
Abstract
We have previously shown that a non-toxic noscapinoid, EM011 binds tubulin without altering its monomer/polymer ratio. EM011 is more active than the parent molecule, noscapine, in inducing G2/M arrest, inhibiting cellular proliferation and tumor growth in various human xenograft models. However, the mechanisms of mitotic-block and subsequent cell death have remained elusive. Here, we show that EM011-induced attenuation of microtubule dynamics was associated with impaired association of microtubule plus-end tracking proteins, such as EB1 and CLIP-170. EM011 treatment then led to the formation of multipolar spindles containing ‘real’ centrioles indicating drug-induced centrosome amplification and persistent centrosome declustering. Centrosome amplification was accompanied by an upregulation of Aurora A and Plk4 protein levels, as well as a surge in the kinase activity of Aurora A, suggesting a deregulation of the centrosome duplication cycle. Cell-cycle phase-specific experiments showed that the ‘cytotoxicity-window’ of the drug encompasses the late S-G2 period. Drug-treatment, excluding S-phase, not only resulted in lower sub-G1 population but also attenuated centrosome amplification and spindle multipolarity, suggesting that drug-induced centrosome amplification is essential for maximal cell death. Subsequent to a robust mitotic arrest, EM011-treated cells displayed diverse cellular fates suggesting a high degree of intraline variation. Some ‘apoptosis-evasive’ cells underwent aberrant cytokinesis to generate rampant aneuploidy that perhaps contributed to drug-induced cell death. These data indicate that spindle multipolarity induction by means of centrosome amplification has an exciting chemotherapeutic potential that merits further investigation.
Similar content being viewed by others


Main
Perturbations of the precise space–time coordination of cell-cycle events lead to genomic instability causing cancerous growth or death of progeny cells.1 Cancer cells illegitimately pass through tightly controlled cell-cycle transitions and gain a selective growth advantage over normal cells. Although strict regulation of centrosome duplication and inheritance is pivotal to the correct execution of cell division, centrosome amplification, either due to the deregulated centrosome duplication or cytokinetic failure, frequently occurs in cancer cells and correlates with chromosomal instability.
Microtubule-binding drugs also produce abnormalities in centriole structure and centrosome duplication2 in response to cell-cycle arrest3 or because of damaged or incompletely replicated DNA during mitosis.4 Interestingly, Drosophila and vertebrate cells in culture harboring extra centrosomes initially form multipolar spindles in early mitosis,5 but eventually divide bipolarly yielding division-competent progeny. The extra centrosomes cluster at two dominant spindle poles and/or become progressively inactivated.6, 7, 8 However, in cells in which extra centrosomes failed to cluster, the resulting multipolar division invariably produced inviable progeny. These observations have raised the possibility that induction of spindle multipolarity could be a potential chemotherapeutic strategy.
Anti-tubulin agents, such as taxanes and vincas are extensively used for clinical treatment of human malignancies.9 However, they either overpolymerize (taxanes) or depolymerize microtubules and form paracrystals (vincas), thereby impairing crucial microtubule functions and causing devastating toxicities.9 Noscapinoids represent a novel class of microtubule-modulating agents that avoid the harsher effects of currently available tubulin-binding chemotherapeutics by preserving the total polymer mass of tubulin.10, 11, 12 The parent molecule, noscapine, a plant-derived antitussive alkaloid, is in Phase I/II clinical trials for treatment of multiple myeloma. In silico molecular modeling efforts to rationally design and chemically synthesize better noscapine analogs have led to the identification of more potent noscapine derivatives.13, 14, 15 In particular, 9–bromonoscapine (EM011) binds tubulin with greater affinity than noscapine, and is effective even against drug-resistant xenograft tumors without any detectable toxicity.16, 17, 18, 19, 20 Therefore, noscapine and its analogs are known as ‘kinder and gentler’ microtubule-modulating agents.21
EM011 treatment causes a G2/M arrest in cancer cells followed by apoptotic cell death.16, 17, 18, 19, 20, 21, 22 However, the relationship between mitotic-block and cell death has remained elusive. In order to understand EM011's mode of action, we examined how this drug induces perturbations in the coordination of mitotic events causing cell-cycle stasis, and then tips the outcomes in favor of cell death rather than continued, increasingly aberrant and aggressive proliferation. We determined the EM011's effects on cell-cycle progression and spindle biogenesis and analyzed how it induces apoptosis. Our data show that EM011 treatment reduces microtubule dynamicity and deregulates the centrosome duplication cycle, causing centrosome amplification and spindle multipolarity. Spindle multipolarity in turn, produces multiple catastrophic cellular outcomes, culminating in apoptosis induction.
Results
EM011 preserves microtubule ultrastructure
Error-free mitotic progression requires precise regulation of microtubule dynamics. As both microtubule-stabilizing and -destabilizing agents suppress spindle dynamics,23, 24 we first asked whether EM011 mediates cytotoxicity by perturbing dynamic instability of interphase microtubules in living GFP-tubulin expressing MCF-7 cells (Supplementary Movie-1 (control) and Movie-2 (treated), time-lapse movies). In contrast to control cells in which microtubules alternated between phases of growth and shortening, with intervening ‘pause’ phases, EM011 treatment attenuated microtubule dynamics as indicated by largely unaltered position of microtubule ends at the cell periphery (Figure 1ai). Figure 1aii shows life-history traces of microtubules from control and treated cells. Supplementary Table 1 shows quantitative analysis of microtubule growth, shortening, frequencies of catastrophe and rescue and average duration of pause phase. We found that EM011 reduced overall dynamicity by ∼72% and increased pause duration by ∼76%. Thus EM011 decreases the number of dynamic events in the life history of a microtubule without affecting its long-term existence.

‘Kinder and gentler’ effects of EM011 on microtubules. Dynamic instability behavior of fluorescently labeled interphase microtubules in the thin lamellar edge of MCF-7 cells expressing GFP-tubulin was imaged by time-lapse microscopy.(ai) Time-lapse sequence of video frames, 3 s apart, showing ends of microtubules near the cell periphery in vehicle-treated control (top) and 10 μM (bottom) EM011-treated MCF-7 GFP-tubulin cells. In the control cell, white and green arrows indicate two microtubules that undergo growing and shortening events, respectively. In the EM011-treated cell, white-arrowhead indicates two microtubule ends the positions of which did not change significantly over time. (aii) The positions of the microtubule ends near the cell periphery were tracked over time to generate life-history plots in the absence (top) or presence (bottom) of 10 μM EM011 from which microtubule dynamic instability variables were derived. The life-history traces of control microtubules show extensive length changes, whereas those of treated cells are nearly flat, indicating marked dampening of dynamics in the presence of EM011. Graphs, lengths of excursions made by individual microtubule ends over time. (b) EM011 does not perturb the morphology of microtubules as assessed by light and electron microscopy. Confocal immunomicrographs (top-panels) of cells with microtubules (green) and DNA (red) showing that 0.01 μM paclitaxel treatment overpolymerized and bundled microtubules in 2 h, evident by dense green sheet-like structures (top-middle). However, a 2-h treatment with 10 μM EM011 had no effect on the overall morphology of microtubules (top-right). The radial-array of interphase microtubules was maintained, and microtubules appeared similar to control microtubules (top-left). Transmission electron microscopy (bottom-panels) revealed that EM011 does not affect the ultrastructure of microtubules as compared with controls (see black-arrows). Paclitaxel-treated microtubules showed bundling (black-arrows, middle bottom-panel) and were closely packed into parallel-sheets. (c) Confocal fluorescence micrographs showing GFP-EB1 localization and CLIP-170 staining patterns (green) and nuclei (blue) in HeLa cells either transfected with GFP-EB1 (top-row) or labeled using anti-CLIP-170 antibody (bottom-row) following treatment with vehicle or 10 μM EM011. Scale bars: 10 μ m. Scale bar for EM image: 250 nm
Unlike taxanes and vincas that often exert extreme effects on microtubules, EM011 preserves the monomer/polymer ratio of tubulin at concentrations as high as 100 μM.11, 13 Using immunofluorescence confocal microscopy and transmission electron microscopy, we found that EM011 did neither perturb the radial-array of microtubules nor their ultrastructural morphology in interphase cells, compared with vehicle-treated cells (Figure 1b). In contrast, 0.01 μM paclitaxel overpolymerized microtubules and bundled them into continuous sheets (Figure 1b). Hence, EM011 treatment merely dampened microtubule dynamics without causing any detectable structural changes to the microtubule cytoskeleton.
EM011 modulates microtubule dynamics by affecting microtubule plus-end proteins
Changes in microtubule dynamicity are generated by microtubule-associated proteins and plus-end-tracking proteins that are crucial for many microtubule-regulated processes. Among the latter, EB1 (end binding protein-1) is a core regulator of microtubule plus-end function.25 We examined whether EM011-induced reduction in microtubule dynamicity correlated with any changes in EB1 localization in HeLa cells. In control cultures, conspicuous GFP-EB1 comets decorated the growing plus-ends of microtubules (Figure 1c) as expected.25 In contrast, EM011 treatment reduced the size of EB1 comets that appeared as fine puncta dispersed throughout the cell (Figure 1c). This phenotype could reflect a tubulin and/or microtubule conformation change that alters the binding affinity or the number of EB1-binding sites on microtubules. Alternatively, EM011 may directly inhibit EB1 binding to microtubules or EB1 stability. In either case, a drastic reduction in the amount of microtubule-bound EB1 is likely to significantly impact the behavior of microtubule plus-ends. Next, we examined how EM011 affected localization of CLIP-170 (cytoplasmic linker protein-170), another microtubule plus-end tracking protein. Inhibition of microtubule dynamics by anti-mitotic drugs can disrupt the microtubule association of CLIP-170.26 We observed that in contrast to vehicle-treated cells in which CLIP-170 localizes to microtubule plus-ends as distinct comets, EM011 treatment abolished CLIP-170's plus-end association and caused its intracellular dispersal (Figure 1c), an effect strikingly reminiscent of EM011's effect on EB1-GFP localization. Thus, EM011 causes dramatic changes in overall microtubule dynamicity and in particular, may impair normal behavior of microtubule plus-ends, without affecting its gross morphology.
EM011 induces mitotic and not G2 arrest
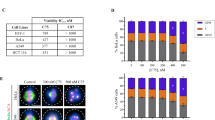
EM011 inhibits proliferation of various cancer cells through induction of G2/M arrest.16, 17, 18, 19, 20, 22 To explore cell-cycle-specific events that result in mitotic-block and subsequent cell death, we chose cervical cancer HeLa cells, owing to their relatively strong checkpoints. A standard sulforhodamine-B assay reported the IC50 of EM011 in HeLa cells to be 4.5 μM (data not shown). We next examined time–dose effects (10 and 25 μM EM011) on cell-cycle progression of HeLa cells flow-cytometrically (Figure 2a) and quantitated the proportion of cells in each cell-cycle phase (Figure 2b). Our results indicated that upon drug treatment, cells transit through S-phase and accumulate in G2/M-phase (with 4N DNA) maximally at 24 h (∼72%) for 10 μM and 18 h (∼80%) for 25 μM EM011 (Figure 2c). Furthermore, two-color flow-cytometry showed that the mitotic population peaked to ∼56% (10 μM EM011, 24 h) and ∼63% (25 μM EM011, 18 h) indicating time-dependent accumulation of mitotic cells at both doses (Figure 2d).

EM011 affects kinetics of cell-cycle progression. (a) Dose-dependent (10 and 25 μM) effects of EM011 over time of treatment. The x-axis shows DNA amount representing different cell-cycle phases (sub-G1, G1, S, G2/M), y-axis shows number of cells that contain that much amount of DNA and z-axis shows time of treatment. (b) Bar-graphical quantitative comparison of percent cells in various cell-cycle phases at two doses at the stated time points. Values for each phase of the cell-cycle in the graph represent average, respectively, of three independent experiments (P<0.05). (c) DNA histograms depicting the time of maximal G2/M arrest for each dose. (d) Corresponding dot-plots clearly showing mitotic cells (cells with 4N DNA content and an elevated MPM-2 signal)
EM011 induces formation of multipolar spindles through centrosome amplification
We next visualized formation of highly aberrant multipolar mitotic spindles in EM011-treated cells using immunofluorescence confocal microscopy. We quantitated the frequencies of abnormal spindles using a well-accepted classification scheme,23 at different drug-dosages over time. 25 μM EM011 induced multipolarity, predominantly with three or four poles (Supplementary Movie-3), at 12 h post-treatment (Figure 3ai, top-panel). However, at 50 μM, multiple poles appeared as early as 3 h and increased in number ranging between 5 to 8 (Figure 3ai, top-panel). Surprisingly, interphase cells in treated cultures displayed normal radial-arrays of microtubules at both 25 and 50 μM EM011 (Figure 3ai, bottom) emanating from centrosomes (Supplementary Figure 1). This is a unique edge of EM011, compared with conventional tubulin-binding drugs that polymerize or depolymerize microtubules at concentrations higher than their IC50.24 Figure 3aii shows quantitation of frequencies of drug-induced spindle abnormalities (Types I–III). All cell-cycle stages appeared normal in control cells (Figure 3b and Supplementary Movie-4). In drug-treated cultures, mitotic cells with bipolar, as well as multipolar spindles (Supplementary Figure 2a), displayed defects in chromosome congression to the mid-plate. Concomitant with the appearance of multipolar spindles, the kinase activity of Aurora B increased (Supplementary Figure 2b), suggesting presence of aberrant kinetochore–microtubule attachments.27 Further evidence for the same came from the strong increase in BubR1 staining in EM011-treated cells (Supplementary Figure 2c, top-row) compared with controls. An increase in BubR1 phosphorylation (Supplementary Figure 2d), implied a lack of tension and/or existence of aberrant kinetochore–microtubule connections that caused spindle checkpoint activation. It is intriguing that 25 and 50 μM drug concentrations trigger conspicuous spindle multipolarity with a striking declustering of supernumerary centrosomes. Although suppression of microtubule dynamics may also be involved in multipolar spindle formation, other mechanisms such as interference with centrosome duplication28 or dispersion of pericentriolar material to generate multiple microtubule-nucleating sites could result in multipolar spindles.29 Microtubule-targeting drugs are also known to influence centrosome structure presumably by disrupting microtubule-dependent events in centrosome biogenesis.2

Frequency of spindle abnormalities induced in HeLa cells by EM011 treatment. (ai) Immunofluorescence confocal micrographs of cells treated with indicated concentrations of EM011. Upper-panel shows the arrest phenotypes of mitotic cells in treated samples, whereas lower-panel shows microtubule arrays in interphase cells in the same samples. (aii) Bar-graphical representation of various spindle types observed in EM011-treated cells at various doses. On the basis of a well-accepted classification nomenclature,23 type I and II spindles are bipolar with one or more uncongressed chromosomes. Type III are monopolar spindles with chromosomes arranged in a ball around the spindle. Multipolar spindles displayed more than two poles (n>2). Columns, mean of three independent experiments upon evaluation of at least 200 cells per condition; error bars, SD (P<0.05). (b) Control cells in interphase were flat, well-spread and uniform in size, with a fine filamentous array of microtubules and a single spherical or ovoid nucleus. In addition, control vehicle-treated cells showed normal bipolar metaphase spindles, and all phases of mitosis appeared to be executed normally. (ci) Confocal micrographs of cells treated with vehicle or 25 μM EM011 for 24 h. Panels show α-tubulin (green), γ-tubulin (red) and DNA (Hoechst 33 258) in control vehicle-treated (top-row) and EM011-treated (bottom-row) cells. A total 200 mitotic cells in several randomly selected fields were counted for the frequency of γ-tubulin spots. (cii) Bar-graph quantitation of the number of mitotic cells harboring the indicated numbers of γ-tubulin spots in vehicle or 24 h EM011-treated cultures. Scale bar=10 μm
Centrosome hyperamplification may also result from a prolonged S-phase30 or cytokinesis failure.31 However, our previously published data from various cell-types and data shown in Figure 2 emphasize that EM011-treated cells do not stall in S-phase. Also, the rapid emergence of multipolar phenotype rules out cytokinesis failure and consequent multinucleation as possible causes. As tightly regulated centrosome duplication is crucial to proper spindle biogenesis, we examined whether centrosomes could be one of the targets of EM011 and whether they contributed to the formation of multipolar spindles. We performed α- and γ-tubulin staining to examine the presence of a true centrosome on each spindle pole. Although majority of vehicle-treated mitotic cells showed only two spots of γ-tubulin per cell, supernumerary centrosomes were present at multiple spindle poles upon EM011 treatment (Figure 3ci and cii) and only ∼15% of EM011-treated mitotic cells showed two γ-tubulin spots per cell. The frequencies of three γ-tubulin spots and four or more γ-tubulin spots per mitotic cell were ∼13-fold and ∼30-fold higher, respectively, in EM011-treated cells than in vehicle-treated cells (Figure 3cii). Thus, EM011 exposure causes centrosome amplification and the centrosomal component γ-tubulin is present at each of the multiple poles.
EM011-induced supernumerary centrosomes contain ‘real’ centrioles
The detection of γ-tubulin at spindle poles alone is not sufficient to unambiguously distinguish between centrosome overduplication and centrosomal matrix fragmentation. To distinguish between centrosome fragmentation and overduplication, we evaluated centriole profiles in EM011-induced supernumerary centrosomes using anti-centrin-2 antibodies. Centrin-2 localizes to distal-lumen of centrioles and their interconnecting fibers.32 Although vehicle-treated mitotic cells contained two centrosomes, each with a pair of centrioles, EM011-treated cells had amplified centrosomes with each γ-tubulin-positive structure containing the centriolar component, centrin-2 (Figure 4a). Majority of vehicle-treated mitotic cells contained only four centrin-2 spots per cell. However, a large proportion of EM011-treated mitotic cells had six or more centrin-2 spots per cell that co-localized with the γ-tubulin-positive spindle poles (Figure 4b). These data strongly suggested that multipolarity was in fact due to abnormal amplification of centrosomes that clearly contained centrioles. Interestingly, EM011 also caused untimely disengagement of centriole pairs and the intercentriolar distance was increased by ∼2.5 fold in drug-treated cells compared with controls (Figure 4c). Occasionally, four spots of centrin-2 were visible in close proximity (Figure 4d). However, alternative possibilities, viz, de novo synthesis or localization of centriolar components,33 cannot be ruled out.

Centrosome amplification in HeLa cells upon EM011 treatment. (a) Immunofluorescence confocal micrographs of cells stained for centrioles in green (centrin-2), centrosomes in red (γ-tubulin) and DNA in blue (Hoechst 33 258). Mitotic cells in vehicle-treated controls had two γ-tubulin-positive spindle poles each with a centriole pair (top-row). The multiple centrosomes induced in 24 h drug-treated cells also had ‘real’ centriole pairs (bottom-row). A total of 200 mitotic cells in several randomly selected fields were counted for the frequency of centrin-2 spots. A quantitative analysis of the proportions of cellular γ-tubulin that was recruited to the spindle poles in these cells revealed that there was no net increase in the amount of γ-tubulin recruited to spindle poles in EM011-treated cells as compared with controls; instead it appeared that the amount of γ-tubulin found at spindle poles in vehicle-treated cells was redistributed among the multiple spindle poles in EM011-treated cells. (b) Bar-graph showing quantitation of the number of mitotic cells harboring the indicated numbers of centrin-2 spots in vehicle or 24 h EM011-treated cultures. (c) EM011 treatment results in disengagement of centriole pairs and increases the intercentriolar distances. Zoom-in images show intercentriolar distance in control (left) and 24 h drug-treated cells (right). Values and error bars shown in the graph represent average and standard deviations, respectively, of three independent experiments (P<0.05). (d) Confocal immunomicrographs showing clusters of centrioles at one of the poles of the multipolar spindles (white arrowheads) in two different cells upon drug treatment. Insets show zoom-in images of the clusters pointed by white arrowheads revealing the presence of three or four centrioles at the spindle pole. Scale bar=10 μm
EM011 deregulates centrosome duplication
To further establish whether EM011 treatment caused misregulation of centrosome duplication cycle, we examined levels of Polo-like kinase 4 (Plk4), a central regulator of centriole biogenesis, the overexpression of which produces supernumerary centrioles. Conversely, RNA interference of Plk4 causes sequential reduction of centriole numbers,34 indicating that intracellular Plk4 abundance must be tightly regulated to prevent untimely centriole reduplication. We found that Plk4 levels increased by 12 h post-treatment (Figure 5a) when supernumerary centrosomes begin to appear, and rose further between 12–24 h of EM011 exposure. Plk4 accumulation, therefore, correlated with the period of centrosome duplication in drug-treated cells and may disrupt control of centriole numbers in these cells. Cyclin E levels also surged during the same time interval (Figure 5a) as increase in Plk4 levels. This was noteworthy as in human cells, centriole overduplication through concurrent formation of multiple daughter centrioles at single maternal centrioles, requires cooperative action of both cyclin E/cdk2 and Plk4.33 Next, we examined the levels of Aurora kinase A, the overexpression of which also leads to centrosome amplification and cellular transformation.35 EM011 treatment caused an increase in Aurora A levels (Figure 5a) and its kinase activity (Figure 5b), concomitant with accumulation of Plk4 and cyclin E. Immunostaining also showed a massive increase in intracellular Aurora A levels in treated cells, compared with controls in which Aurora A was clearly restricted to centrosomes (Figure 5c). Taken together, our data show that EM011-triggered induction of spindle multipolarity through centrosome amplification and declustering is accompanied by an accumulation of cyclin E, Plk4 and Aurora A kinases, factors that could be responsible for deregulation of centrosome duplication.

(a) Representative immunoblots showing Plk4, Aurora A and cyclin E levels in cells treated with 25 μM EM011 for the indicated time periods. β-actin was used as a loading control.(b) Immunoblot for phospho-histone-H3 showing the increased kinase activity of Aurora A kinase immunoprecipitated from 12 h EM011-treated cells. (c) Confocal immunomicrographs of cells stained for Aurora A in red and DNA in blue (DAPI). Aurora A localizes to centrosomes in vehicle-treated cells (top-row), but EM011 treatment for 12 h results in a massive intracellular accumulation of Aurora A. (d) EM011-induced mitotic arrest is followed by an exit from mitosis. Representative dual-color dot plots indicating that 36-h drug treatment results in the depletion of MPM-2-positive cells and a concomitant increase in cells containing 4N DNA, suggesting mitotic exit without cytokinesis. (e) Confocal immunomicrographs depicting representative mitotically arrested cells (left) at 24 h that eventually slip out of mitosis at 36 h and appear as conspicuously large multinucleated cells (right). Scale bar=10 μm
EM011-induced mitotic arrest is followed by an exit from mitosis
Among the possible outcomes of drug-induced mitotic arrest, one involves an erroneous exit from mitosis in the presence of drug, and subsequent transition into interphase. We thus evaluated the fate of chronically arrested mitotic cells. Our data show that although ∼56% cells were mitotically arrested at 24 h, ∼51% of cells exited mitosis without cytokinesis at 36 h after treatment (Figure 5d). These cells were still tetraploid (4N), but were no longer MPM-2 positive, implying that they had exited mitosis. As multinucleation and tetraploidy are the hallmarks of mitotic slippage, it is likely that several aberrant mitotic cells had slipped out of mitosis (Figure 5d and e) without proceeding through normal anaphase and cytokinesis. Upon exit from mitosis, these cells decondense their chromatin forming multinucleated, pseudo G1-like tetraploid cells (Figure 5e, right).
EM011 induces chronic mitotic delay followed by cell death by multiple fates
Next, we reduced temporal heterogeneity to closely monitor EM011-induced responses by synchronizing HeLa cells at the G1/S boundary. Upon G1/S release, we determined the average time control cells spend in each cell-cycle phase (Supplementary Figure 3) and examined immunostained vehicle- and drug-treated cells for their spindle and nuclear morphologies (Figure 6ai). At 8 h post release, most drug-treated cells showed numerous microtubule nucleation sites (due to centrosome amplification) evident as distinct bright spots (Figure 6ai). These typically aberrant G2 cells then rounded up into abnormal multipolar mitotic cells at about 9–10 h and accumulated up to 20 h post release (Figure 6ai). At 12 h of EM011-treatment post release, several γ-tubulin spots (3, 4 or 5) were evident (Figure 6aii (b–d)). This was in stark contrast to vehicle-treated cells that underwent a normal anaphase at 7–8 h, cytokinesis at 8–9 h and progressed into the next interphase at about 9 h post release (Figure 6aiii a–d).

EM011 induces a prolonged mitotic arrest in synchronous HeLa cells. (a,i) Representative confocal immunomicrographs of cells showing microtubules (green) and DNA (red) upon treatment with EM011 for the noted hours post-G1/S release. It is clearly evident that G2 cells begin to show numerous ‘nucleation sites’ that are perhaps the cause of multipolarity that sets in during mitosis. Multiple poles (up to six) can be seen in mitotically arrested cells. Mitotic cells continue to accumulate until about 20 h of drug-treatment post release(ii). Drug-treated cells (12 h treatment post release) showed several γ-tubulin spots (b shows three, c shows four and d shows five spots) indicating centrosome hyperamplification compared with control vehicle-treated mitotic cells. (iii) Vehicle-treated cells display normal kinetics of cell-cycle progression and all hallmarks of a typical mitotic process were visible. (b,i) Histogram overlays representing progression of cell cycle of synchronous HeLa cells over noted time points for drug- and vehicle-treated cells post release. (ii) Bar-graphical quantitation of G2 and mitotic cells from a two-color flow cytometric experiment using MPM-2 and PI staining for EM011- and vehicle-treated cells post release for various time points. Values for the G2 and M phase in the graph represent average, respectively, of three independent experiments (P<0.05). (iii) Line-graph depicting percent G2/M and sub-G1 cell populations from drug- and vehicle-treated cells over time post release. The values and error bars shown in the line-graph represent average and standard deviations, respectively, of three independent experiments (P<0.05). (iv) Representative immunoblots showing cyclin B1, cleaved caspase-3 and cleaved PARP levels in synchronized cells that were EM011- or vehicle-treated over the noted time post release. β-actin was used as a loading control. Scale bar=10 μm
Next, we quantitated cells in various cell-cycle phases upon drug-treatment post release over time by dual-color flow cytometry (Figure 6bi). Upon G1/S release, both vehicle- and drug-treated cells displayed comparable kinetics of progression through S and G2 phases indicating that EM011 did not affect cell-cycle progression through these phases (Figure 6bi). However, at ∼9 h post release, drug-treated cells begin to arrest in mitosis with 4N DNA and positive MPM-2 staining, in contrast to vehicle-treated cells that had transited through mitosis and completed their cell cycle. Drug-treated cells remain stuck in mitosis with MPM-2-positive population peaking at 16 h to ∼85% (Figure 6bii). A sub-G1 population at 20 h that continued to accumulate until 24 h indicated apoptosis (Figure 6biii). Cyclin B1 accumulation commenced at 6 h of drug-treatment post release, and continued to increase until 20 h post release (Figure 6biv), and declined thereafter (perhaps due to mitotic slippage). Apoptosis induction was confirmed by detection of cleaved caspase-3 and PARP expression (Figure 6biv) between 12–24 h post release.
Following mitotic arrest, EM011-treated cells display variable cellular outcomes
Consequent to mitotic arrest, cells are consigned to variable fates on the basis of two competing networks, one being cyclin B1 driven and the other being caspase-3 dictated.36 As our immunoblotting data (in Figure 6biv) reflect average protein levels within entire cell populations, we comprehended drug-induced outcomes at single-cell level by double immunostaining cells for microtubules and DNA. Drug-treated synchronized cells were microscopically observed after 20 h of drug-exposure post release. This time point was chosen as ∼80–90% cells accumulate in mitosis at 16 h post release. In agreement with the notion of intraline variation, we observed that mitotically arrested cells displayed substantial variation in their ultimate fates. First, cell death was initiated in ∼10–20% of mitotically arrested cells (Figure 7A, a–d). Figure 7A, a–d show mitotic cells with tubulin accumulation in the blebs. Second, ∼40% (n=200) cells exited mitosis without cytokinesis into a multinucleate G1-like interphase (Figure 7A, e). We also found multinucleated cells succumbing to cell death as indicated by the appearance of highly fragmented nuclei (Figure 7A, f–g). We speculate that the unsilenced spindle checkpoint complex might trigger apoptosis in these pseudo-4N G1-like interphase cells. Occasionally, multinucleated cells initiated cell death by blebbing (Figure 7A, h). Some cells showed condensed chromatin, reminiscent of apoptotic bodies (Figure 7A, i–j). We also noted asymmetric cell divisions of apoptosis-reluctant cells into three or four daughter cells generating high-grade aneuploidy (Figure 7A, k–l). Such divisions often led to asymmetric distribution of cytoplasm (anisocytosis) and DNA (anisokaryosis), which perhaps explain a sub-G1 peak because cells would then have an average of 1.33N DNA instead of 2N DNA and might be inviable. These observations support the idea that high-grade aneuploidy may have a tumor-suppressive and death-promoting role.37

(A) Synchronized HeLa cells displayed profound intraline variation in cellular outcomes post mitotic arrest upon EM011 treatment. Mitotic arrest terminated in one of the four ways: cells died while still in mitosis, with blebbing and eventual lysis (a–d); cells slipped from mitosis without cytokinesis, respread and reformed a multinucleate cell (e), cells with micronuclei also evident in some cases (f, g) and blebs beginning to appear in others (h); some cells that slipped simply remained in G1-like interphase, often growing larger, which we interpret as cell-cycle arrest in G1 with 4N DNA. These cells underwent death from the polyploid interphase as evidenced by small fragmented DNA, reminiscent of apoptotic bodies (i, j); cells that escaped mitotic death and slippage aggressively moved ahead to divide asymmetrically into three or four aneuploid daughter cells with abnormal cytoplasmic and nuclear divisions resulting perhaps in inviable karyotypes (k–n) that were death bound as evident by blebbing (o, p). Induction of apoptosis was visualized using immunofluorescence microscopy by increased expression of cleaved caspase-3 (B) and cleaved PARP (C) in drug-treated cells compared with controls upon 20-h treatment post release. Scale bar=10 μm
Some drug-treated cells underwent abnormal cytokinesis upon mitotic exit, generating multinucleated daughters containing varying numbers of micronuclei (Figure 7A, m–n) or occasionally blebs, indicating early apoptosis (Figure 7A, o–p). Such abnormal cytokinesis was almost never observed in control cultures, indicating that this does not occur spontaneously in HeLa cells. Furthermore, apoptosis induction was observed by cleaved caspase-3 and PARP immunostaining in synchronized cells that were drug treated for 20 h post release (Figure 7B and C). These data thus suggest that EM011-induced high-grade aneuploidy produces an intensely unstable inviable karyotype facilitating cell death.
Induction of spindle multipolarity is crucial for optimal cell death
In order to understand further EM011's mode of action, we determined its ‘cytotoxicity-window’ by evaluating the cell-cycle phase during which treatment resulted in maximum multipolarity and cell death. We also examined whether spindle multipolarity induction was necessary for maximal cell death. To this end, we designed a phase-specific treatment regimen and exposed synchronized HeLa cells to 25 μM EM011 during the duration of each cell-cycle phase, viz, S, G2 or M-phase, followed by EM011 wash off (Figure 8a). Cells were allowed to continue cell cycle for another 8 h before fixation for flow cytometry and immunostaining. The duration of each cell-cycle phase was determined on the basis of the cell-cycle progression kinetics in synchronized HeLa cells (Supplementary Figure 3). We observed that the cytotoxic effect of EM011 was maximum (∼30% sub-G1) when the drug was applied between 7–11 h post release (i.e., during duration of G2) (Figure 8b,c). Considerable cell death was also observed in S-phase-treated cells (∼15% sub-G1) unlike cells treated in M-phase (∼4% sub-G1) (Figure 8c). To define more precisely the phase-specific drug sensitivity, cell-cycle phases were subdivided into early, mid and late phases, and drug treatment was performed as before (Figure 8d). This detailed set-up revealed that the most ‘critical-window’ for drug treatment corresponds to mid and late S and early G2 phases, but not mitosis, as cells appeared quite resistant to cell death when treated in mitosis (Figure 8e). Moreover, cells treated specifically in S-phase showed multipolar mitotic spindles, whereas cells left untreated in S-phase and treated thereafter rarely showed multipolarity in mitosis (Figure 8f). This correlation between enhanced spindle multipolarity and apoptosis in S/G2-phase-treated cells tempts us to propose that the deregulation of centrosome duplication cycle and the resulting multipolar spindle morphology are crucial for activating apoptosis in these cells.

Phase-specific treatment of HeLa cells revealing the specific ‘cytotoxic-window’ of EM011. (a) Schematic representing the first set of drug-treatments specific for S, G2 and M-phase. (b) Histogram overlays of cell-cycle profiles of cells treated with specific durations representing full cell-cycle phases (profiles are color-coded as per the Scheme in a). (c) Quantitation of sub-G1 population of HeLa cells treated with EM011 in a phase-specific manner, revealing G2 as the most sensitive phase to drug treatment. The values and error bars shown in the bar-graph represent average and standard deviations, respectively, of three independent experiments (P<0.05). (d) Schematic representation of the second set of drug-treatments with the respective cell-cycle phases subdivided into early (E), mid (M) and late (L) phases. (e) Quantitation of sub-G1 population after the noted treatments was performed using flow-cytometric analysis, revealing the precise ‘cytotoxicity-window’ of EM011. Values and error bars shown represent average and SDs, respectively, of three independent experiments (P<0.05). (f) Confocal immunomicrographs depicting representative mitotic spindle morphology of cells that were drug-treated only in S-phase compared with cells that were not treated in the S-phase
Discussion
In this study, we show that EM011, a novel noscapinoid, induces a time-dependent robust mitotic arrest through attenuation of microtubule dynamics, and triggers loss of EB1 and CLIP-170 comets from microtubule plus-ends. As EM011 leaves interphase microtubular virtually unperturbed albeit with reduced dynamicity, it spares the vast majority of post-mitotic cells in vivo, in stark contrast to the non-specific effects of taxanes and vincas. We envision that dampening of microtubule dynamics impairs the ability of mitotic microtubules to capture chromosomes effectively, resulting in a large number of uncongressed chromosomes, increased Aurora B kinase activity, and spindle checkpoint activation in EM011-treated cells. Spindle checkpoint activation in turn prevents anaphase onset and cyclin B destruction, as multipolar cells accumulate in metaphase.
A cell, however, cannot remain arrested in metaphase indefinitely. Our data uncover a high-degree of inherent intraline variation as HeLa cells exhibit diverse fates consequent to EM011-induced mitotic arrest. Mitotic arrest concluded in several different ways perhaps depending on a complex interplay between cell cycle (cyclin B1 and BubR1) and apoptosis (caspase-3 and PARP) regulatory molecules. Some cells showed aberrant multipolar spindles along with extensive plasma membrane blebbing indicating mitotic death. However, we cannot exclude the possibility that cells that succumb to apoptosis during mitotic arrest may have suffered an insult before mitosis. Nonetheless, the appearance of significant sub-G1 population only after 20 h of drug exposure makes any early insult an unlikely cause in apoptosis induction in these cells. Our data instead strongly argue that prolonged metaphase arrest compels cells to seek escape routes that culminate in cell death. Mitotic slippage constitutes one such route in which cyclin B1 levels begin to decline eventually leading to an inappropriate exit from cell cycle. We found that a significant proportion of drug-treated cells slipped out of mitosis without cytokinesis, re-spread and formed massive G1-like interphase cells, often with several micronuclei. We also found an apoptosis-reluctant population that progressed through the cell cycle after mitotic stall to undergo aberrant cytokinesis generating three or four aneuploid daughter cells created by multiple cytokinetic furrows. This supports the emerging paradigm that highlights tremendous complexity in cellular responses to tubulin-binding anticancer drugs consequent to mitotic arrest.
The most remarkable feature of EM011-induced mitotic arrest is hyperamplification of centrosomes and multipolar spindle formation. Although other anti-microtubule drugs, such as like taxanes and nocodazole, also generate multiple spindle poles at low concentrations, they primarily do so by causing pericentriolar matrix fragmentation and not by producing new centrioles. In contrast, EM011-induced spindle poles contained the hallmark components of true centrioles γ-tubulin and centrin-2. Furthermore, accumulation of cells with multipolar spindles was accompanied by a surge in Plk4, cyclin E and Aurora A levels in drug-treated cells, suggesting deregulation of centrosome duplication, with lethal consequences. These findings strongly correlate with our observation that EM011 exposure during S-phase was crucial for maximal apoptosis induction. Thus EM011 treatment not only reduces microtubule dynamicity but also leads to a derailment of centrosome duplication cycle, causing inappropriate centrosome amplification.
Previous studies in vertebrate cells carrying extra centrosomes have shown that even though these cells initially form multipolar spindles, supernumerary centrosomes eventually aggregate at two poles resulting in bipolar division. These cells, therefore, rely on centrosome clustering for remaining viable.6, 7, 8 In contrast, EM011 causes centrosome hyperamplification and spindle multipolarity. EM011-induced centrosome declustering is an extraordinary and unique effect of this drug that perhaps compels cells, after a prolonged mitotic arrest, to undergo multipolar and asymmetric divisions, with catastrophic consequences. Although it is presently unresolved whether dampening of microtubule dynamics and centrosome hyperamplification represent two independent cellular activities of EM011, or whether mechanistic links exist between the two processes, we propose that EM011-induced attenuation of microtubule dynamics directly incapacitates a cellular mechanism for suppressing multipolar mitoses, forcing the cell into a ‘suicidal’ multipolar division. Such drugs that interfere with centrosome clustering mechanisms could potentially be selectively lethal to tumor cells that are inherently vulnerable to forming extra centrosomes or those that already possess additional centrosomes. In fact, EM011 displays remarkable tumor selectivity in various mice models16, 17, 18, 19, 20 with no detectable toxicity to tissues with frequently proliferating normal cells. Furthermore, in contrast to the effects of EM011 on HeLa cells as reported in our study herein, the drug had no discernable effect on centrosome numbers or bipolar spindle morphology in non-malignant near-normal breast epithelial cells.38
Several studies have also demonstrated that multipolar division results in cell death from high-grade aneuploidy39 arising from intolerable amounts of chromosome missegregation. Our data, therefore, focus the spotlight once again on the induction of spindle multipolarity through centrosome amplification and declustering as a possible new chemotherapeutic approach. Tumors, however, comprise cellular subpopulations that are heterogeneous for karyotype, growth rate, metastasis, antigenicity and sensitivity to chemotherapeutics agents.40 Such clonal heterogeneity and clonal evolution during progression of human cancers present a major clinical challenge for the development of powerful and selective chemotherapeutics. Undoubtedly, further work is warranted to more fully explore the chemotherapeutic potential presented by centrosome amplification and declustering activities of tubulin-active drugs.
Materials and Methods
Cell lines, culture and drug treatment
HeLa cells were grown in DMEM, containing 10% FBS. EM011 was synthesized from noscapine as previously described11 and 50 mM stock solutions were made in DMSO. For GFP-EB1 localization studies, HeLa cells were transfected with the GFP-EB1 construct for 48 h after which cells were treated either with EM011 or vehicle for 2 h before imaging. For CLIP-170 staining, HeLa cells were treated with vehicle or EM011 for 2 h before fixation.
Image acquisition and analysis of microtubule dynamics in living MCF-7 cells
Cells were prepared for the analysis of interphase microtubule dynamics as described previously.19 Briefly, MCF-7 human breast cancer cells stably expressing GFP-tubulin were grown for 24 h on polylysine coverslip-bottom MatTek plates (MatTek, Ashland, MA, USA) and then incubated in presence or absence of EM011 for 2 h. Control cells were incubated with equivalent volume of DMSO alone. Time-lapse images of each cell were acquired at 3 s intervals using a Hamamatsu ORCA-II digital camera (Middlesex, NJ, USA) driven by Metamorph software (Universal Imaging, Media, PA, USA) on an UltraView ERS fluorescence microscope with a heated chamber. The criteria for microtubule dynamics analysis are described.19
Cell synchronization
HeLa cells were synchronized at G1/S by treatment with 2 mM thymidine for 19 h, followed by incubation in thymidine-free medium for 9 h and then incubation with 2 mM thymidine again for 16 h. Cell synchronization at G1/S boundary was confirmed by flow cytometry. Blocked cells were then released into thymidine-free medium, EM011-treated and collected at the indicated time points after G1/S release.
Immunofluorescence microscopy, cell-cycle analysis, immunoprecipitation, kinase assays and immunoblotting
Cells were cultured to ∼70% confluence and medium was replaced with fresh medium containing either vehicle (0.1% DMSO) or 10 μM or 25 μM EM011 for the noted times, followed by processing for immunofluorescence microcopy, flow cytometry or immunoblotting. Mitotic index was determined by using MPM-2 antibody as described.22 A detailed analysis of γ-tubulin immunofluorescence signal intensity at the spindle poles was performed utilizing Metamorph analysis software to determine the proportion of cellular γ-tubulin that was recruited to the spindle poles in these cells. The final output for both vehicle- and EM011-treated cells was the integrated intensity based on total stained area and staining intensity at individual pixels. To examine Aurora A and B kinase activities, Aurora A or Aurora B antibodies were used to selectively immunoprecipitate Aurora A or B proteins from control and EM011-treated HeLa cell lysates. The resulting immunoprecipitate was incubated with pure histone-H3 protein in the presence of ATP and kinase buffer. The kinase assay reaction allowed immunoprecipitated Aurora A or B to phosphorylate histone-H3 in vitro, the extent of which was measured by immunoblotting using phospho-histone-H3 antibody. Antibodies for Aurora A, Aurora B, Plk4, cyclin B1, phospho-histone-H3, cleaved caspase-3 and cleaved PARP were from Cell Signaling (Beverly, MA, USA). Antibodies against γ-tubulin, α-tubulin and β-actin were from Sigma (St. Louis, MO, USA). Anti-BubR1 antibody was from BD Biosciences (Pharmingen, San Jose, CA, USA). Horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Alexa 488- or 555-conjugated secondary antibodies were from Invitrogen (Carlsbad, CA, USA). Dilution range for the antibodies was from 1 : 200 to 1 : 1000. Histone-H3 protein was from Millipore (Billerica, MA, USA).
Electron microscopy
Monolayer cells on coverslips were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) overnight. After two buffer washes, cells were post-fixed in 1% osmium tetroxide and 1.5% potassium ferrocyanide, followed by ethanol dehydration and embedding in Eponate-12 resin. 60–70 nm sections were cut parallel to monolayer surface, counter stained with 4% aqueous uranyl acetate and lead citrate and observed on a Hitachi H7500 transmission electron microscope.
Abbreviations
- EB1:
-
end binding protein-1
- CLIP-170:
-
cytoplasmic linker protein-170
- Plk4:
-
polo-like kinase-4
References
Kops GJ, Weaver BA, Cleveland DW . On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer 2005; 5: 773–785.
Zuckerberg C, Solari AJ . Centriolar changes induced by vinblastine sulphate in the seminiferous epithelium of the mouse. Exp Cell Res 1973; 76: 470–475.
Keryer G, Ris H, Borisy GG . Centriole distribution during tripolar mitosis in Chinese hamster ovary cells. J Cell Biol 1984; 98: 2222–2229.
Hut HM, Lemstra W, Blaauw EH, Van Cappellen GW, Kampinga HH, Sibon OC . Centrosomes split in the presence of impaired DNA integrity during mitosis. Mol Biol Cell 2003; 14: 1993–2004.
Nigg EA . Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2002; 2: 815–825.
Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A et al. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008; 133: 1032–1042.
Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev 2008; 22: 2189–2203.
Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS . Spindle multipolarity is prevented by centrosomal clustering. Science 2005; 307: 127–129.
Tuxen MK, Hansen SW . Neurotoxicity secondary to antineoplastic drugs. Cancer Treat Rev 1994; 20: 191–214.
Ye K, Ke Y, Keshava N, Shanks J, Kapp JA, Tekmal RR et al. Opium alkaloid noscapine is an antitumor agent that arrests metaphase and induces apoptosis in dividing cells. Proc Natl Acad Sci USA 1998; 95: 1601–1606.
Zhou J, Gupta K, Aggarwal S, Aneja R, Chandra R, Panda D et al. Brominated derivatives of noscapine are potent microtubule-interfering agents that perturb mitosis and inhibit cell proliferation. Mol Pharmacol 2003; 63: 799–807.
Ke Y, Ye K, Grossniklaus HE, Archer DR, Joshi HC, Kapp JA . Noscapine inhibits tumor growth with little toxicity to normal tissues or inhibition of immune responses. Cancer Immunol Immunother 2000; 49: 217–225.
Aneja R, Vangapandu SN, Lopus M, Viswesarappa VG, Dhiman N, Verma A et al. Synthesis of microtubule-interfering halogenated noscapine analogs that perturb mitosis in cancer cells followed by cell death. Biochem Pharmacol 2006; 72: 415–426.
Aneja R, Lopus M, Zhou J, Vangapandu SN, Ghaleb A, Yao J et al. Rational design of the microtubule-targeting anti-breast cancer drug EM015. Cancer Res 2006; 66: 3782–3791.
Aneja R, Vangapandu SN, Lopus M, Chandra R, Panda D, Joshi HC . Development of a novel nitro-derivative of noscapine for the potential treatment of drug-resistant ovarian cancer and T-cell lymphoma. Mol Pharmacol 2006; 69: 1801–1809.
Aneja R, Zhou J, Vangapandu SN, Zhou B, Chandra R, Joshi HC . Drug-resistant T-lymphoid tumors undergo apoptosis selectively in response to an antimicrotubule agent, EM011. Blood 2006; 107: 2486–2492.
Aneja R, Zhou J, Zhou B, Chandra R, Joshi HC . Treatment of hormone-refractory breast cancer: apoptosis and regression of human tumors implanted in mice. Mol Cancer Ther 2006; 5: 2366–2377.
Aneja R, Liu M, Yates C, Gao J, Dong X, Zhou B et al. Multidrug resistance-associated protein-overexpressing teniposide-resistant human lymphomas undergo apoptosis by a tubulin-binding agent. Cancer Res 2008; 68: 1495–1503.
Aneja R, Asress S, Dhiman N, Awasthi A, Rida PC, Arora SK et al. Non-toxic melanoma therapy by a novel tubulin-binding agent. Int J Cancer 2010; 126: 256–265.
Aneja R, Miyagi T, Karna P, Ezell T, Shukla D, Vij Gupta M et al. A novel microtubule-modulating agent induces mitochondrially driven caspase-dependent apoptosis via mitotic checkpoint activation in human prostate cancer cells. Eur J Cancer 46: 1668–1678.
Heidemann S . Microtubules, leukemia, and cough syrup. Blood 2006; 107: 2216–2217.
Karna P, Sharp SM, Yates C, Prakash S, Aneja R . EM011 activates a survivin-dependent apoptotic program in human non-small cell lung cancer cells. Mol Cancer 2009; 8: 93.
Jordan MA, Toso RJ, Thrower D, Wilson L . Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA 1993; 90: 9552–9556.
Dhamodharan R, Jordan MA, Thrower D, Wilson L, Wadsworth P . Vinblastine suppresses dynamics of individual microtubules in living interphase cells. Mol Biol Cell 1995; 6: 1215–1229.
Honore S, Pagano A, Gauthier G, Bourgarel-Rey V, Verdier-Pinard P, Civiletti K et al. Antiangiogenic vinflunine affects EB1 localization and microtubule targeting to adhesion sites. Mol Cancer Ther 2008; 7: 2080–2089.
Tanenbaum ME, Galjart N, van Vugt MA, Medema RH . CLIP-170 facilitates the formation of kinetochore-microtubule attachments. EMBO J 2006; 25: 45–57.
Tien JF, Umbreit NT, Gestaut DR, Franck AD, Cooper J, Wordeman L et al. Cooperation of the Dam1 and Ndc80 kinetochore complexes enhances microtubule coupling and is regulated by aurora B. J Cell Biol 2010; 189: 713–723.
Brinkley BR . Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol 2001; 11: 18–21.
Sellitto C, Kuriyama R . Distribution of pericentriolar material in multipolar spindles induced by colcemid treatment in Chinese hamster ovary cells. J Cell Sci 1988; 89 (Pt 1): 57–65.
Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR . Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol 1995; 130: 105–115.
Meraldi P, Honda R, Nigg EA . Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 2002; 21: 483–492.
Bornens M . Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol 2002; 14: 25–34.
Duensing A, Liu Y, Perdreau SA, Kleylein-Sohn J, Nigg EA, Duensing S . Centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal templates. Oncogene 2007; 26: 6280–6288.
Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA . The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 2005; 7: 1140–1146.
Lukasiewicz KB, Lingle WL . Aurora A, centrosome structure, and the centrosome cycle. Environ Mol Mutagen 2009; 50: 602–619.
Gascoigne KE, Taylor SS . Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008; 14: 111–122.
Weaver BA, Silk AD, Cleveland DW . Low rates of aneuploidy promote tumorigenesis while high rates of aneuploidy cause cell death and tumor suppression. Cell Oncol 2008; 30: 453.
Jaiswal AS, Aneja R, Connors SK, Joshi HC, Multani AS, Pathak S et al. 9-bromonoscapine-induced mitotic arrest of cigarette smoke condensate-transformed breast epithelial cells. J Cell Biochem 2009; 106: 1146–1156.
Lentini L, Iovino F, Amato A, Di Leonardo A . Centrosome amplification induced by hydroxyurea leads to aneuploidy in pRB deficient human and mouse fibroblasts. Cancer Lett 2006; 238: 153–160.
Heppner GH . Tumor heterogeneity. Cancer Res 1984; 44: 2259–2265.
Acknowledgements
We thank the technical assistance of Yendi Linares. This work was supported by grants to RA from the National Cancer Institute at the National Institutes of Health (1R00CA131489) and the Department of Defense (PC073104).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by V De Laurenzi
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Karna, P., Rida, P., Pannu, V. et al. A novel microtubule-modulating noscapinoid triggers apoptosis by inducing spindle multipolarity via centrosome amplification and declustering. Cell Death Differ 18, 632–644 (2011). https://doi.org/10.1038/cdd.2010.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.133
Keywords
This article is cited by
-
YLT-11, a novel PLK4 inhibitor, inhibits human breast cancer growth via inducing maladjusted centriole duplication and mitotic defect
Cell Death & Disease (2018)
-
Paclitaxel and the dietary flavonoid fisetin: a synergistic combination that induces mitotic catastrophe and autophagic cell death in A549 non-small cell lung cancer cells
Cancer Cell International (2016)
-
A FRET biosensor reveals spatiotemporal activation and functions of aurora kinase A in living cells
Nature Communications (2016)
-
Centrosome amplification, chromosomal instability and cancer: mechanistic, clinical and therapeutic issues
Chromosome Research (2016)
-
Centrosome-declustering drugs mediate a two-pronged attack on interphase and mitosis in supercentrosomal cancer cells
Cell Death & Disease (2014)
