trithorax group involved in cancer is mixed-lineage leukemia 1 (MLL1) (also known as HRX (human trithorax) or ALL-1 (acute lymphocytic leukemia-1)), which is involved in chromosomal rearrangements in a variety of leukemias, including the majority of infant leukemias.26, 27, 28, 29 MLL1 encodes a large protein (3969 amino acids) with several functional domains, including a SET (Su(var), enhancer of zeste, trithorax) domain that contains histone methyltransferase (HMT) activity.29, 30, 31, 32 The loss of regulatory domains and aberrant downstream activity are common among MLL1 fusion proteins26, 27 in leukemia. MLL1 has not been associated with brain tumors to date, but MLL2 and MLL3 are mutated or amplified in medulloblastomas and GBMs.33, 34 Methylation of specific histone lysine residues may either activate or silence gene expression. Bivalent histone methylation is commonly found in stem cell populations and causes genes to be silenced, but poised for activation by MLL1 or other chromatin-modifying proteins, as observed in normal neurogenesis from postnatal neural progenitor cells.35 MLL1 directly functions as a histone 3 lysine 4 (H3K4) methyltransferase, but can also recruit histone 3 lysine 27 (H3K27) demethylases in its complex.30, 36 Although the MLL1 complex may regulate large numbers of genes in some cell types during embryonic development and hematopoiesis, the number of target genes may be much smaller in adult tissues.36 The significance of MLL1 has been largely investigated in leukemia models using gene fusions,26, 28, 31 which transform hematopoietic progenitors and can aberrantly promote expression of tumorigenic target genes such as the homeobox gene HoxA9. Little is known about wild-type MLL1, but a recent report demonstrated that DNA damage leads to MLL1 phosphorylation and checkpoint regulation.32 Further, the core stem cell regulators p53 and β-catenin associate with MLL1 during transcriptional activation.37 Thus, MLL1 and its family members are critical cellular regulators of normal stem cell biology (particularly in the brain) and oncogenesis that integrate a variety of pathways in transcriptional regulation.
Based on this background, we hypothesized that MLL1 may be implicated in the hypoxia response of GBMs and in regulating the tumorigenicity of GSCs. In this study, we examined the relationship between hypoxia and the histone modifier MLL1. We sought to better understand how MLL1 might regulate HIF2α expression and downstream hypoxia responses that have been previously described as important to the tumorigenic phenotype. Finally, we interrogated the effect on tumor propagation following modulation of MLL1 in GSCs.
Results
Hypoxia increases expression of HMT MLL1
As studies have defined the ability of hypoxia to regulate epigenetic modifiers,20 we interrogated the importance of epigenetic regulators on the hypoxia-induced GSC phenotype. One epigenetic modifier previously implicated in leukemia stem cells is the HMT MLL1.26, 28, 29, 31 To determine if MLL1 could play a role in GBM hypoxia responses, we evaluated the transcriptional regulation of MLL1 to environmental oxygen using xenograft-derived 4121 and 387 non-stem cells, which had been previously characterized to gain GSC characteristics after exposure to hypoxia.15 MLL1 mRNA levels were significantly increased under atmospheric hypoxia (1% O2) or treatment with the hypoxia mimetic DFX (Figures 1a and b). These results were confirmed in newly derived CW619 non-stem tumor cells exposed to DFX (Figure 1c). The ability of both 1% O2 and DFX to induce hypoxic gene responses was confirmed by significant induction of HIF2α (Figures 1d–f) and VEGF (Figures 1g–i) as in our prior report.10 These data support the hypothesis that microenvironmental hypoxia induces the epigenetic-modifying protein, MLL1.

MLL1 is a hypoxia-responsive gene in non-stem glioblastoma cells. In all, 4121 or 387 non-stem tumor cells were cultured with 1% oxygen for 4 days or treated with 200 μM DFX for 24 h as indicated. To confirm these data, a freshly isolated human specimen, CW619, was treated with 200 μM DFX for 24 h. Following hypoxic treatment, total RNA was harvested, cDNA generated by reverse transcription, and mRNA evaluated for (a–c) MLL1, (d–f) HIF2A, and (g–i) VEGF. HIF2A and VEGF levels were used as internal hypoxic controls. *P<0.001; **P<0.05
HIF1α and HIF2α are necessary for MLL1 induction
HIFα isoforms have differential effects and expression within glioma.10 Overexpression of non-degradable HIF2α promotes tumorigenicity in non-stem GBM cells, and HIF2α is preferentially expressed in the GSCs in response to hypoxia.15, 16 Although HIF2α is preferentially expressed in GSCs, HIF1α is also present in GBM subpopulations. To determine if HIF1α or HIF2α is required for MLL1 hypoxic regulation, 4302 (Figure 2a), 387 (Figure 2b), and 4121 (Figure 2c) non-stem glioma cells were treated transduced with HIF1A or HIF2A shRNA, and then subjected to hypoxia via DFX. We confirmed specificity of the HIF shRNA constructs (Supplementary Figures 1A–D). Both HIFs modulated MLL1, suggesting that MLL1 is a general hypoxic target (Figures 2a–c). To rule out off-target effects of the HIF shRNA, we utilized an HIF2α small-molecule inhibitor (inhibitor 77, generated by the Iliopoulos Laboratory, Charlestown, MA, USA) that prevents HIF2A translation, thereby reducing HIF2α protein levels.38 To verify the efficacy of the drug, U87 MG glioma cells stably expressing HREs driving luciferase expression were treated with the HIF2α inhibitor (Figure 2d). HRE activity under moderate hypoxia (2% O2) was decreased by greater than 80% when cells were treated with HIF2α inhibitor, whereas more modest effects (50% inhibition) were observed with severe hypoxia (1% O2) (Figure 2d). These data were consistent with evidence that HIF2α, but not HIF1α, is stabilized at oxygen tensions greater than 1%, and both HIF1α and HIF2α proteins are stable at 1% O2.10 Attenuated responses to hypoxia were associated with the ability of the HIF2α inhibitor to reduce HIF2α protein expression as expected (Figure 2e). After confirming that the HIF2α inhibitor was effective in glioma cells, we measured inhibitor effects on MLL1 expression. Unfractionated 4121 cells were exposed to hypoxia induced by DFX treatment in the presence and absence of HIF2α inhibitor. Treatment with the HIF2α inhibitor did not alter HIF2A transcript levels (Figure 2f), but caused a complete loss of the hypoxic response of MLL1 mRNA levels (Figure 2g). We next determined if MLL1 was a direct transcriptional target of the HIFs. Promoter analysis of MLL1 revealed a lack of consensus HRE binding sites typically found in HIF targets, like VEGF (data not shown). To further evaluate HIF binding, ChIP was performed on HIF2α and the presence of MLL1 DNA was analyzed. In comparison to VEGF-positive control, we observed no significant difference in HIF2α binding to MLL1 DNA in 21 or 1% O2 cultured cells, which suggests that MLL1 is not a direct transcriptional target of the HIFs (Supplementary Figure 1E). These data define MLL1 as a novel hypoxia response gene indirectly regulated by the HIFs.

HIF1α and HIF2α are required for MLL1 hypoxic response. (a–c) In all, 4302, 387, or 4121 non-stem glioma cells were stably transduced with HIF1A or HIF2A shRNA, and then treated with 200 μM DFX for 24 h. Following treatment, RNA was harvested and cDNA generated by reverse transcriptase for analysis by RT-qPCR. (d) U87 cells expressing an HRE-driven luciferase expression vector were pretreated with or without HIF2α inhibitor, and subsequently exposed to 24 h of culture at 1, 2, or 21% oxygen tension with concurrent inhibitor treatment. Relative luciferase units were subsequently measured using a luminometer. (e) HIF2α inhibitor prevents the expression of HIF2α protein. Bulk glioma cells isolated from a 4121 xenograft were pretreated with HIF2α inhibitor for 24 h before concurrent inhibitor and DFX treatment. Total cell lysates were analyzed via immunoblotting using antibodies against HIF2α and tubulin as a loading control. (f and g) HIF2α inhibitor does not alter HIF2A transcript levels, but reduces MLL1 expression. Following dissociation of bulk tumor cells from a 4121 xenograft, cells were pretreated with HIF2α inhibitor for 24 h before incubation with DFX and concurrent inhibitor treatment. Total RNA was harvested and analyzed via RT-qPCR for HIF2A (f) or MLL1 (g). *P<0.001; **P<0.05
MLL1 can be efficiently targeted with shRNA
To further define the functional roles of MLL1 in GBM, we characterized the ability of multiple shRNAs directed against MLL1 in comparison to an NT control to decrease the expression of MLL1 in GBM cells (data not shown). We identified two different shRNAs against MLL1, designated shRNA1 and shRNA2, which were effective in reducing the expression of MLL1 mRNA in 4121 non-stem glioma cells with 21% O2 and under hypoxia (Figure 3a). The reduction of MLL1 mRNA was sufficient to decrease MLL1 protein expression (Figure 3b), suggesting the shRNAs may inhibit MLL1-mediated downstream effects. As MLL1 is an H3K4 methyltransferase, we measured global H3K4 triple methylation by western blot with MLL1 knockdown. No difference was seen in global H3K4m3 levels (data not shown), similar to reports suggesting that H3K4 methyltransferases contain familial redundancy, and knockout of specific factors only affects a small subset of genes.36 However, we did confirm that targeting MLL1 had functional consequences as MLL1 shRNA decreased the expression of HoxA9, a downstream target of MLL1 (Figure 3c). These data demonstrate the ability to reduce effectively MLL1 expression and function through the introduction of specific shRNAs.

Targeting MLL1 via shRNA inhibits the expression of HIF2A and downstream hypoxic responses. (a and c) Following stable incorporation of either NT or MLL1 shRNA, 4121 non-stem glioma cells were treated with 200 μM DFX for 24 h. Total RNA was harvested and levels of MLL1 (a) or HoxA9 (c) were measured using RT-qPCR. (b) Total cell lysates were harvested using RIPA buffer and immunoblotted for MLL1 protein and tubulin as a loading control. (d) In all, 387 non-stem glioma cells were treated with 200 μM DFX for 24 h to stabilize HIF protein. Cell lysates were harvested and then immunoblotted for MLL1, HIF1α, or HIF2α. Actin was used as loading control. (e) One thousand 387 non-stem glioma cells were plated on treated coverslips following transduction with NT or MLL1 shRNA. Cells were treated with 200 μM DFX for 24 h and then fixed. MLL1 or HIF2α was labeled with antibody and then detected by immunofluorescence. (f–i) Following treatment with NT or MLL shRNA, 4121 non-stem glioma cells were treated for 24 h with 200 μM DFX. Harvested mRNA was then analyzed using RT-qPCR to measure relative mRNA of (f) HIF2A, (g) HIF1A, (h) VEGF, or (i) PGK1. (j) U87 cells expressing an HRE-containing promoter-driving luciferase were treated with NT or MLL1 directed shRNA and relative luciferase units subsequently measured using a luminometer. *P<0.001; **P<0.05
Loss of MLL1 reduces HIF2α expression and hypoxia-induced signaling
To further elucidate the role of MLL1 in hypoxia responses, we determined the effect of MLL1 knockdown on non-stem tumor cells in which hypoxia was previously characterized to promote phenotypic changes.15 We first examined the effect on HIF1α and HIF2α protein levels following MLL1 knockdown. Surprisingly, although both HIFs are required for hypoxic regulation of MLL1, knockdown of MLL1 has a specific effect on HIF2α. Inhibition of MLL1 via shRNA in 387 non-stem glioma cells induced a decrease of HIF2α protein levels as assessed by immunoblotting (Figure 3d) and immunofluorescence (Figure 3e and Supplementary Figure 2A), but did not affect HIF1α levels (Figure 3d). Our results demonstrated co-distribution of MLL1 and HIF2α as well as loss of HIF2α expression in cells without MLL1. Hypoxia-induced transcription of HIF2A (Figure 3f and Supplementary Figure 2B) was significantly downregulated by MLL1 inhibition as well. Although these data suggest that MLL1 regulates HIF1A transcript (Figure 3g and Supplementary Figure 2C), >85% reduction in HIF2A transcript levels under hypoxia with MLL1 targeting (Figure 3f) demonstrates more potent and specific effects on HIF2A versus HIF1A. To further elucidate the effects of MLL1 targeting on hypoxia signaling, we evaluated the mRNA expression of known HIF target genes. We found that levels of VEGF (Figure 3h and Supplementary Figure 2D) and PGK1 (Figure 3i and Supplementary Figure 2E) are both potently reduced by MLL1 knockdown. We also confirmed that targeting MLL1 decreased overall HIF transcriptional activity by evaluating HRE activity via luciferase expression (Figure 3j).
We next performed ChIP to determine the mechanism by which MLL1 regulated HIF2A transcription. We hypothesized that the mode of action of MLL1 on HIF2A was due to changes in histone modification at the HIF2A promoter. By analyzing the relative levels of regions proximal to the HIF2A start site on triple-methylated H3K4 and H3K27, we found that inhibition of MLL caused a loss of H3K4m3 (a pro-transcriptional signal) and an increase in H3K27m3 (a transcriptional repressive signal; Supplementary Figure 3). This is similar to previously published results that were observed in MLL1-knockout mice that MLL1 target genes had increased H3K27m3 marks upon disruption of MLL1.35 These data demonstrate that loss of MLL1 reduces HIF2A transcription, likely by post-translational modifications of histone moieties near the HIF2A start site.
MLL1 is preferentially expressed in GSCs
As GSCs possess elevated levels of HIF2α10 that may be regulated by MLL1, we hypothesized that MLL1 is preferentially expressed in GSCs. Our laboratory and others have previously determined that GSCs can be prospectively enriched from many human glioma xenografts and patient specimens using cell surface marker CD133 (Prominin-1). We previously employed CD133 enrichment to derive cultures that fulfill GSC functional characteristics (self-renewal, multilineage differentiation, stem cell marker expression, and tumor propagation). In our models, CD133-positive GBM cells have an increased ability to form neurospheres that express CSC markers, including Nestin and Olig2 (Supplementary Figure 4). Using matched GSCs and non-stem GBM cells (4121 and 387 tumor specimens), we utilized a PCR screen for epigenetic modifiers to determine if MLL1 or other factors displayed preferential expression. Out of 96 epigenetic regulators, only MLL1 demonstrated consistent preferential expression in the GSCs (Figure 4a). The differential mRNA expression translated into increased MLL1 protein in GSCs as verified by immunoblotting (Figure 4b). As these data suggested an important role for MLL1 in GSCs, we examined additional samples that were enriched or depleted for GSCs. Using real-time PCR, preferential mRNA expression of MLL1 was confirmed in 4121 and 387 GSCs and GSCs isolated from the additional xenograft 4302 (Figure 4c). Enrichment for the GSC subpopulation was confirmed through elevated Olig2 mRNA (Figure 4d). We further assessed MLL1 expression in GSCs by immunofluorescence. In GSCs grown in suspension culture, MLL1 demonstrated high levels of colocalization with validated GSC marker, CD133, in three separate specimens (Figure 4e). These findings implicate MLL1 in glioma maintenance.

MLL1 expression is elevated in glioma stem cells compared to non-stem glioma cells. Patient-derived glioma specimens were dissociated and enriched for GSCs as described previously.9 (a) A PCR screen for epigenetic modifiers determined that out of 96 epigenetic regulators, only MLL1 demonstrated consistent preferential expression in the GSCs when RNA was isolated from 4121 and 387 GSCs, and non-stem cell fractions were compared. (b) These data were confirmed with total lysates immunoblotted for MLL1. (c) Real-time PCR demonstrated that MLL1 mRNA was consistently upregulated in GSC fractions. (d) The cancer stem cell nature of the GSC fractions was confirmed through elevation of Olig2 mRNA. All mRNA levels were normalized to actin. *P<0.001; **P<0.05. (e) Colocalization of MLL1 and putative GSC marker, CD133, was visualized by staining of sectioned neurospheres in three different patient-derived specimens
Hypoxia regulates MLL1 expression in GSCs
Our data demonstrated that MLL1 was preferentially expressed in GSCs and hypoxia regulated in glioma cells. We therefore sought to determine whether MLL1 was regulated by hypoxia in GSCs. Minimally cultured 387 and 4302 GSCs were exposed to hypoxic conditions induced through culture with 1% O2 or exposure to DFX. Both treatments significantly increased MLL1 mRNA in the GSCs (Figure 5b) and induced other hypoxic gene responses as confirmed by the induction of HIF2α (Figures 5c and d) and VEGF (Figures 5e and f). To confirm a role for HIF2α in MLL1 regulation, we also utilized the HIF2α small-molecule inhibitor, which had no effect on hypoxia-induced HIF2α mRNA (Figure 5g), but significantly reduced MLL1 mRNA levels (Figure 5h). These data supported our earlier findings in unfractionated tumor cells (Figure 2g). Taken together, these data demonstrated that MLL1 is hypoxia responsive and an HIF2α target gene in GSCs.

MLL1 is regulated by hypoxia and HIF2α in GSCs. In all, 387 or 4302 GSCs were cultured in Neurobasal media with 1% oxygen for 4 days or treated with 200 μM DFX for 24 h as indicated. Following hypoxic treatment, total RNA was harvested, cDNA generated by reverse transcription, and mRNA evaluated for (a and b) MLL1, (c and d) HIF2A, and (e and f) VEGF. HIF2A and VEGF levels were used as internal hypoxic controls. (g and h) Following enrichment for 4121 GSCs, cells were pretreated with HIF2α inhibitor for 24 h before incubation with DFX and concurrent inhibitor treatment. Total RNA was harvested and analyzed via RT-qPCR for HIF2A (g) or MLL1 (h). *P<0.001; **P<0.05
Targeting MLL1 in GSCs reduces VEGF expression and GSC-mediated endothelial cell growth
As GSCs contribute to tumor growth through the hypoxia-responsive gene VEGF to promote tumor angiogenesis,39 we determined the contribution of MLL1 to VEGF production. Knockdown of MLL1 in 4121 (Figure 6a) or 387 (Figure 6b) GSCs led to a significant decrease of secreted VEGF. Conditioned media from the same patient-derived specimens was also applied to HUVECs and the relative HUVEC growth was assessed by incorporation of tritiated thymidine (Figure 6c). Conditioned media from MLL1-depleted glioma cells displayed significantly reduced ability to stimulate HUVEC growth (Figure 6c). These data indicate that hypoxic induction of MLL1 contributes to the angiogenic nature of glioblastoma cells. MLL1 is not only important for VEGF regulation in GSCs, but targeting of MLL1 has downstream biological consequences on GSC growth as well.

Targeting MLL1 reduces GSC-mediated VEGF production and endothelial cell proliferation. Following puromycin selection of infected GSCs isolated from (a) 4121 or (b) 387 xenografts, fresh GSC cell culture media without growth factors were added for 48 h. Conditioned media were passaged through a 0.22 μm syringe filter and then used to measure VEGF production by ELISA. (c) Following a similar media collection as detailed above and as shown in the diagram on the left, HUVECs were grown in the conditioned media for 24 h before the addition of tritiated thymidine for 4 h. Thymidine incorporation was measured using a scintillation counter. *P<0.001; **P<0.05
Loss of MLL1 inhibits GSC growth and self-renewal
Central to the CSC paradigm is the notion that the stem-like subpopulation exhibits sustained proliferation and self-renewal that contributes to tumor propagation.6 We therefore examined whether MLL1 contributes to the growth and self-renewal capacity of GSCs in vitro. Following stable incorporation of MLL1 directed shRNA or NT control into GSCs isolated from 4121 (Figure 7a) or 387 (Figure 7b) xenografts, we sequentially measured GSC growth. Depletion of MLL1 significantly reduced GSC growth within 3 days and led to a greater than fivefold reduction in the number of cells at 7 days in both GSC samples. We also compared the effects of MLL1 knockdown on growth rates of non-stem tumor cultures (Supplementary Figure 4B). Although we observed statistically significant growth inhibition in the non-stem tumor cells, the extent of the reduction in cell numbers was always greater in the GSCs. These data demonstrate that MLL1 is important in both glioma subpopulations tested, but MLL1 is especially critical for the growth of GSCs.

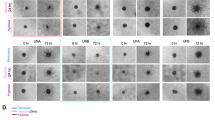
MLL1 knockdown decreases the growth and neurosphere formation capacity of glioma stem cells. (a and b) Following shRNA viral infection and drug selection, (a) 4121 or (b) 387 GSCs were plated at a density of 1000 cells per well in 96-well plates. Cell growth was measured using a Cell Titer Glo Kit (Promega) at the indicated times. (c) In all, 4121 GSCs were sorted into a 96-well plate at a density of 10 cells per well. Cells were left in culture for 14 days in normal GSC media and the number of spheres per well counted. Images in right panels were taken on day 14 by wide-field microscope. *P<0.001; **P<0.05
As an in vitro measure of a stem cell-like behavior, neurosphere formation demonstrates the ability to proliferate and is also utilized as a surrogate measure of self-renewal. We measured the ability of GSCs to form neurospheres following MLL1 inhibition. GSCs stably expressing MLL1 shRNA formed significantly fewer neurospheres compared to cells infected with NT shRNA (Figure 7c). MLL1-depleted cells rarely formed small collections of cells, which contrasted sharply with the large, tight spheres typical of normal GSCs (Figure 7c). Taken together, these data suggest that MLL1 is an important factor in the biological function of the CSC subpopulation in GBM.
Inhibition of MLL1 reduces tumorigenic capacity
Orthotopic transplantation is the gold standard for determining tumor propagation potential of putative GSCs. To measure the effect of MLL1 inhibition on tumor formation, GSCs stably expressing MLL1 shRNA and GFP driven by the same promoter (as a visual marker for shRNA expression) were intracranially implanted into immunocompromised hosts. Upon the development of neurological deficit, tumor-bearing mice were examined for the presence of GBM. GSCs transduced with MLL1 shRNA formed tumors at a significantly greater latency (Figure 8a). Furthermore, tumors that arose from the shRNA cohort had very few cells expressing the shRNA construct as assessed by GFP marker expression (Figure 8b). These data suggest that MLL1 plays an important role in GSC-driven tumor propagation by modulating the growth characteristics of GSCs.

MLL1 inhibition reduces tumor propagation in mice. (a) In all, 387 GSCs bearing stable NT or MLL1 shRNA tagged with GFP was implanted into the right frontal lobe of athymic nu/nu mice. Mice were monitored for signs of neurological deficits. *P<0.01. (b) Following euthanasia, whole mouse brains were fixed and frozen. A total of 10 μM sections were stained for GFP to assess the presence of cells transduced with NT or MLL1 shRNAs. Tumors from shRNA-bearing mice (n=3 per group) contained fewer GFP-positive cells, indicating that tumors forming in the cohort were likely from an contaminating GFP-negative cells
Discussion
Cancer cell heterogeneity is increasingly appreciated as an important component of tumor propagation and recurrence following therapy. Previously GSCs were thought to undergo unidirectional lineage commitment and irreversible differentiation. However, aberrant differentiation is a hallmark of cancer, suggesting that a rigid cellular hierarchy is likely not present. Several publications have demonstrated the ability of the non-stem tumor bulk to become more tumorigenic and exhibit functional characteristics of GSCs in the presence of microenvironmental stimuli. For example, publications from our lab and others have demonstrated that culture under low oxygen conditions promotes the ability of non-stem tumor cells to form tumors and upregulates genes typically associated with GSCs, such as Nanog and c-Myc.15, 16, 17 Whether these biological phenomena are products of selection or true reprogramming similar to induced pluripotency remains unclear and warrants further investigation. As bevacizumab and other targeted therapies may function as anticancer agents through disruption of the tumor microenvironment, increasing our understanding of the microenvironmental factors regulating the GSC phenotype will be vital for defining therapeutic interventions to target this highly tumorigenic subpopulation.
Although recent experimental evidence suggests plasticity in the GSC phenotype, the mechanisms through which associated molecular and biological changes could arise in the absence of novel genetic mutations remain unknown. In the iPS field where phenotypic plasticity has been well characterized, epigenetic regulation is recognized as a gatekeeper for downstream global genomic changes.23 In particular, methylation status of H3K4 and H3K27 are well known markers for the transcriptional state of the cell. H3K4m3 proximal to the gene promoter, for example, associates with actively transcribed genes.36 These histone modifications are regulated primarily by histone-modifying enzymes, of which there are many families that are evolutionarily well-conserved. Extrinsic and intrinsic communication between histone modifiers and the cell environment is not well understood. Through the field of induced pluripotency, hypoxia is being appreciated as a possible regulator of histone modifier activity.20 However, the contribution of specific epigenetic-modifying proteins to the GSC phenotype has not been well studied. Relatively few publications have addressed the epigenomic phenotype of the heterogeneous tumor subpopulations, although it is now becoming a larger focus of research.
In this study, we sought to elucidate the relationship between hypoxia and MLL1, a HMT that has been previously associated with tumorigenicity in leukemia. We first demonstrated that restricted oxygen can modulate the expression of MLL1 in GSC and non-stem tumor populations. This is a particularly novel finding as HMTs were not typically thought to be hypoxia-responsive. Notably, this increase in MLL1 transcript levels requires functional HIF1α and HIF2α, as shRNA and pharmacological HIF inhibition abrogated the MLL1 hypoxic response. Interestingly, the HIF regulation of MLL1 does not appear to be through direct binding of the HIF2α protein. ChIP of HIF2α did not display significant binding to the MLL1 promoter and we also observed a lack of any consensus HIF binding sequences in the MLL1 promoter. Previous publications have shown that hypoxia regulates the demethylating families of enzymes (specifically Jumonji proteins40), but this effect was thought to be restricted to epigenetic proteins that suppress gene expression. Owing to the complexity of the hypoxic response in GSCs, we evaluated the importance of MLL1 within the HIF transcriptional pathway. Surprisingly, depletion of MLL1 preferentially diminished HIF2A. To further elucidate this mechanism, we measured changes in the relative abundance of H3K4m3 and H3K27m3 on the HIF2A promoter. Inhibition of MLL led to a decrease in H3K4m3 and an increase in H3K27m3, which suggests that MLL1 reduces HIF2A transcription via chromatin modification. However, it is important to note that there may be still undiscovered regulatory mechanisms of HIF2α and further studies are needed. Our novel data suggest that loss of MLL1 reduces HIF2α levels and downstream hypoxic responses in glioma cells. We confirmed loss of HIF response by evaluating HRE transcriptional activity as well as downstream function of well-characterized HIF targets such as VEGF. Indeed, cells depleted of MLL1 demonstrated significant reduction in VEGF production, even when stimulated by hypoxia. Although it is not yet clear what MLL1-dependent effects are mediated through HIF2α, these data suggest that critical aspects of the GSC phenotype, high rates of proliferation and self-renewal, depend on MLL1. This extends in vivo where inhibition of MLL1 caused a marked decrease in tumor propagation.
Taken together, our data provide the first evidence that epigenetic modifier, MLL1, is a hypoxia-responsive gene. We also demonstrate for the first time that MLL1 has a significant role in glioma biology through the regulation of HIF2α and downstream HIF2α targets. Specifically, hypoxia-driven upregulation of the pro-angiogenic factor, VEGF, was significantly inhibited in MLL1-depleted cells. Our data further demonstrate the importance of HMT and MLL1 activity in the ability of GSCs to propagate tumors in vivo. These data suggest that epigenetic-modifying proteins play a vital role in promoting tumor growth through the regulation of the hypoxic response in GSCs and that the ability to alter the epigenetic landscape of tumor cells could yield novel therapeutic opportunities.
Materials and Methods
Isolation and culture of CSCs and non-stem cancer cells
Cultures enriched or depleted for GSCs were isolated from primary human brain tumor patient specimens or human glioblastoma xenografts as previously described in accordance with a Duke University, University Hospital, Seidman Cancer Center, or Cleveland Clinic Foundation Institutional Review Board-approved protocol concurrent with national regulatory standards, with patients signing informed consent as described previously.7 GSCs were cultured in Neurobasal media with B27 (without vitamin A; Invitrogen, Carlsbad, CA, USA), basic fibroblast growth factor (bFGF, 10 ng/ml), and epidermal growth factor (10 ng/ml). After trypsinization, non-stem glioma cells were cultured overnight in Dulbecco's minimal essential media (DMEM) and 10% fetal bovine serum (FBS) to allow cell attachment and survival. Then, in some cases, DMEM medium was removed and the cells cultured in supplemental Neurobasal medium in order for experiments to be performed in identical media. To induce hypoxia, cells were cultured in multi-gas chambers (Sanyo, Wood Dale, IL, USA). Nitrogen gas was supplied to the chambers to compensate for the reduced percentage of oxygen. Alternatively, cells were treated by 200 mM hypoxia-mimetic deferoxamine mesylate (DFX, Sigma-Aldrich, St. Louis, MO, USA).
Semiquantitative PCR
Total RNA was harvested from cells using RNAeasy Kit (Qiagen, Germantown, MD, USA). PCR was performed on cDNA generated by Superscript III reverse transcriptase (Invitrogen) or qScript Reverse Transcriptase (Quanta Biosciences, Gaithersburg, MD, USA) and subsequent semiquantitative PCR was performed using SYBR Green Master Mix (Qiagen). Primers used were as follows: MLL1 forward, 5′-CAGATAAAGTCCAGGAAGCTCG-3′ and reverse, 5′-GTAATTTCGACAGTGCTTGGC-3′; HIF1α forward, 5′-TCCATGTGACCATGAGGAAA-3′ and reverse, 5′-CCAAGCAGGTCATAGGTGGT-3′; vascular endothelial growth factor (VEGF) forward, 5′-AGTCCAACATCACCATGCAG-3′ and reverse, 5′-TTCCCTTTCCTCGAACTGATTT-3′; HIF2α forward, 5′-CCACCAGCTTCACTCTCTCC-3′ and reverse, 5′-TCAGAAAAAGGCCACTGCTT-3′; HoxA9 forward 5′-AATGCTGAGAATGAGAGCGG-3′ and reverse, 5′-GGGTCTGGTGTTTTGTATAGGG-3′; phosphoglycerate kinase 1 (PGK1) forward, 5′-GCTTCTGGGAACAAGGTTAAAG-3′ and reverse, 5′-CTGTGGCAGATTGACTCCTAC-3′; and Olig2 forward, 5′-AGCTCCTCAAATCGCATCC-3′ and reverse, 5′-ATAGTCGTCGCAGCTTCG-3′. All data were normalized to actin transcript levels.
HIF2α small-molecule inhibition
U87 or 4121 cells were treated with 10 μM (final concentration) of HIF2α inhibitor 77 or vehicle control for 24 h before hypoxic stimulation.38 Media were changed every 24 h, with inhibitor or control added following media change.
Luciferase assay
U87 cells bearing the hypoxia response element (HRE) driving luciferase expression construct were cultured in DMEM with 10% FBS. Following 24 h of pretreatment with HIF2α inhibitor, the cells were treated with 1, 2, or 21% oxygen and the inhibitor concurrently. After 24 h of treatment, total cell lysates were harvested by passive lysis buffer containing luciferin (Promega, Madison, WI, USA). Luciferase activity was measured using a luminometer (Perkin-Elmer, Waltham, MA, USA).
Immunoblotting
Before lysis, cells were washed once with cold phosphate-buffered saline (PBS). Total cell lysates were prepared by the addition of RIPA lysis buffer (Sigma-Aldrich) containing protease inhibitors (Sigma-Aldrich) and placed at −20 °C for no less than 1 h. Brief sonication with a wand-style sonicator (Fisher Scientific, Pittsburgh, PA, USA) at 10% duty cycle was used to ensure complete lysis of the nuclear membrane. Total protein was separated on bis-acrylamide gels and transferred to polyvinylidene fluoride membranes. Primary antibodies for MLL1 (1 : 500; Abcam, Cambridge, MA, USA), TUBULIN (1 : 10 000; Millipore, Billerica, MA, USA), or HIF2α (1 : 1000; Novus, Littleton, CO, USA) were incubated overnight at 4°C. Secondary antibodies conjugated to horseradish peroxidase (Jackson Labs, Bar Harbor, ME, USA) or infrared epitopes (Licor, Lincoln, NE, USA) were incubated for 1 h at room temperature, protected from light.
Immunofluorescent imaging
Brain sections or cells were fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich) for 15 min at room temperature. After fixation, samples were washed with PBS. Before blocking, sodium citrate boiling was performed for 5 min to enhance antigen retrieval. Samples were blocked in buffer containing 10% normal goat serum, 0.1% Triton X-100, and 0.01% sodium azide in PBS for 30 min at room temperature. Following blocking, primary antibody, green fluorescent protein (GFP, 1 : 500; Aves Labs, Tigard, OR, USA), MLL1 (1 : 100; Abcam), CD133 (1 : 500; Abcam), or HIF2α (1 : 500; Millipore), was added and incubated at 4 °C overnight. The next day, secondary antibodies conjugated to fluorescent isotopes (Alexa Fluor; Invitrogen) were added to cells at appropriate dilutions. After incubation, Hoescht 33342 nuclear stain was added for 5 min. Images were taken on a wide-field inverted fluorescent or confocal microscope (Leica, Buffalo Grove, IL, USA).
Targeting of MLL1 by RNA interference
To screen RNA interference (RNAi) vectors, 293T cells were transfected with control or MLL1 short hairpin RNA (shRNA) containing lentiviral plasmids with packaging DNA and resulting virus containing media used to infect glioma cells. A non-targeting (NT) vector designed to activate the RISC and RNAi pathways, but not target any human genes, was used as control (Sigma-Aldrich). shRNA plasmids (Sigma-Aldrich) used for experiments were as follows: shRNA 1 sequence, targeting the coding sequence: CCGGGATTATGACCCTCCAATTAAACTCGAGTTTAATTGGAGGGTCATAATCTTTTTG; shRNA 2 sequence, targeting the 3′-untranslated region: CCGGTGCCTG GAAGGAGCCTATTATCTCGAGATAATAGGCTCCTTCCAGGCATTTTTG.
Vectors contained either a puromycin or GFP selection marker. HIF1A and HIF2A shRNA constructs were designed as previously described.10
VEGF ELISA
GSCs were plated at equal density on reduced growth factor extracellular matrix (Geltrex; Invitrogen)-treated tissue culture dishes in standard GSC media without bFGF or EGF. Following 24 h of culture, media were harvested, filtered, and VEGF quantified using a QuantiGlo chemiluminescent EISA kit (R&D Systems, Minneapolis, MN, USA) with a luminometer (Perkin-Elmer).
Thymidine incorporation
Human umbilical vein endothelial cells (HUVECs; ATCC, Manassas, VA, USA) were plated at a density of 10 000 cells per well in six-well dishes in appropriate media and allowed to recover overnight. The following day, the media were replaced with conditioned media collected from GSCs transduced with MLL1 shRNA or NT shRNA (as described above), and HUVECs cultured with conditioned media for an additional 24 h. Tritiated thymidine was then added for 6 h, followed by quantification using a scintillation counter.
Cell titer assay
GSCs and non-stem GBM cells were trypsinized and plated into 96-well plates containing appropriate growth media at a density of 1000 cells per well on day 0. Adenosine triphosphate levels were measured over time using a Cell Titer Glo kit (Promega) and a luminometer (Perkin-Elmer).
Neurosphere culture assay
GSCs were cultured in Neurobasal media with B27 in the presence of growth factors for at least 24 h before plating. Cells were sorted by flow cytometry into 96-well plates (Sarstedt, Newton, NC, USA) at 10 cells per well. Wells were serially observed over 14 days and the number of neurospheres per well was counted. Images of neurospheres were taken using a wide-field microscope (Leica).
Chromatin immunoprecipitation
Following transduction with shRNA and hypoxia, unfractionated GBM cells were grown on Geltrex in 10 cm tissue culture treated dishes. Chromatin immunoprecipitation (ChIP) was performed according to the manufacturer's instructions (Millipore). Briefly, cells were fixed for 10 min at room temperature in 1% PFA (125 mM, final concentration). Glycine was added for 5 min at room temperature to quench unreacted PFA. Cells were collected and lysed in a 1% sodium dodecyl sulfate solution. Cells were subjected to sonication at 30% duty cycle using a wand-style sonicator (Fischer Scientific) for 15 s, and then put on ice for no less than 60 s. This cycle was repeated five times for each sample. Following sonication, 100 μl aliquots were diluted into 900 μl ChIP dilution buffer (Millipore) and pre-cleared with 60 μl protein A-agarose/salmon sperm DNA beads. Five micrograms of primary antibody (histone 3 lysines 4 (H3K4m3) or 27 (H3K27m3), Millipore) were added overnight. The following day, the bead–antibody complexes were washed, crosslinks reversed, and DNA purified by phenol–chloroform extraction. DNA was analyzed by RT-qPCR utilizing primers designed to amplify regions of HIF2A, HoxA9, MLL1, or VEGF DNA less than 1000 base pairs from the transcriptional start site (Qiagen).
Orthotopic transplantation assays
Intracranial implantation of GBM cells was performed in accordance with Cleveland Clinic Foundation Institutional Animal Care and Use Committee-approved protocols concurrent with national regulatory standards.7, 10 GSCs bearing MLL1 shRNA or NT control were implanted into the right frontal lobes of athymic nu/nu mice. Mice were monitored daily for signs of neurological deficit. At the development of neurological impairment, the mice were perfused with 4% PFA, brains removed, and placed in 4% PFA. Brains were then cryo-protected in a 30% sucrose solution and frozen. Immunofluorescent imaging was performed as described above.
Statistical analysis
Descriptive statistical analysis was generated for all repeated quantitative data with inclusion of means and standard error. Significance was tested by Student's t-test using SigmaStat 3.5 (Chicago, IL, USA).
Abbreviations
- CSC:
-
cancer stem cell
- DFX:
-
deferoxamine mesylate
- GSC:
-
glioma stem cell
- H3K4:
-
histone 3 lysine 4
- H3K27:
-
histone 3 lysine 27
- HIF:
-
hypoxia-inducible factor
- HMT:
-
histone methyltransferase
- HRE:
-
hypoxia response elements
- MLL1:
-
mixed-lineage leukemia 1
- PGK1:
-
phosphoglycerate kinase 1
- VEGF:
-
vascular endothelial growth factor
- bFGF:
-
basic fibroblast growth factor
- DMEM:
-
Dulbecco's minimal essential media
- NT:
-
non-targeting
- GFP:
-
green fluorescent protein
References
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009; 10: 459–466.
Hesselager G, Holland EC . Using mice to decipher the molecular genetics of brain tumors. Neurosurgery 2003; 53: 685–694;discussion 695.
Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One 2009; 4: e7752.
Network CGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–1068.
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9: 157–173.
Vescovi AL, Galli R, Reynolds BA . Brain tumour stem cells. Nat Rev Cancer 2006; 6: 425–436.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444: 756–760.
Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 2007; 67: 4010–4015.
Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010; 6: 421–432.
Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009; 15: 501–513.
Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006; 5: 67.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458: 780–783.
Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 2006; 13: 1238–1241.
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11: 69–82.
Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN . The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009; 8: 3274–3284.
Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain J Neurol 2010; 133: 983–995.
Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009; 28: 3949–3959.
Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, Macswords J, Lathia JD et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 2011; 18: 829–840.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872.
Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S . Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009; 5: 237–241.
Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009; 27: 40–48.
Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE et al. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 2008; 3: e3769.
Guenther MG, Frampton GM, Soldner F, Hockemeyer D, Mitalipova M, Jaenisch R et al. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell 2010; 7: 249–257.
Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G . BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci 2009; 29: 8884–8896.
Bruggeman SW, Hulsman D, Tanger E, Buckle T, Blom M, Zevenhoven J et al. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell 2007; 12: 328–341.
Krivtsov AV, Armstrong SA . MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 2007; 7: 823–833.
Mohan M, Lin C, Guest E, Shilatifard A . Licensed to elongate: a molecular mechanism for MLL-based leukaemogenesis. Nat Rev Cancer 2010; 10: 721–728.
Somervaille TC, Cleary ML . Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006; 10: 257–268.
Somervaille TC, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM et al. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009; 4: 129–140.
Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 2006; 13: 713–719.
Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009; 113: 2375–2385.
Liu H, Takeda S, Kumar R, Westergard TD, Brown EJ, Pandita TK et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature 2010; 467: 343–346.
Huntsman DG, Chin SF, Muleris M, Batley SJ, Collins VP, Wiedemann LM et al. MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene 1999; 18: 7975–7984.
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011; 331: 435–439.
Lim DA, Huang YC, Swigut T, Mirick AL, Garcia-Verdugo JM, Wysocka J et al. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009; 458: 529–533.
Wang P, Lin C, Smith ER, Guo H, Sanderson BW, Wu M et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol 2009; 29: 6074–6085.
Sierra J, Yoshida T, Joazeiro CA, Jones KA . The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev 2006; 20: 586–600.
Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A et al. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol cell 2008; 32: 838–848.
Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 2006; 66: 7843–7848.
Uemura M, Yamamoto H, Takemasa I, Mimori K, Hemmi H, Mizushima T et al. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin Cancer Res 2010; 16: 4636–4646.
Acknowledgements
We thank Cathy Shemo, Moneen Morgan, and Sage O’Bryant for flow cytometry assistance; Linda Vargo for histology assistance; and the members of the Rich laboratory for technical assistance and critical review of the manuscript. Work in the Rich laboratory is supported by the McDonnell Foundation and NIH Grants NS054276, CA129958, CA116659, and CA154130. AH is supported by the National Brain Tumor Society and the NIH Grant CA151522. JDL is supported by an American Brain Tumor Association Basic Research Fellowship (sponsored by the Joelle Syverson Fund) and a National Service Research Award (NCI F32 CA142159). OI is supported by RO1 CA122591. AS is supported by CA137443, CA116257, HHS N2612010, the Kimble Fund and Cristal Chair at CWRU. Studies were also supported by the Cleveland Clinic Foundation Tissue Procurement Service and the Tissue Procurement and Biorepository Cores at University Hospitals and the Case Comprehensive Cancer Center, and we also thank S Staugatis, R Weil, and M McGraw for their assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by R De Maria
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Heddleston, J., Wu, Q., Rivera, M. et al. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ 19, 428–439 (2012). https://doi.org/10.1038/cdd.2011.109
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.109
Keywords
This article is cited by
-
MLL1 inhibits the neurogenic potential of SCAPs by interacting with WDR5 and repressing HES1
International Journal of Oral Science (2023)
-
Functional importance of glucose transporters and chromatin epigenetic factors in Glioblastoma Multiforme (GBM): possible therapeutics
Metabolic Brain Disease (2023)
-
ABTC-0904: targeting glioma stem cells in GBM: a phase 0/II study of hedgehog pathway inhibitor GDC-0449
Journal of Neuro-Oncology (2023)
-
Gene regulation by histone-modifying enzymes under hypoxic conditions: a focus on histone methylation and acetylation
Experimental & Molecular Medicine (2022)
-
Alcohol-abuse drug disulfiram targets pediatric glioma via MLL degradation
Cell Death & Disease (2021)
