
Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a potentially useful anticancer agent with exquisite selectivity for cancer cells. Unfortunately, many cancers show or acquire resistance to TRAIL. In this study we report that TRAIL activates a TGF-β-activated kinase 1 → mitogen-activated protein kinase (MAPK) kinase 3 (MKK3)/MKK6 → p38 pathway in prostate cancer cells that transcriptionally upregulates expression of the antiapoptotic BCL-2 family member MCL-1. TRAIL alone triggered robust formation of the ‘death-inducing signaling complex’ (DISC), activation of the initiator caspase-8, and truncation of the BH3-only protein BID (tBID). Nevertheless, simultaneous disruption of the p38 MAPK pathway was required to suppress MCL-1 expression, thereby allowing tBID to activate the proapoptotic BCL-2 family member BAK and stimulate mitochondrial outer membrane permeabilization (MOMP). Release of the inhibitor-of-apoptosis (IAP) antagonist, Smac/DIABLO, from the intermembrane space was sufficient to promote TRAIL-induced apoptosis, whereas release of cytochrome c and activation of the apoptosome was dispensable. Even after MOMP, however, mitochondrial-generated reactive oxygen species (ROS) activated a secondary signaling pathway, involving c-Jun N-terminal kinases (JNKs), that similarly upregulated MCL-1 expression and partially rescued some cells from death. Thus, stress kinases activated at distinct steps, before and after mitochondrial injury, mediate TRAIL resistance through maintenance of MCL-1 expression.
Similar content being viewed by others


Main
The tumor necrosis factor (TNF) family of cytokines has critical roles in inflammation and immune surveillance.1 Among them, TNF-related apoptosis-inducing ligand (TRAIL; also known as Apo2L) has emerged as a promising anticancer therapy, because of its remarkable capacity to induce apoptosis (programmed cell death) in cancer cells with little to no toxicity to normal cells.2 Membrane-bound TRAIL initiates the extrinsic (death receptor) pathway in target cells by binding to its trimerized receptors, TRAIL-R1 and TRAIL-R2 (also known as DR4 and DR5), resulting in receptor aggregation and recruitment of the adapter protein Fas-associated death domain (FADD) and procaspase-8. This complex of TRAIL receptors, FADD, and procaspase-8 is often referred to as the ‘death inducing signaling complex’ (DISC), and recruitment of procaspase-8 to the DISC leads to its dimerization and subsequent activation.3
In some cells (designated type I cells), the apoptotic signal from active caspase-8 is sufficient to activate the downstream effector procaspase-3 and induce apoptosis. However, in other cells (designated type II cells), there is insufficient activation of procaspase-3 – or caspase-3 is inhibited by an inhibitor-of-apoptosis (IAP) protein such as X-linked IAP (XIAP) – and hence the apoptotic signal must be further amplified by engaging the intrinsic (mitochondrial) pathway.4, 5 In this instance, caspase-8 cleaves and activates the BH3-only protein BID, which in turn activates the proapoptotic BCL-2 family members, BAX or BAK, and induces mitochondrial outer membrane permeabilization (MOMP). After MOMP, additional apoptogenic proteins are released into the cytoplasm, including the IAP antagonist, second mitochondrial activator of caspases (Smac; also known as DIABLO), and cytochrome c, the latter of which activates yet another caspase-activating complex, known as the Apaf-1•caspase-9 apoptosome.
In addition to inducing apoptosis, TRAIL receptor stimulation also leads to activation of various signaling pathways, including activation of the IκB kinase (IKK) complex, mitogen-activated protein kinases (MAPKs), and c-Jun N-terminal kinases (JNKs).6, 7, 8 Although not required for apoptosis, TRAIL receptors reportedly undergo internalization and caspase-8-dependent formation of a secondary signaling complex.6, 9 This intracellular complex contains receptor-interacting protein 1 (RIP1) and TNF receptor-associated factor 2 (TRAF2), which then signal for the activation of IKK and the stress kinases, p38 MAPKs and JNKs, through ill-defined mechanisms.6 Notably, TNF induces robust activation of these pathways, and in particular, activation of the transcription factor NF-κB by IKK suppresses TNF-induced apoptosis in most cell types through upregulation of various antiapoptotic genes.10 TRAIL, in comparison, generally activates these pathways to a lesser extent and their importance in regulating TRAIL-induced cell death remains unclear.6
Despite its promise as an anticancer agent, a significant problem associated with TRAIL-based therapy is that many tumors possess or acquire resistance to TRAIL. Expression of TRAIL ‘decoy’ receptors, TRAIL-R3 and TRAIL-R4, has been proposed to mediate TRAIL resistance by competing with TRAIL-R1 and TRAIL-R2 for TRAIL,11 and at high expression levels, cellular FLICE-like inhibitory protein (c-FLIP) inhibits apoptosis by competing with caspase-8 for binding to FADD.12, 13 Finally, overexpression of antiapoptotic BCL-2 family members, including BCL-2, BCL-xL, and MCL-1, is associated with TRAIL resistance,14, 15, 16 particularly in type II cells, because of their abilities to prevent MOMP and the release of cytochrome c or Smac/DIABLO.4
Gene ablation studies indicate that MCL-1 is essential for peri-implantation, the development and maintenance of B and T lymphocytes, and the survival of hematopoietic cells.17, 18 MCL-1 is highly regulated at the transcriptional level in hematopoietic cells through the transcription factors SRF/ETS, STAT3, CREB, and PU.1,19, 20, 21, 22 and at the post-translational level through a complex interplay involving three kinases (ERK, JNK, and GSK-3β) and at least two E3 ubiquitin ligases (MCL-1 ubiquitin ligase E3 (MULE) and β-TrCP).23, 24, 25, 26, 27, 28 ERK-mediated phosphorylation of human MCL-1 at Thr-163 prolongs its half-life,25 although recent studies by Davis and colleagues27, 28 indicate that JNK phosphorylates mouse MCL-1 at Thr-144 (analogous to Thr-163 in human MCL-1), which in turn enhances its phosphorylation by GSK-3β at Ser-140.28 GSK-3β-mediated phosphorylation of mouse/human MCL-1 at Ser-140/Ser-159 then promotes its ubiquitination by E3 ligases and subsequent degradation by the 26S proteasome.
In this study, we explored the mechanisms responsible for TRAIL resistance in prostate cancer cells. Remarkably, we found that TRAIL activates a TGF-β-activated kinase 1 (TAK1) → MKK3/MKK6 → p38 pathway that transcriptionally upregulates the expression of MCL-1 and suppresses BAK activation, MOMP, and cell death, despite caspase-8 activation and robust BID cleavage. Disruption of the p38 MAPK signaling pathway downregulated MCL-1 and sensitized cells to TRAIL-induced MOMP and apoptosis. However, reactive oxygen species (ROS), generated by injured mitochondria, activated a secondary JNK pathway in some cells that upregulated MCL-1 expression and afforded partial protection from death. Thus, we show for the first time that stress kinases activated by TRAIL at distinct steps in the extrinsic pathway mediate TRAIL resistance through maintenance of MCL-1 expression levels.
Results
TRAIL activates an antiapoptotic TAK1 → MKK3/MKK6 → p38 MAPK signaling pathway in resistant prostate cancer cells
In an effort to determine the mechanism(s) of TRAIL resistance in human prostate cancer cells, DU145 cells were exposed to recombinant TRAIL and examined for activation of various stress and growth-related kinases (Figure 1a). Notably, p38-α and its downstream target, MAPK-activated protein kinase 2 (MK2), were phosphorylated within 2 h of treatment (before cell death), whereas other stress kinases, such as JNKs, were only weakly activated at later time points (Figure 1a). ERKs were constitutively active in naive cells, but TRAIL did not enhance their activation, nor did it activate AKT/PKB (Figure 1a; data not shown). The importance of p38-α activation was evident when cells were cotreated with TRAIL and the selective p38-α and p38-β inhibitor, SB203580, as the combination led to a significant increase in apoptosis (Figure 1b). TRAIL activated the initiator caspase-8 and the effector caspase-3 (as determined by caspase processing and DEVDase activity), particularly in the presence of SB203580 (Figure 1c). Cell death was fully inhibited by benzyloxycarbonyl-Val-Ala-Asp-(OMe)fluoromethyl ketone (z-VAD-fmk); however, this polycaspase inhibitor had no effect on the activation of p38-α, further establishing p38-α activation as an early upstream event (Figures 1b and c, top panel, lanes 5–8). This was an important observation, because although few studies have addressed how TRAIL activates p38-α, the adapter protein RIP1 and caspase-8 activity were previously thought to be essential.6 The sensitization of DU145 cells to TRAIL was due to the specific inhibition of p38 MAPKs, as transfection with a constitutively active form of p38-α (CA) largely rescued cells from TRAIL/SB203580-induced cell death, but only when accompanied by a mutation in the ‘gate-keeper’ residue (T106M), which lowers the affinity of SB203580 for the ATP-binding pocket (Figures 1d–f).29

TRAIL activates a TAK1 → MKK3/MKK6 → p38 MAPK pathway that suppresses apoptosis. (a) DU145 prostate cancer cells were treated with recombinant TRAIL (500 ng/ml) for 0.5–8 h, and cells were immunoblotted for various active phosphorylated kinases, including p-p38α (Thr-180/Tyr-182), p-MK2 (Thr-222), p-JNK (Thr-183/Tyr-185), and p-ERK (Thr-202/Tyr-204). (b, c) Cells were treated with TRAIL±SB203580 (50 μM) ±z-VAD-fmk (50 μM) and assayed for cell death by annexin V/PI staining and flow cytometry. Each bar represents the mean of three separate experiments±S.E.M. Cells were also immunoblotted for phosphorylated p38, as well as active caspase-8 (p18 large subunit) and caspase-3 (p20, p19, and p17 large subunits). Caspase-3/7 DEVDase activity was also measured, as described in the Materials and Methods. (d, e) DU145 cells were transiently transfected with expression plasmids encoding EGFP, constitutively active (D176A/F327S) p38-α (EGFP-p38α-CA), or p38α-CA containing a T106M ‘gate-keeper’ mutation (EGFP-p38α-CA (T106M)). Cells were then exposed to TRAIL for 8 h, stained with Hoechst 33258, and assayed for apoptosis by measuring the percentage of GFP+ cells with condensed nuclei using fluorescence microscopy. Each bar represents the mean of three separate experiments±S.E.M. Note that EGFP-p38α-CA undergoes autophosphorylation and phosphorylates endogenous p38-α, but is sensitive to SB203580, whereas EGFP-p38α-CA (T106M) is only weakly inhibited by SB203580. (f) As shown in the crystal structure (PDB: 1A9U; rendered with UCSF chimera), the fluorophenyl ring in SB203580 binds into a hydrophobic groove in the ATP binding pocket of p38-α, with one edge of the ring lying between the side chains of Thr-106 and Leu-104.55 Substitution of Thr-106, that is, the ‘gate-keeper’, with a bulkier and more hydrophobic methionine residue (naturally found in p38-γ and p38-δ) reduces the affinity of SB203580 for p38-α and p38-β.29 (g, h) Cells were cotreated with TRAIL±5Z-7-oxozeaenol (5Z-7-oxo; 1 μM) ±z-VAD-fmk (50 μM) and were blotted for phosphorylated p-TAK1 (Thr-184/Thr-187) and p-p38 (Thr-180/Tyr-182). The asterisk denotes a nonspecific band. In addition, cells were treated with TRAIL±5Z-7-oxozeaenol or SB203580 and were assayed for cell death by annexin V/PI staining and flow cytometry. Each bar represents the mean of three separate experiments±S.E.M. (i) DU145 cells were transiently transfected with pEGFP for 24 h, along with empty vectors (pcDNA3-FLAG; pcDNA3-HA; and pcDNA6-Myc), or those expressing dominant-negative mutants of TAK1 (K63A), MKK3 (S189A/T193A), MKK6 (S207A/T211A), or p38-α (D168A). Cells were then exposed to TRAIL for 8 h, stained with Hoechst 33258, and assayed for apoptosis by measuring the percentage of GFP+ cells with condensed nuclei. Each bar represents the mean of three separate experiments±S.E.M. Cell lysates were also immunoblotted for FLAG-TAK1-DN, HA-MKK3-DN, HA-MKK6-DN, Myc-p38-DN, and endogenous p-p38α
As TNF reportedly activates p38-α through the activation of TAK1 or apoptosis signal-regulating kinase 1 (ASK1),30, 31 we next sought to determine whether either of these MAP3Ks was essential for TRAIL-mediated activation of p38-α. Exposure of DU145 cells to TRAIL resulted in the phosphorylation/activation of TAK1 within ∼30–60 min, before (or concomitant with) activation of p38-α (Figures 1a and g, inset), and did so in a caspase-independent manner (Figure 1h, lanes 1, 2, and 4), whereas ASK1 failed to undergo phosphorylation at Thr-845 (data not shown). 5Z-7-oxozeaenol, a resorcylic lactone of fungal origin, has been reported to selectively inhibit TAK1.32 Therefore, we pretreated cells with 5Z-7-oxozeaenol and found that it inhibited TRAIL-induced activation of TAK1 and p38-α (Figure 1h, lanes 1–3 and 5), and correspondingly, potentiated TRAIL-induced apoptosis at levels comparable to TRAIL plus SB203580 (Figure 1g). Finally, to confirm our results with 5Z-7-oxozeaenol, we determined the effects of dominant-negative TAK1 and p38-α mutants on TRAIL-induced activation of p38-α and apoptosis in DU145 cells (Figure 1i). Dominant-negative MKK3 and MKK6 mutants were also examined, as these two MAP2Ks have redundant but essential roles in TNF-dependent activation of p38 MAPKs.33 As expected, disruption of the TAK1 → MKK3/MKK6 → p38 MAPK pathway, at any step, suppressed the phosphorylation/activation of p38-α and resulted in significant sensitization of cells to TRAIL-induced cell death (Figure 1i).
p38 MAPKs suppress TRAIL-induced apoptosis downstream of the DISC by inhibiting MOMP and the release of Smac/DIABLO
Our initial results suggested a model in which TRAIL receptor stimulation resulted in the activation of a pro-survival TAK1 → MKK3/MKK6 → p38 MAPK pathway and a pro-death TRAIL-R1/TRAIL-R2 → FADD → caspase-8 pathway (Figure 2a). It remained unclear, however, precisely how p38-α activation prevented apoptosis. To address this question, we first analyzed the effects of SB203580 on formation of the TRAIL DISC. As shown in Figure 2b, SB203580 had no effect on recruitment of the adapter protein FADD to TRAIL-R2, or the subsequent recruitment and activation of the initiator procaspase-8. Cell surface expression of TRAIL-R1 was very low when compared with TRAIL-R2, such that only TRAIL-R2 was detected within the DISC (data not shown). As previously noted, c-FLIP overexpression can inhibit the activation of procaspase-8 within the DISC and is often cited as a cause of TRAIL resistance. However, SB203580 had no effect on the basal levels of c-FLIP in whole lysates (Figure 2c, compare lanes 1 and 2), and despite repeated attempts, c-FLIP could not be detected within the DISC (Figure 2b). In contrast, an examination of mitochondria revealed that inhibition of p38 MAPKs was required for TRAIL-induced MOMP, as determined by the mitochondrial release of cytochrome c and Smac/DIABLO into the cytosol and a loss in mitochondrial inner membrane potential (Δψm) (Figure 2d).

p38 MAPKs suppress TRAIL-induced apoptosis downstream of the DISC by inhibiting MOMP and the release of Smac/DIABLO. (a) Scheme of the antiapoptotic TAK1 → MKK3/MKK6 → p38 MAPK and proapoptotic FADD → caspase-8 → caspase-3 pathways initiated by TRAIL. (b) DU145 cells were treated with biotinylated-TRAIL (bTRAIL; 500 ng/ml) ±SB203580 (50 μM), and DISC analyses were performed as described in the Materials and Methods. Note that the unstimulated control (U/S) was obtained by adding bTRAIL to lysed control cells to rule out nonspecific ligand interactions, and the pulldown of endogenously biotinylated acetyl-CoA carboxylase (ACC) was used as a loading control. (c) Cells were treated with TRAIL±SB203580±z-VAD-fmk for 0.5–2 h and immunoblotted for c-FLIP. (d) Cells were treated with TRAIL±SB203580, and mitochondrial pellets and supernatants were immunoblotted for cytochrome c or Smac/DIABLO. Cells were also assayed for changes in Δψm by flow cytometry using the fluorescent dye TMRE (25 nM). (e, f) DU145 cells, infected with empty virus or virus expressing a dominant-negative caspase-9 (C287A), were treated with TRAIL±SB203580 and assayed for cell death by annexin V/PI staining and flow cytometry. Each bar represents the mean of three separate experiments±S.E.M. To confirm the effectiveness of the dominant-negative caspase-9, lysates from untreated cells were assayed for apoptosome activity in response to the addition of cytochrome c and dATP. (g) DU145 cells were transiently transfected with pEGFP for 24 h, along with pEBB-ubiquitin or pEBB-ubiquitin-Smac/DIABLO, the latter of which undergoes cotranslational processing to liberate mature Smac into the cytosol35 (see inset for expression of cytosolic Smac/DIABLO). Cells were then exposed to TRAIL±SB203580 for 8 h, stained with Hoechst 33258, and assayed for the percentage of GFP+ apoptotic cells. Each bar represents the mean of three separate experiments±S.E.M. NS, not statistically significant. (h) DU145 cells were treated with TRAIL±SB203580 and immunoblotted for changes in XIAP expression over a time course (0.5–8 h)
Cytochrome c induces formation of the apoptosome and sequential activation of caspases-9 and -3.34. However, in the case of death receptor stimulation, it is often the release of Smac/DIABLO, rather than cytochrome c, that is critical for cell killing.4 In these instances, active caspase-8 processes procaspase-3, but caspase-3 is inhibited by IAPs, and cytosolic Smac/DIABLO is required to antagonize IAPs and relieve the inhibition of caspase-3.4 We therefore stably infected cells with a lentivirus expressing dominant-negative caspase-9 (C287A), a potent inhibitor of the apoptosome,34 or transiently transfected cells with a ubiquitin-Smac/DIABLO fusion construct that produces mature cytosolic Smac/DIABLO.35 Overexpression of dominant-negative caspase-9 had no effect on TRAIL/SB203580-induced apoptosis in DU145 cells (Figure 2e), although it clearly inhibited the activation of endogenous caspase-9 in cytochrome c/dATP-activated lysates (Figure 2f). Smac/DIABLO, on the other hand, potentiated TRAIL-induced cell death, even in the absence of SB203580 (Figure 2g). A recent study suggests that loss of XIAP can sensitize BID-deficient hepatocytes and pancreatic β-cells to FasL/CD95L-induced apoptosis in vivo, essentially phenocopying MOMP and the release of Smac/DIABLO.36 However, SB203580 had no effect on the expression levels of XIAP (Figure 2h, lanes 1 and 2). Thus, the TAK1 → MKK3/MKK6 → p38 MAPK pathway seemed to suppress TRAIL-induced apoptosis in large part by preventing MOMP and the release of Smac/DIABLO.
p38 MAPKs inhibit TRAIL-induced MOMP, in spite of BID activation, through transcriptional upregulation of MCL-1
Death receptors normally engage the mitochondrial pathway through caspase-8-dependent cleavage and activation of the proapoptotic BH3-only protein BID.37 Although controversial, truncated BID (tBID) and other BH3-only proteins then directly activate the multidomain proapoptotic BCL-2 family members, BAX or BAK (direct activation model), and/or antagonize the antiapoptotic Bcl-2 family members, BCL-2, BCL-xL, or MCL-1, thereby relieving their inhibition of BAX or BAK (indirect activation model).38, 39 We treated DU145 cells with TRAIL±SB203580 and examined them for BID cleavage and downstream BAK activation. Remarkably, exposure to TRAIL alone induced near-complete BID cleavage (Figure 3a, lane 5), but in the absence of SB203580, tBID failed to induce the conformational change in BAK required for its activation (Figure 3b, compare TRAIL with TRAIL+SB). Thus, consistent with our previous DISC results, TRAIL receptor stimulation induced significant caspase-8 activation and BID cleavage, but tBID was unable to induce BAK oligomerization and MOMP unless the antiapoptotic p38 MAPK pathway was disrupted (Figures 2b and d; 3a and b).

TRAIL stimulates BID cleavage, but p38 inhibition is essential to suppress MCL-1 expression, facilitate BAK activation and MOMP, and induce apoptosis. (a, b) DU145 cells were treated with TRAIL (500 ng/ml) ±SB203580 (50 μM) ±z-VAD-fmk (50 μM), immunoblotted for BID cleavage, and assayed for BAK activation by flow cytometry using an epitope-specific antibody that recognizes its active conformation. The cells in (b) were stably transfected with a scrambled shRNA plasmid and served as controls for (k); identical results were obtained in untransfected cells. (c, d) Cells were treated with TRAIL±SB203580 and assayed for expression of antiapoptotic multidomain BCL-2 family members, MCL-1, BCL-2, and BCL-xL, at the protein (immunoblotting) and/or transcriptional (qRT-PCR) levels. (e, f) Time course experiments were performed (as described above) to assess temporal changes in MCL-1 expression after treatment with TRAIL±SB203580. (g) DU145 cells were transiently transfected with pcDNA3-MCL-1 for 24 h, after which cycloheximide (CHX; 1 μM) was added, along with either SB203580 (50 μM) or the ERK inhibitor U0126 (10 μM). Turnover was then assessed by immunoblotting MCL-1 over a 3-h time course. (h) DU145 cells were transiently transfected with pEGFP or pEGFP-MCL-1 for 24 h (see inset for immunoblot of expressed proteins), exposed to TRAIL±SB203580 for 8 h, stained with Hoechst 33258, and assayed for the percentage of GFP+ apoptotic cells. Each bar represents the mean of three separate experiments±S.E.M. (i) DU145 cells were transfected with pSuper-scramble or pSuper-MCL-1 to stably knockdown the expression of MCL-1 by RNAi (see inset for knockdown of MCL-1). Cells were then exposed to TRAIL±SB203580 and assayed for cell death by annexin V/PI staining and flow cytometry. Each bar represents the mean of three separate experiments±S.E.M. (j) DU145 cells were transiently transfected with pmCherry, pmCherry-NOXA, or pmCherry-BAD for 24 h (see inset for immunoblot of expressed proteins), exposed to TRAIL±SB203580 for 8 h, stained with Hoechst 33258, and assayed for the percentage of mCherry+ apoptotic cells. Each bar represents the mean of three separate experiments±S.E.M. (k) DU145 cells were depleted of MCL-1 by RNAi and assayed for BAK activation after treatment with TRAIL±SB203580, as described in (b). (l) DU145 cells were transiently transfected with EGFP or tBID-EGFP for 24 h (see inset for immunoblot of expressed proteins), exposed to DMSO or SB203580 for 8 h, stained with Hoechst 33258, and assayed for the percentage of GFP+ apoptotic cells. Each bar represents the mean of three separate experiments±S.E.M.
Given the aforementioned results, we considered that p38 MAPKs might regulate the expression of BCL-2, BCL-xL, or MCL-1. Indeed, after treatment with SB203580, basal MCL-1 (but not BCL-2 or BCL-xL) expression levels were reduced, both at the protein and transcriptional levels (Figure 3c, lanes 1 and 2; Figure 3d), whereas TRAIL treatment alone led to an increase in MCL-1 expression (Figure 3c, lanes 1 and 3; Figure 3d). Surprisingly, when used in combination, MCL-1 levels were largely maintained, but it was important to note that these assays were performed at 8 h, when the combination of TRAIL plus SB203580 induced significant apoptosis (Figure 3c, lane 4; Figure 3d). Subsequent time course experiments revealed that SB203580 suppressed the basal and upregulated expression of MCL-1 by TRAIL during the first 2–3 h of treatment and that MCL-1 levels did not rebound until later (⩾4 h) after the activation of caspases (Figure 3e, lanes 4–8; Figure 3f). To confirm that the loss of MCL-1 expression was due to a decrease in transcription and was not the result of an increase in protein turnover, we transiently expressed MCL-1 from a CMV promoter and followed the turnover of MCL-1 in the presence of cycloheximide, an inhibitor of protein synthesis. As ERK-dependent phosphorylation of MCL-1 at Thr-163 has been shown to regulate the turnover of MCL-1,25 we compared the effects of SB203580 with the ERK inhibitor U0126. As shown in Figure 3g, whereas treatment with U0126 led to rapid turnover of MCL-1 (lower panels), inhibition of p38 MAPKs with SB203580 had no such effect (upper panels). Thus, collectively, the data argued strongly that p38 MAPK activity regulated the expression of MCL-1 at the transcriptional level.
We next confirmed the importance of MCL-1 for mediating TRAIL resistance using several approaches. First, we found that ectopic expression of EGFP-MCL-1 significantly inhibited cell death induced by TRAIL plus SB203580 (Figure 3h), whereas selective downregulation of endogenous MCL-1 by RNA interference (RNAi) sensitized cells to TRAIL alone, at a level comparable to scrambled control cells treated with TRAIL plus SB203580 (Figure 3i). Second, as the BH3-only proteins, NOXA and BAD, selectively antagonize MCL-1 and BCL-2/BCL-xL/BCL-w, respectively,40 we expressed NOXA or BAD as mCherry fusion proteins and found that only NOXA sensitized DU145 cells to TRAIL (Figure 3j). Thus, although TRAIL receptor stimulation led to near-complete cleavage of BID, the intracellular concentration of tBID was apparently insufficient to fully antagonize MCL-1 and alleviate the inhibition of BAK by MCL-1; or alternatively, MCL-1 sequestered all of the available tBID and prevented its activation of BAK. Perhaps more consistent with the latter interpretation, knockdown of MCL-1 by RNAi failed to induce spontaneous BAK activation, whereas treatment of MCL-1-deficient cells with TRAIL alone (which triggered BID cleavage) induced significant BAK activation, similar to that observed in TRAIL/SB203580-treated vector control cells (compare Figure 3k with b). Similarly, ectopic expression of NOXA alone failed to induce apoptosis (Figure 3j), whereas overexpression of tBID alone induced apoptosis, and when combined with SB203580 (which inhibited basal p38 activity and downregulated basal MCL-1 levels), killed ∼90% of transfected cells (Figure 3l). In short, the data indicated that disruption of the TAK1 → MKK3/MKK6 → p38 MAPK pathway, after TRAIL receptor stimulation, prevented the upregulation of MCL-1 and facilitated tBID-induced BAK activation, MOMP, and cell death.
Inhibition of p38 MAPKs potentiates TRAIL-induced apoptosis in BAX-expressing and androgen receptor-positive prostate cancer cells
Although downregulation of MCL-1 by SB203580 sensitized DU145 cells to TRAIL, these prostate cancer cells do not express BAX, the androgen receptor, or prostate-specific antigen, all of which are commonly observed in primary prostate cancers.41, 42 We therefore expanded our studies to include DU145 cells, stably transfected with BAX, and LNCaP prostate cancer cells, which naturally express both BAX and BAK and are androgen receptor and prostate-specific antigen positive.41, 42 As shown in Figure 4a, re-expression of BAX in DU145 cells sensitized them to TRAIL, but cotreatment with SB203580 still resulted in a further increase in cell death. This observation was not surprising given that MCL-1 sequesters tBID, and tBID is reportedly required to activate both BAK and BAX.39 LNCaP cells also showed resistance to TRAIL, and inhibition of p38 MAPKs with SB203580 similarly downregulated MCL-1 expression and sensitized these cells to TRAIL-induced apoptosis (Figures 4b and c).

Inhibition of p38 MAPKs potentiates TRAIL-induced apoptosis in androgen receptor-positive and BAX-expressing prostate cancer cells. (a) BAX-deficient DU145 cells, and those stably expressing BAX (see inset for immunoblot of expressed proteins), were treated with TRAIL (500 ng/ml)±SB203580 (50 μM) and assayed for cell death by annexin V/PI staining and flow cytometry. (b, c) LNCaP prostate cancer cells, which express both BAX and BAK, as well as the androgen receptor and prostate-specific antigen,41, 42 were similarly immunoblotted for MCL-1 and assayed for their sensitivity to TRAIL±SB203580
TRAIL-induced MOMP activates a ROS-dependent JNK pathway that upregulates MCL-1 expression and partially rescues cells from apoptosis
As already noted, SB203580 inhibited TRAIL-activated p38 MAPKs, thereby preventing the expression of MCL-1 in DU145 cells during the first 2–3 h after treatment with TRAIL and sensitized the cells to apoptosis (Figures 1 and 3e). MCL-1 expression levels, however, partially rebounded ⩾4 h later, concomitant with caspase activation (Figures 3e and f; data not shown). To characterize this resurgence in MCL-1 expression and to assess its potential effect on cell death, we treated DU145 cells with TRAIL±SB203580 and once again assayed them for the activation of several kinases, including JNKs, ERKs, and AKT. Interestingly, JNKs were robustly activated, as determined by autophosphorylation and downstream phosphorylation of c-Jun, but only in cells cotreated with TRAIL plus SB203580 (Figure 5a, lane 4; Figure 5b, lane 4). More importantly, a cell-permeable peptide inhibitor of JNK (derived from JNK interaction protein) inhibited JNK activity (Figure 5e inset, lanes 3 and 4), prevented entirely the rebound in MCL-1 expression (Figure 5c, lanes 6 and 8; Figure 5d), and further sensitized the cells to apoptosis (Figure 5e; compare TRAIL+SB+JIP with TRAIL+SB, P<0.05). In contrast, in LNCaP cells, JNK activity was not upregulated after treatment with TRAIL plus SB203580 (Figure 5b, lane 8), MCL-1 levels failed to fully rebound (Figure 4c), and LNCaP cells were correspondingly more sensitive to apoptosis than DU145 cells (Figure 4b, TRAIL+SB; P<0.05).

TRAIL plus SB203580 activates a secondary JNK pathway that upregulates MCL-1 expression and partially protects cells from apoptosis. (a) DU145 cells were treated with TRAIL (500 ng/ml) ±SB203580 (50 μM) and immunoblotted for active p-JNK (Thr-183/Tyr-185), p-ERK (Thr-202/Tyr-204), and p-AKT (Ser-473). (b) DU145 and LNCaP prostate cancer cells treated with TRAIL (500 ng/ml) ± SB203580 (50 μM) and the activity of JNKs determined by immunoblotting for phospho-c-Jun (Ser-73). (c–e) DU145 cells were treated with TRAIL±SB203580±JIP peptide (10 μM) and assayed for MCL-1 expression at the protein (immunoblotting) and transcriptional (qRT-PCR) levels. Apoptosis was determined by annexin V/PI staining and flow cytometry. Each bar represents the mean of three separate experiments±S.E.M. Note that in the inset to (e), the inhibitory effect of JIP was confirmed, as it inhibited JNK-dependent phosphorylation of c-Jun
ROS have been implicated in the TNF-dependent activation of JNKs through activation of ASK1 and/or inhibition of MAPK phosphatases (MKPs),31, 43 leading us to speculate that TRAIL might similarly stimulate the production of ROS and activate JNKs. Indeed, TRAIL treatment triggered the generation of ROS in DU145 cells, but only in the presence of SB203580 (Figure 6a), and the ROS scavenger, catalase, inhibited both the activation of JNKs and the resurgence in MCL-1 expression (Figure 6c, lanes 6 and 7). In contrast, TRAIL plus SB203580 failed to stimulate the production of ROS in LNCaP cells, in accordance with its inability to activate JNK in these cells (Figures 5b and 6b). Given that caspase-dependent cleavage of BID and downregulation of MCL-1 was required to induce MOMP after exposure to TRAIL plus SB203580 (Figure 3) and that mitochondrial perturbation often leads to the production of ROS,44 we speculated that mitochondrial-generated ROS might be responsible for the activation of JNKs. As predicted, in DU145 cells, inhibition of caspases with z-VAD-fmk inhibited the production of ROS, the activation of JNKs, and the resurgence in MCL-1 expression after treatment with TRAIL plus SB203580 (Figure 6c, lanes 6 and 8; Figure 6d).

Mitochondrial-generated ROS are responsible for JNK activation and the resurgence in MCL-1 expression after treatment with TRAIL plus SB203580. (a, b) DU145 and LNCaP prostate cancer cells were treated with TRAIL±SB203580 and assayed for ROS production by flow cytometry using the fluorescent dye DCFDA. (c, d) DU145 cells were treated with TRAIL±SB203580±the ROS scavenger catalase (CAT; 100 U) or the polycaspase inhibitor z-VAD-fmk (50 μM), immunoblotted for MCL-1 expression, and assayed for ROS production by flow cytometry using the fluorescent dye DCFDA. (e–h) DU145 cells deficient in BID or MCL-1 were treated with TRAIL±SB203580 and assayed for ROS production or Δψm by flow cytometry using the fluorescent dyes DCFDA (10 μM) and TMRE (25 nM), respectively. Each bar represents the mean of three separate experiments±S.E.M. (i, j) DU145 cells were transiently transfected with pHyPer-Mito, a mitochondrial-localized oxidant-activated fluorescent protein, and treated with TRAIL±SB203580 to assay for mitochondrial-specific generation of ROS. Each bar represents the mean of three separate experiments±S.E.M.
However, TNF reportedly stimulates ROS production upstream of mitochondria.31, 43, 45 Therefore, rather than inhibiting the cleavage of all caspase-8 substrates, some of which might theoretically mediate ROS production upstream of mitochondria, we instead knocked down the expression of BID to selectively prevent tBID-induced MOMP. Remarkably, partial depletion of BID was sufficient to suppress TRAIL/SB203580-induced mitochondrial injury (as determined by Δψm) and the generation of ROS (Figures 6e and f), whereas knockdown of MCL-1 sensitized cells to TRAIL alone, resulting in a loss in Δψm with a concomitant increase in ROS (Figures 6g and h). Finally, to this point, all of our ROS assays were performed using an oxidant-sensitive fluorescent dye, DCFDA (2′,7′-dichlorodihydrofluorescein diacetate), which is largely cytosolic. Therefore, to more directly assay mitochondria for ROS production, we transfected cells with a mitochondrial-localized circularly permuted yellow fluorescent protein, inserted into the regulatory domain of the oxidant-sensitive E. coli transcription factor OxyR (pHyPer-Mito).46 As shown in Figures 6i and j, control cells and those treated with SB203580 or TRAIL alone showed little activation of the pHyPer-Mito probe. In stark contrast, cells cotreated with TRAIL plus SB203580 – a combination that induced MOMP, cytochrome c release, and a loss in Δψm (Figure 2d) – showed a profound increase in mitochondrial fluorescence (Figures 6i and j).
Taken together, the data in DU145 cells supported a model in which TRAIL plus SB203580 induced BID activation and downregulation of MCL-1, resulting in MOMP, cytochrome c release, a loss in Δψm, increased ROS production, activation of JNKs, and ultimately, a resurgence in the expression of MCL-1 (Figure 7). Thus, even during times of profound stress, for example, after mitochondrial injury, the affected cells initiated a complex secondary survival pathway that converged upon MCL-1, reemphasizing the importance of MCL-1 as a regulator of TRAIL-induced cell death.

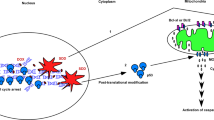
Model of TRAIL-activated proapoptotic and antiapoptotic pathways. TRAIL stimulates its receptors, TRAIL-R1 and/or TRAIL-R2, resulting in recruitment of the adapter protein FADD and activation of procaspase-8 within the DISC. Caspase-8 subsequently processes procaspase-3, but active caspase-3 is rapidly inhibited by IAPs, including XIAP. Caspase-8 also cleaves and activates BID, and tBID would normally induce BAK-dependent MOMP and release the IAP antagonist Smac/DIABLO. However, TRAIL receptor stimulation simultaneously activates an antiapoptotic TAK1 → MKK3/MKK6 → p38 MAPK signaling pathway that transcriptionally upregulates MCL-1, which in turn sequesters/neutralizes tBID. Disruption of this signaling pathway at any step sensitizes cells to TRAIL, but remarkably, even after mitochondrial injury, mitochondrial-generated ROS activate a secondary JNK pathway that similarly leads to transcriptional upregulation of MCL-1 and partial protection from cell death. As shown in the inset, ROS are most likely produced as a result of inefficient electron transfer, after the release of cytochrome c, or perhaps in response to caspase-dependent cleavage of complex I proteins including NDUFS144
Discussion
TNF activates various antiapoptotic signaling pathways that typically subvert an otherwise proapoptotic extrinsic pathway, resulting in minimal (if any) death in most cell types.10 TRAIL, in comparison, induces apoptosis selectively in tumor cells, but unfortunately ∼50% of tumors possess or acquire resistance to TRAIL. Previous studies have shown that TRAIL, similar to TNF, activates ERK, JNK, and p38 MAPKs, but the magnitude of the responses is generally modest, and it remains unclear whether these pathways have significant roles in mediating TRAIL resistance. In this study, we have shown for the first time that stimulation of TRAIL receptors activates a TAK1 → MKK3/MKK6 → p38 MAPK pathway in prostate cancer cells that is largely responsible for the transcriptional upregulation of MCL-1 and subsequent resistance to apoptosis (Figures 1, 3, and 4). Importantly, exposure to SB203580 had no effect on c-FLIP expression levels, and TRAIL treatment alone led to robust DISC formation, caspase-8 activation, and near-complete BID cleavage, ruling out a prominent role for c-FLIP in mediating TRAIL resistance (Figure 2). Endogenous tBID however did not induce apoptosis unless MCL-1 expression levels were downregulated through simultaneous inhibition of the TRAIL-activated p38 pathway, knockdown of MCL-1 by RNAi, or antagonism of MCL-1 by ectopically expressed NOXA (Figure 3).
The fact that overexpression of tBID could induce apoptosis, whereas NOXA could not, suggests that there was simply insufficient generation of endogenous tBID after TRAIL treatment to fully antagonize MCL-1 and directly activate BAK. However, our data do not fully exclude either the direct or indirect activation models as previously posited, and indeed, recent studies suggest that these models are not mutually exclusive, in that BIM (which similar to tBID is proposed to be a ‘direct activator’) antagonizes antiapoptotic BCL-2 family members and directly activates BAX in vivo.47 Regardless, our studies reveal the critical importance of maintaining MCL-1 expression levels to suppress TRAIL-induced apoptosis, a fact that is further emphasized by our discovery that even after MOMP, mitochondrial-generated ROS stimulated a secondary JNK pathway that also transcriptionally upregulated the expression of MCL-1 and partially rescued a fraction of DU145 cells from death (Figures 5 and 6). It remains unclear why TRAIL plus SB203580 did not trigger the production of ROS or engage the secondary ROS → JNK → MCL-1 pathway in LNCaP cells. However, in DU145 cells, mitochondria were clearly the source of ROS, as caspase-8 inhibition and BID knockdown suppressed (whereas MCL-1 knockdown enhanced) losses in Δψm and ROS production (Figure 6). Mitochondrial-generated ROS are most likely produced at complex III, because of inefficient electron transfer after the release of cytochrome c, or perhaps at complex I, because of caspase-dependent cleavage of NDUFS4 (Figure 7).44
The notion that cells may survive after MOMP is controversial in cancer cells, although normal postmitotic neurons and cardiomyocytes seem to survive MOMP so long as caspases are inhibited.48 We cannot rule out the possibility that low levels of tBID might induce mild injury to mitochondria, in a small fraction of cells, resulting in ROS production, JNK activation, and upregulation of MCL-1, before (or in the absence of) frank rupture of the outer mitochondrial membrane. Irrespective, our results are in stark contrast to TNF signaling, in which ROS are produced by NADPH oxidases upstream of mitochondria and in turn activate ASK-1 or inactivate MKPs, resulting in prolonged JNK activation, MOMP, and apoptosis.31, 43, 45 At this stage, the transcription factor(s) responsible for p38-α and JNK-dependent upregulation of MCL-1, after treatment with TRAIL and TRAIL plus SB203580, respectively, remain unknown and are currently under investigation. STAT3 and PU.1 have previously been implicated in MCL-1 expression; however, PU.1 is generally restricted to hematopoietic tissues, and dominant-negative mutants of STAT1, STAT3, and ATF2 (a p38-α and JNK-targeted transcription factor) failed to potentiate TRAIL-induced apoptosis (data not shown).
Finally, there are several clinical implications that arise from our studies, particularly related to the use of small-molecule BCL-2 antagonists and Smac mimetics. There is currently legitimate excitement surrounding the use of BCL-2 antagonists, such as ABT-737, in the treatment of tumors made resistant to conventional chemotherapy through overexpression of antiapoptotic BCL-2 family members. Similar to BAD, however, ABT-737 selectively targets BCL-2, BCL-xL, and BCL-w, but not MCL-1, and thus is unlikely to synergize with TRAIL in the treatment of MCL-1-overexpressing tumors. One potential approach for bypassing the antiapoptotic BCL-2 family members altogether could be to use Smac mimetics, which antagonize IAPs and liberate active caspases-3 and -7 after their initial processing by caspase-8. Indeed, consistent with previous studies in other cancer cell types, from our laboratory and others, we found that release of Smac (rather than cytochrome c) was essential for TRAIL-induced apoptosis in DU145 prostate cancer cells (Figure 2). Smac mimetics however reportedly induce apoptosis by stimulating autoubiquitination and degradation of cellular IAP1 (cIAP1) and cIAP2, resulting in activation of the noncanonical NF-κB pathway and upregulation of TNF. Consequently, it remains unclear how effective these agents will be in vivo or whether they will show significant side effects due to the production of TNF. Our studies suggest that TRAIL might instead be used effectively in combination with clinically available p38 MAPK inhibitors for the treatment of cancers expressing MCL-1.
Materials and Methods
Reagents and antibodies
SB203580 (cat. no. 1020) was obtained from Tocris Bioscience (Ellisville, MO, USA), and 5Z-7-oxozeaenol (cat. no. NP-009245) was purchased from AnalytiCon Discovery GmbH (Potsdam, Germany). TAT-TI-JIP153–163 (cat. no. 420134) and U0126 (cat. no. 662005) were obtained from Calbiochem (Gibbstown, NJ, USA). Antibodies to p-p38 MAPK (Thr-180/Tyr-182; cat. no. 9211), p-ERK (Thr-202/Tyr-204; cat. no. 9101), p-JNK (Thr-183/Tyr-185; cat. no. 9251), p-c-Jun (pSer-73; cat. no. 9165), p-MAPKAPK2 (pThr-222; cat. no. 3316), p-AKT (Ser-473; cat. no. 9271), p-TAK1 (Thr-184/Tyr-187; cat. no. 4531), BCL-2 (cat. no. 2876), BCL-xL (cat. no. 2762), BID (cat. no. 2002), myc tag (cat. no. 2276), EGFP (cat. no. 2555), and active caspase-3 (cat. no. 9662) were purchased from Cell Signaling Technology (Danvers, MA, USA). c-FLIP antibody (cat. no. ALX-804-127-C100) was obtained from Alexis Biochemicals (San Diego, CA, USA), and caspase-8 and Smac/DIABLO antibodies were generously provided by Dr. X-M Sun and Professor GM Cohen (MRC Toxicology Unit, Leicester, UK). Antibodies to FADD (cat. no.610399) and cytochrome c (cat. no.556433) were purchased from BD Biosciences (San Jose, CA, USA), and antibodies to MCL-1 (cat. no. sc-12756) and actin (cat. no. CP01-100UL) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Oncogene Research Products (Cambridge, MA, USA), respectively. TRAIL-R2 antibody (cat. no. 2019) was obtained from ProSci Incorporated (Poway, CA, USA), and antibodies to FLAG (cat. no. F3165) and Hemagglutinin (cat. no. MMS-101P) tags were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Covance (Princeton, NY, USA), respectively. RFP/mCherry antibody (cat. no. PM005) was obtained from MBL International (Woburn, MA, USA).
Cloning
The human MCL-1 cDNA encompassing the entire coding region (1.1 kb) was amplified by reverse transcription-PCR (RT-PCR) with the following two primers (MCL1F: 5′-CCAAGATCTATGTTTGGCCTCAAAAGAAACGC-3′; MCL1R: 5′-CCGGAATTCCTATCTTATTAGATATGCCAAACC-3′), digested, and cloned into the BglII–EcoRI sites (underlined) of pEGFP-C1 (Clontech, Mountain View, CA, USA). The cDNAs of human BAD and NOXA were obtained by RT-PCR with the following primers (BADF: 5′-CCGCTCGAGCTATGTTCCAGATCCCAGAGTTTG-3′; BADR: 5′-CGCGGATCCTCACTGGGAGGGGGCGGAG-3′; NOXAF: 5′-CCGCTCGAGCTATGCCTGGGAAGAAGGCGCG-3′; NOXAR: 5′-CGCGGATCCTCAGGTTCCTGAGCAGAAGAG-3′), digested, and cloned into the XhoI–BamHI sites (underlined) of pmCherry-C1 (Clontech). For the generation of shRNAs, oligonucleotides to MCL-1 (sense: 5′-GATCCCCAGTCCGATTACCGCGTTTCTTCAAGAGAGAAACGCGGTAATCGGACTTTTTTTC-3′; antisense: 5′-TCGAGAAAAAAAGTCCGATTACCGCGTTTCTCTCTTGAAGAAACGCGGTAATCGGACTGGG-3′) or a scrambled control (sense: 5′-GATCCCCACCGTCGATTTCACCCGGGTTCAAGAGACCCGGGTGAAATCGACGGTTTTTTTC-3′; antisense: 5′-TCGAGAAAAAAACCGTCGATTTCACCCGGGTCTCTTGAACCCGGGTGAAATCGACGGTGGG-3′) were annealed to generate sticky ends (underlined) and immediately cloned into the BglII–XhoI sites of pSuper.retro.puro (OligoEngine, Seattle, WA, USA). The boxed sequence corresponds to the 9-nt hairpin that is generated after transcription.
A cDNA encoding p38-α (kindly provided by Dr. Kevin N Dalby, University of Texas at Austin, Austin, TX, USA) was subcloned into pcDNA6 with an N-terminal myc tag (Invitrogen, Carlsbad, CA, USA), and a kinase-dead (D168A) mutant was subsequently incorporated by site-directed mutagenesis. A constitutively active mutant of p38-α (D176A/ F327S; obtained from Professor David Engelberg, Hebrew University, Israel)49 was subcloned into the XhoI–BamHI sites of the pEGFP-C1 vector (Clontech), and an SB203580-resistant mutation (T106M) was incorporated by site-directed mutagenesis.29 Dominant-negative mutants of TAK1 (K63A), MKK3 (S189A/T193A), and MKK6 (S207A/T211A) were generously provided by Dr. Xin Lin (University of Texas, M.D. Anderson Cancer Center, Houston, TX, USA) and Dr. Jiahuai Han (Scripps Research Institute, La Jolla, CA, USA).50, 51 pEBB-ubiquitin-Smac/DIABLO, which is designed to express a mature form of Smac in the cytosol, was provided by Dr. Colin S Duckett (University of Michigan Medical School, Ann Arbor, MI, USA).35 Wild-type caspase-9 was cloned into the FG9 lentiviral vector (kindly provided by Dr. Casey W. Wright, UT-Austin, Austin, TX, USA), and a dominant-negative mutation (C287A) was incorporated by site-directed mutagenesis.
Cell culture and transfections
DU145 prostate cancer cells were grown in RPMI-1640, supplemented with 5% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA, USA), 5% Fetalplex (Gemini Bio-products, West Sacramento, CA), 1% penicillin–streptomycin (100 units/ml), and 2 mM glutamine. Cells were maintained at 37°C in humidified air containing 5% CO2 and were routinely passaged every 3 days. For transient transfections, cells were transfected with 1 μg/ml plasmid DNA using TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI, USA), and in the case of stable shRNA knockdowns (pSuper-mcl-1), cells were selected for 10 days in 1 μg/ml of puromycin and then isolated as individual clones. For siRNA experiments, DU145 cells were transiently transfected with 40 nM of BID siRNA (nucleotides 35–53: 5′-GGGATGAGTGCATCACAAA-3′)52 or control siRNA (5′-TTCTCCGAACGTGTCACGT-3′) (Shanghai GenePharma Co., Shanghai, China) using TransIT-TKO Transfection Reagent.
Quantitation of apoptosis
Cells were harvested by trypsinization, washed with PBS, and resuspended in annexin-V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) containing annexin-V-fluorescein isothiocyanate (FITC) and propidium iodide (PI; Roche Applied Sciences, Indianapolis, IN, USA). Annexin V was expressed, labeled with FITC, and purified in-house. FITC- and/or PI-labeled cell populations were analyzed by flow cytometer (Beckman-Coulter, Fullerton, CA, USA).
DEVDase assay
Cells were pretreated with SB203580 (50 μM) for 2 h, cotreated with TRAIL (500 ng/ml) for an additional 8 h, and collected for analysis. The cells were then washed twice with PBS, resuspended in lysis buffer (50 mM Tris, pH 7.5, 1 mM EDTA, 10 mM EGTA, and 10 μM digitonin), and incubated for 10 min at 37°C. Cytosolic fractions were obtained by centrifugation at 15 000 × g for 10 min and were subsequently incubated for 30 min at 37°C with an equal volume of assay buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 0.05% NP-40, and 5 mM MgCl2) containing the fluorescent substrate, Asp-Glu-Val-Asp-7-amino-4-methylcoumarin (DEVD-AMC). DEVDase activities were measured (λex/λem=360/450 nm) in a 96-well plate format using a Wallac Victor3 1420 Multilabel counter (PerkinElmer, Waltham, MA, USA). To confirm the effectiveness of our dominant-negative (DN) caspase-9 (C287A) construct, lysates were prepared from cells stably expressing caspase-9-DN and activated by adding cytochrome c (10 μM), dATP (2 mM), and MgCl2 (2 mM). After a 30-min incubation at 37°C, DEVD-AMC was added and all samples were assayed for DEVDase activity.
Digitonin-based subcellular fractionation
Cells, treated with TRAIL±SB203580 (as described above), were assayed for MOMP. In brief, cells were washed with PBS and lysed for 10 min on ice in 100 μl of digitonin lysis buffer (75 mM KCl, 1 mM NaH2PO4, 8 mM Na2HPO4, 250 mM sucrose, and 60 μg/ml of digitonin) containing protease inhibitors (1 mM PMSF, 2 μg/ml of aprotinin, 2 μg/ml of leupeptin, and 2 μg/ml of pepstatin). The cells were then pelleted by centrifugation at 15 000 × g for 10 min, and the supernatant (cytosol) was obtained. The remaining pellet (mitochondria) was then resuspended in lysis buffer (20 mM Tris-HCl, 135 mM NaCl, 10% glycerol, 50 mM NaF, 5 mM Na3VO4, and 0.2% NP-40) with protease inhibitors for 20 min on ice. Finally, the supernatant and pellet fractions were immunoblotted with antibodies to Smac/DIABLO and cytochrome c.
Isolation of TRAIL signaling complexes
Cells were pretreated with SB203580 (50 μM) for 2 h, followed by a 30-min treatment with biotinylated TRAIL (bTRAIL; 500 ng/ml). The cells were then washed three times with ice-cold PBS to remove any unbound ligand and lysed for 30 min on ice in 3 ml of lysis buffer (30 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 10 mM glycerophosphate, 1 mM sodium orthovanadate, and 5 mM NaF) with protease inhibitors (1 mM PMSF, 2 μg/ml of aprotinin, 2 μg/ml of leupeptin, and 2 μg/ml of pepstatin). Lysates were cleared by centrifugation at 13 000 × g for 30 min and bTRAIL complexes precipitated overnight at 4°C using streptavidin-conjugated to Sepharose beads (Amersham Biosciences, Pittsburgh, PA, USA). After overnight incubation, the beads were washed four times with lysis buffer, and 60 μl of 2 × Laemmli sample buffer were added to the beads.
Quantitative RT-PCR
Cells were plated at a density of 0.6 × 106 cells. After 24 h, the cells were treated with different combination treatments at the indicated time points. Total RNA was extracted using Qiagen RNeasy Mini kit (Valencia, CA, USA), and reverse transcription was performed from 2 μg of total RNA using oligo-dT and AMV reverse transcriptase (Promega, Madison, WI, USA), according to the manufacturer's instructions. The primer sequences were designed as follows: qMCL1F: 5′-AAGCCAATGGGCAGGTCT-3′; qMCL1R: 5′-TGTCCAGTTTCCGAAGCAT-3′; qGAPDHF: 5′-TGCACCACCAACTGCTTAGC-3′; qGAPDHR: 5′-GGCATGGACTGTGGTCATGAG-3′.53 Quantitative RT-PCR was performed with SYBR Green dye using an ABI 7900HT (Perkin-Elmer Applied Biosystems, Foster City, CA, USA), according to the manufacturer's instructions. PCR reactions were performed in triplicate, and the relative amount of MCL-1 cDNA was calculated by the comparative CT method.54 CT values obtained for the different samples were normalized to corresponding CT values of GAPDH.
Western blot analysis
After SDS-PAGE, proteins were transferred to Hybond-N Nitrocellulose (Amersham Biosciences). Membranes were blocked in Tris-buffered saline (TBS) containing 5% non-fat dry milk and 0.1% Tween-20 (TBS-T), before incubation with the primary antibody for 1 h. The membranes were then washed with TBS-T followed by exposure to the appropriate horseradish peroxidase-conjugated secondary antibody for 1 h. Immunostained proteins were visualized on Kodak X-ray film using the enhanced chemiluminescence (ECL) detection system (Amersham Biosciences).
ROS quantification
To measure ROS, cells were pretreated with SB203580 (50 μM) for 2 h±zVAD-fmk (50 μM) for 1 h. TRAIL (500 ng/ml) was then added for 8 h and incubated with 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA, 10 μM; Molecular Probes, Carlsbad, CA, USA) for 30 min. Excess DCFDA was removed by washing the cells twice with PBS at room temperature, and labeled cells were then trypsinized, rinsed, and resuspended in PBS. Oxidation of DCFDA to the highly fluorescent 2′,7′-dichloro-fluorescein (DCF) is proportionate to ROS generation and was analyzed by flow cytometry. To measure mitochondrial-generated ROS, pHyPer-Mito, a mitochondrial-localized oxidant-activated fluorescent protein, was transfected into cells and cells were treated with TRAIL±SB203580 (as described above). Fluorescence intensity, corresponding to mitochondrial-generated H2O2, was analyzed by flow cytometry and images were captured using fluorescence microscopy.
Determination of BAK activation
To detect activated BAK, control and treated cells were fixed for 5 min in 0.25% paraformaldehyde, washed twice with PBS, and incubated for 30 min at room temperature in a PBS buffer containing 100 μg/ml digitonin and a conformation-specific mouse anti-BAK antibody (1 : 30; AM03, Calbiochem). Cells were then washed, incubated with 0.25 μg of Alexa Fluor 488 goat anti-mouse antibody (Invitrogen) for 30 min in the dark, washed again, and analyzed using flow cytometry.
Abbreviations
- Apaf-1:
-
apoptotic protease-activating factor 1
- ASK1:
-
apoptosis signal-regulating kinase 1
- DISC:
-
death-inducing signaling complex
- FADD:
-
Fas-associated death domain
- c-FLIP:
-
FLICE-inhibitory protein
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
mitogen-activated protein kinase
- MK2:
-
MAPK-activated protein kinase 2
- MKK3:
-
MAP kinase kinase 3
- MKK6:
-
MAP kinase kinase 6
- MOMP:
-
mitochondrial outer membrane permeabilization
- MULE:
-
MCL-1 ubiquitin ligase E3
- RIP1:
-
receptor-interacting protein 1
- ROS:
-
reactive oxygen species
- TAK1:
-
TGF-β-activated kinase 1
- TNF:
-
tumor necrosis factor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- TRAF2:
-
TNF receptor-associated factor 2
- MCL-1:
-
myeloid cell leukemia/lymphoma 1
- IAP:
-
inhibitor of apoptosis
- XIAP:
-
X-linked IAP
- z-VAD-fmk:
-
benzyloxycarbonyl-Val-Ala-Asp-(OMe)fluoromethyl ketone
References
Grivennikov SI, Kuprash DV, Liu ZG, Nedospasov SA . Intracellular signals and events activated by cytokines of the tumor necrosis factor superfamily: from simple paradigms to complex mechanisms. Int Rev Cytol 2006; 252: 129–161.
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M et al. Tumoricidal activity of tumor necrosis factor-related apoptosis- inducing ligand in vivo. Nat Med 1999; 5: 157–163.
Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000; 12: 599–609.
Bratton SB, Cohen GM . Death receptors leave a caspase footprint that Smacs of XIAP. Cell Death Differ 2003; 10: 4–6.
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998; 17: 1675–1687.
Varfolomeev E, Maecker H, Sharp D, Lawrence D, Renz M, Vucic D et al. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J Biol Chem 2005; 280: 40599–40608.
Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S et al. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol Cell Biol 2000; 20: 6638–6645.
Harper N, Farrow SN, Kaptein A, Cohen GM, MacFarlane M . Modulation of tumor necrosis factor apoptosis-inducing ligand-induced NF-kappa B activation by inhibition of apical caspases. J Biol Chem 2001; 276: 34743–34752.
Kohlhaas SL, Craxton A, Sun XM, Pinkoski MJ, Cohen GM . Receptor-mediated endocytosis is not required for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J Biol Chem 2007; 282: 12831–12841.
Wajant H, Pfizenmaier K, Scheurich P . Tumor necrosis factor signaling. Cell Death Differ 2003; 10: 45–65.
LeBlanc HN, Ashkenazi A . Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ 2003; 10: 66–75.
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190–195.
Geserick P, Drewniok C, Hupe M, Haas TL, Diessenbacher P, Sprick MR et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL- and CD95L-mediated apoptosis. Oncogene 2008; 27: 3211–3220.
Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007; 12: 66–80.
Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, Bayer E et al. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene 2000; 19: 5477–5486.
Fulda S, Meyer E, Debatin KM . Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002; 21: 2283–2294.
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ . Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003; 426: 671–676.
Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005; 307: 1101–1104.
Wang JM, Lai MZ, Yang-Yen HF . Interleukin-3 stimulation of mcl-1 gene transcription involves activation of the PU.1 transcription factor through a p38 mitogen-activated protein kinase-dependent pathway. Mol Cell Biol 2003; 23: 1896–1909.
Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF . The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol 1999; 19: 6195–6206.
Puthier D, Bataille R, Amiot M . IL-6 up-regulates mcl-1 in human myeloma cells through JAK / STAT rather than ras / MAP kinase pathway. Eur J Immunol 1999; 29: 3945–3950.
Epling-Burnette PK, Zhong B, Bai F, Jiang K, Bailey RD, Garcia R et al. Cooperative regulation of Mcl-1 by Janus kinase/STAT and phosphatidylinositol 3-kinase contribute to granulocyte-macrophage colony-stimulating factor-delayed apoptosis in human neutrophils. J Immunol 2001; 166: 7486–7495.
Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 2007; 27: 4006–4017.
Zhong Q, Gao W, Du F, Wang X . Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121: 1085–1095.
Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW . MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 2004; 23: 5301–5315.
Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW et al. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J Biol Chem 2007; 282: 18407–18417.
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR . Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21: 749–760.
Morel C, Carlson SM, White FM, Davis RJ . Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol 2009; 29: 3845–3852.
Eyers PA, van den IP, Quinlan RA, Goedert M, Cohen P . Use of a drug-resistant mutant of stress-activated protein kinase 2a/p38 to validate the in vivo specificity of SB 203580. FEBS Lett 1999; 451: 191–196.
Cheung PC, Campbell DG, Nebreda AR, Cohen P . Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J 2003; 22: 5793–5805.
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001; 2: 222–228.
Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M et al. A resorcylic acid lactone 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem 2003; 278: 18485–18490.
Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev 2003; 17: 1969–1978.
Malladi S, Challa-Malladi M, Fearnhead HO, Bratton SB . The Apaf-1procaspase-9 apoptosome complex functions as a proteolytic-based molecular timer. EMBO J 2009; 28: 1916–1925.
Hunter AM, Kottachchi D, Lewis J, Duckett CS, Korneluk RG, Liston P . A novel ubiquitin fusion system bypasses the mitochondria and generates biologically active Smac/DIABLO. J Biol Chem 2003; 278: 7494–7499.
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009; 460: 1035–1039.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Fletcher JI, Huang DC . Controlling the cell death mediators Bax and Bak: puzzles and conundrums. Cell Cycle 2008; 7: 39–44.
Chipuk JE, Green DR . How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 2008; 18: 157–164.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403.
Tang DG, Li L, Chopra DP, Porter AT . Extended survivability of prostate cancer cells in the absence of trophic factors: increased proliferation, evasion of apoptosis, and the role of apoptosis proteins. Cancer Res 1998; 58: 3466–3479.
Mitchell S, Abel P, Ware M, Stamp G, Lalani E . Phenotypic and genotypic characterization of commonly used human prostatic cell lines. BJU Int 2000; 85: 932–944.
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M . Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005; 120: 649–661.
Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 2004; 117: 773–786.
Kim YS, Morgan MJ, Choksi S, Liu ZG . TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 2007; 26: 675–687.
Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods 2006; 3: 281–286.
Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA et al. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 2009; 186: 355–362.
Wright KM, Deshmukh M . Restricting apoptosis for postmitotic cell survival and its relevance to cancer. Cell Cycle 2006; 5: 1616–1620.
Askari N, Diskin R, Avitzour M, Capone R, Livnah O, Engelberg D . Hyperactive variants of p38α induce, whereas hyperactive variants of p38γ suppress, activating protein 1-mediated transcription. J Biol Chem 2007; 282: 91–99.
Blonska M, Shambharkar PB, Kobayashi M, Zhang D, Sakurai H, Su B et al. TAK1 is recruited to the tumor necrosis factor-alpha (TNF-alpha) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-kappaB activation. J Biol Chem 2005; 280: 43056–43063.
Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ et al. MAPKK-independent activation of p38α mediated by TAB1-dependent autophosphorylation of p38α. Science 2002; 295: 1291–1294.
Shelton SN, Shawgo ME, Robertson JD . Cleavage of Bid by executioner caspases mediates feed forward amplification of mitochondrial outer membrane permeabilization during genotoxic stress-induced apoptosis in Jurkat cells. J Biol Chem 2009; 284: 11247–11255.
Yuan H, Kamata M, Xie YM, Chen IS . Increased levels of Wee-1 kinase in G(2) are necessary for Vpr- and gamma irradiation-induced G(2) arrest. J Virol 2004; 78: 8183–8190.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001; 25: 402–408.
Wang Z, Canagarajah BJ, Boehm JC, Kassisa S, Cobb MH, Young PR et al. Structural basis of inhibitor selectivity in MAP kinases. Structure 1998; 6: 1117–1128.
Acknowledgements
We wish to thank Dr. X-M Sun and Professor GM Cohen for generously providing caspase-8 and Smac/DIABLO antibodies. We are also grateful to Dr. Kevin N Dalby, Dr. Colin S Duckett, Professor David Engelberg, Dr. Xin Lin, Dr. Jiahuai Han, Dr. Dean G Tang, and Dr. Casey W Wright for providing plasmids and cell lines. This work was supported in part by grants from The American Cancer Society RSG-05-029-01-CCG and the NCI/NIH CA129521 (to SBB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Silke
Rights and permissions
About this article
Cite this article
Son, J., Varadarajan, S. & Bratton, S. TRAIL-activated stress kinases suppress apoptosis through transcriptional upregulation of MCL-1. Cell Death Differ 17, 1288–1301 (2010). https://doi.org/10.1038/cdd.2010.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.9
Keywords
This article is cited by
-
Unexpected inhibition of the lipid kinase PIKfyve reveals an epistatic role for p38 MAPKs in endolysosomal fission and volume control
Cell Death & Disease (2024)
-
TRAIL receptors promote constitutive and inducible IL-8 secretion in non-small cell lung carcinoma
Cell Death & Disease (2022)
-
Bak instead of Bax plays a key role in metformin-induced apoptosis s in HCT116 cells
Cell Death Discovery (2021)
-
Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs)
Apoptosis (2017)
-
UMI-77 primes glioma cells for TRAIL-induced apoptosis by unsequestering Bim and Bak from Mcl-1
Molecular and Cellular Biochemistry (2017)
