
Abstract
Lysosomal cathepsins have recently been reported to play crucial roles in the regulation of the mitochondrial death cascade by an unclear mechanism leading to mitochondrial membrane permeabilization. Glycosaminoglycans (GAG) are a family of ionic polysaccharides present at the lysosomal compartment and shown to inhibit lysosomal cathepsin activities. The implication of this family of polysaccharides in the regulation of the pre-mitochondrial death cascade has still not been considered. Here, we demonstrate in a model of skin fibroblasts submitted to oxidative stress that a GAG–mimetic protects the lysosome from membrane disruption, reduces intracellular ROS levels, and inhibits mitochondrial membrane potential collapse, cytochrome c release and caspases-9 and -3 activations without affecting the extrinsic pathway of apoptosis. Heparan sulfate and chondroitin sulfate, but not heparin, showed also protecting effects when assessing key points of the intrinsic pathway of apoptosis. We suggest the existence of molecular links between endogenous GAGs and the regulation of apoptosis.
Similar content being viewed by others


Main
Sulfated glycosaminoglycans (GAGs) are a family of biologically relevant polysaccharides known to regulate a number of fundamental biological processes, as cell growth, migration, adhesion, and differentiation, through their interaction with particular proteins known as heparin-binding proteins (HBP). Growing number of evidences indicates that the specificities of these interactions are related to the presence of particular and finely shaped sulfated sugar sequences fashioned during the GAGs biosynthesis and catabolism giving a great diversity and potential specificity of their structures.1
There are now increasing evidences out-lighting the existence of a biological machinery that initiates and regulates programmed cell death between lysosomes and mitochondria. A crucial step of this novel death cascade involves lysosomal membrane permeabilization (LMP) associated to a release of cathepsins to the cytosol which occurs before cytochrome c release and caspase activation.2 Lysosomal cathepsins B, D, and L have been shown to translocate from the lysosomal lumen to the cytosol in response to a variety of signals such as TNF receptor ligation,3, 4 p53 activation,5 and oxidative stress.6 Interestingly, recent studies suggest that once at the cytosol, lysosomal enzymes can induce mitochondrial dysfunction through the activation of a Bax/Bak-dependent pathway.7 The fine mechanisms associated with these processes are largely unknown and information concerning the processing and activation of lysosomal proteases remains poor.8 However, implication of GAGs in the pro-cathepsin B activation9 and in the inhibition of cathepsins enzymatic activities10 suggest a potential involvement of these complex polysaccharides in cell death processes. After their excretion to the extracellular level, in where they excerpt a number of functions, GAGs are partially processed by endo-β-glucuronidases, as hyaluronidase and heparanase, and endocyted. Lysosomal GAGs are then partially degraded by sulfatases and exo-glycosidases to produce oligosaccharides that might also interact with intracellular HBP and modify their activities or fates. We postulate that intracellular GAGs may be implicated in the control of cell death. Because of the kinetics of their lysosomal degradation, glycanase-resistant GAGs analogs might facilitate the study of a potential implication of GAGs in the programmed cell death processes.
It has been shown that a family of polysaccharides structurally and functionally related to GAGs protect heparin-binding growth factors against proteolytic degradation, enhance their bioavailability11, 12 and directly inhibit activities of plasmin, neutrophil elastase, and cathepsin G.13 In vivo, some of these compounds modified inflammation kinetics and enhanced tissue repair and regeneration in several models including periodontis, mucositis, and burns.14, 15, 16, 17 Therefore they were also named RGTA® for ReGeneraTing Agents. Here, we postulate that GAG–mimetics can participate to regulation of apoptosis in a mechanism implicating cathepsins activation and lysosomal membrane permeabilization. As oxidative stress plays an important role in tissue injury, we studied here the protective effect of a GAG–mimetic, named OTR4120, against oxidative stress-induced apoptosis. A model of acute hydrogen peroxide (H2O2) stress in primary culture of skin fibroblast was used to evaluate the effect of the compound on lysosome and mitochondrial membrane permeabilizations, in intracellular ROS production, and on caspase-9, -3 and -8 activities. We characterized stressed and non-stressed cells by assessment of aconitase activity and membrane lipid peroxidation in treated and untreated stressed cells and controls. Cytosolic cathepsin D activity was evaluated and the effect of the GAG–mimetic in the in vitro cathepsin activity was assessed. The GAG–mimetic interaction with TNF-α was evaluated to exclude the extrinsic pathway of apoptosis. Finally, the protective effect of natural GAGs including heparan sulfate (HS), chondroitin sulfate (CS), and heparin (Hep) were evaluated by cytosolic cytochrome c release and caspase activities.
Results
Cell status
Stress was induced in fibroblasts by 30 min treatment with H2O2. After removal of the oxidant, cells were treated, or not, by OTR4120 and their functional status was established by measuring membrane lipid peroxidation, aconitase and SOD activities. Lipid peroxidation was evaluated by measuring malondialdehyde (MDA) levels in cell membranes just after the stress period, and 1 and 24 h later in the presence and in the absence of the GAG–mimetic (Figure 1a). Compared to control cells (unstressed fibroblasts), MDA content was increased by 1.6-folds in cells at the end of the stress period and 3.6-folds after 24 h. OTR4120 treatment was not significantly efficient 1 h after the stress but after 24 h lipid peroxidation increase was totally inhibited. The enzymatic activity of aconitase, a ROS-sensitive mitochondrial enzyme, was reduced by 50% after 30 min of H2O2 treatment (Figure 1b). Interestingly, OTR4120 restored this activity close to control values after only 1 h of treatment. The activities of antioxidant enzymes SOD were measured in cytosolic and mitochondrial extracts. Results showed that SOD activities were increased in both fractions with a stronger effect in the cytosolic one. However, the mimetic showed only a slight effect on their activities.

(a) Peroxidation of fibroblast membrane lipids was assessed in cell membrane suspensions by measuring MDA formation (530 nm) in the presence of thiobarbituric acid (1%) and trichloroacetic acid (3%). MDA amount was quantified after 30 min of stress, and 1 and 24 h after the end of stress with and without GAG–mimetic treatment (1 μg/ml). Results are expressed as the percentage of MDA levels in non-stressed cells and represent three independent experiments in duplicate, presented as mean±S.E.M. *P<0.05; **P<0.01 and ***P<0.005 versus control; δδδP<0.005 versus stress after 24 h. (b) Aconitase activity was performed at 37°C in TRIS buffer (50 mM, pH 7.4) with cis-aconitate as substrate (0.4 mM) and at 240 nm using a spectrophotometer Jasco® V-530 (JP). Then the rate was measured during 5 min and expressed in percentage of aconitase activity in unstressed cell. Results represent 3 independent experiments in duplicate and are presented as mean±S.E.M. **P<0.01 versus control unstressed cell; δP<0.05 and δδP<0.01 versus stressed cells. (c and d) SOD activity in mitochondrial and cytosolic fractions, respectively. The reduction of nitroblue tetrazolium, by free O2·− anions produced by the xanthine and xanthine oxidase coupled system and corresponding to the opposite of SOD activity, was measured at 560 nm. Results are expressed in percentage of SOD activity in non-stressed cell and represent 3 independent experiments in duplicate presented as mean±S.E.M. ***P<0.005 versus control
Mitochondrial fragmentation and assembling
Changes in mitochondrial morphology induced by oxidative stress in fibroblasts treated or not by the GAG–mimetic were examined with the fluorescent probe MitoTracker Green FM dye. These changes, known to begin at the very early stages of the cell death cascade18 were observed at 1, 3, 6 and 24 h after H2O2 stress (Figure 2a). Disintegration of the tubular mitochondrial network and formation of punctuate staining were observed (arrows in Figure 2a). The fragmentation of mitochondria, observed from 1 h after H2O2 stimuli, was accompanied by an intracellular migration of mitochondria from the cell periphery to the nuclei. The treatment by OTR4120 after H2O2 withdrawal markedly reversed the formation of roundish mitochondria and prevented nucleus migration.


Mitochondrial fragmentations and membrane potential disruption induced by H2O2 in skin fibroblasts. (a) Mitochondrial morphology was visualized by MitoTracker Green FM staining in different groups of fibroblasts: in unstressed and untreated cells (control), treated with OTR4120 (1 μg/ml) alone, or stress with H2O2 (5 mM, 30 min), or by H2O2+OTR4120, separately. Pictures were taken using confocal scanning microscopy at different times (1, 3, 6 and 24 h) after the end of stress. (b) Representative images of mitochondrial membrane potential (Δψm) in different treatment groups. Cells were co-incubated with JC-1, which exists as a green fluorescent monomer at low membrane potential. At higher membrane potentials, JC-1 forms red-fluorescent ‘J-aggregates’ exhibiting a broad excitation and emission maximum at ∼590 nm (red). Δψm was measured at different times after H2O2 withdrawal in each group. (c) Quantification of the ratio of red-to-green fluorescence is representative of the relative mitochondrial membrane potential. Thus, the ratio of red-to-green fluorescence was calculated for each group at different times after H2O2 withdrawal with an Olympus Fluoview 5.0 software. Results are expressed as the means±S.E.M. and represent at least 100 cells in three independent experiments. (d) Western blot analysis of cytochrome c release in cytosolic extracts 1 and 24 h after the end of stress. Non-stressed cells and/or OTR4120 untreated cells (1 μg/ml) were represented by ‘−’. Cytosolic cytochrome c amount was measured by ‘Image J’ software quantification and expressed in % of cytochrome c released in cytosol related to control cells. This immunoblot represents three independent experiments and is expressed as the mean±S.E.M. δδδP<0.01 versus control unstressed cell; ***P<0.01 versus stressed cells
Mitochondrial membrane potential and cytochrome c release
Changes in the mitochondrial membrane potential (Δψm) were detected in cells by using the JC-1 probe. In stressed cells, a maximum decrease in the ratio of the intensities of J-aggregated sub-populations was observed 1 h after H2O2 withdrawal, indicating a decrease in Δψm (Figure 2b). This low Δψm was found to be stable after 24 h in stressed OTR4120 non-treated cells. Treatment with 1 μg/ml of OTR4120 gradually restored Δψm in 24 h (Figure 2c). The release of cytochrome c into the cytosol can be detected in stressed cells 1 h after the end of stress. Treatment with OTR4120 inhibited this release between 1 to 24 h. Our data also demonstrated that OTR4120 had no obvious effects on Δψm and cytochrome c release of control fibroblasts (data not shown).
Caspase-9 and -3 activations
To further verify the role of OTR4120 in apoptosis induced by H2O2 in skin fibroblasts, we measured caspase-9 and -3 activities at different times after oxidative stress induction. Caspase-9 activity reached a peak level about 6 h after H2O2 withdrawal (Figure 3a). This was followed by a caspase-3 maximum activation at about 24 h after H2O2 withdrawal (Figure 3b). In the presence of OTR4120, a significant decrease in both caspase activations was observed in the stressed cells. The recorded levels were similar to those measured on the non-stressed cells (Figure 3a and b). This was confirmed by Western Blot analysis (Figure 3a and b). This indicates that OTR4120 inhibits the downstream caspase activation characteristic of the intrinsic pathway of apoptosis.

Caspases-9 and -3 activities modification by OTR4120 treatment in fibroblasts. Cells were submitted to H2O2 (0.5 mM, 30 min) and treated or not by OTR4120 (1 μg/ml). Results are expressed in % of unstressed control group and represent three independent experiments in triplicate. Results are presented as the mean±S.E.M. **P<0.01 versus control unstressed cell. (a) Caspase-9 activation was measured at different times after the end of the stress using specific fluorescent substrate Ac-LEHD-AFC. Caspase-9 immunoblot in cytosolic extracts 6 h after the end of stress. Non-stressed cells and/or OTR4120 untreated cells (1 μg/ml) were represented by ‘−’. This immunoblot was representative of three independent experiments. (b) Caspase-3 activity was measured at different times after H2O2 withdrawal in each group using Ac-DEVD-AFC as substrate. Caspase-3 immunoblot in cytosolic extract 24 h after the end of stress. Unstress cells and/or untreated cells by OTR4120 (1 μg/ml) were represented by ‘−’. This immunoblot was representative of three independent experiments
TNF-α activity and caspase-8 activation
To exclude the possibility of an interaction of the GAG–mimetic with the extrinsic pathway of apoptosis, the effects of OTR4120 on the TNF-α apoptosis-induced pathway was measured by three separate experiments. Firstly, binding experiments between mrTNF-α and OTR4120 were performed by an ELISA-based assay with immobilized OTR4120 and the soluble protein. mrTNF-α immunolabeling showed that the GAG–mimetic was not able to bind mrTNF-α even in a low detectable extent indicating a very low or null affinity (data not shown). A positive control (FGF-2) was used to validate the presence of the mimetic on the plate surface. Secondly, the TNF-α mediated cytotoxicity assayed in the cell line L929, routinely used to assess TNF-α activity, was not modified by treatment with OTR4120 (Figure 4a). Finally and importantly, caspase-8 was not activated in the model of fibroblasts submitted to oxidative stress, treated or not with OTR4120, indicating that in this model the extrinsic pathway of apoptosis is not activated in any of the working conditions (Figure 4b).

Effect of OTR4120 on the extrinsic pathway of apoptosis. (a) Skin fibroblasts were submitted to H2O2 (0.5 mM, 30 min) and treated or not by OTR4120 (1 μg/ml). Caspase-8 activation was measured at different times after the end of the stress using the specific fluorescent substrates Ac-IETD-AFC. Results are expressed in % of the unstressed control group response and represent 3 independent experiments in triplicate. Results are presented as means±S.D. (b) L929 were used for their sensitivity to TNF-α mediated cytotoxicity. Cells were incubated with various concentrations of rmTNFα (pg/ml to ng/ml) and with different concentrations of OTR4120 (ng/ml to 1 μg/ml). After 24 h of incubation, the cell viability was measured with MTT assay. Mean percentage cytotoxicity was determined to allow normalization of data in each experiment. Error bars indicate S.E.M. Results represent three independent experiments in triplicate
ROS generation
The intracellular ROS level was tested in the H2O2 stressed cells by using the H2DCF-DA fluorescent dye soon after and 1 h after the oxidant withdrawal. Intracellular ROS level was significantly higher (P<0.001), closed to 10-fold greater, than the basal ROS level in unstressed and OTR4120-untreated cells (Figure 5a and b). One hour after the stress withdrawal, the ROS production fold into decline with time from 28% reduction at 3 h, 68% at 6 h and 78% at 24 h. By comparison, OTR4120 treatment markedly dropped this level (P<0.001), by reducing the ROS production from about 29% at 1 h, 71% at 3 h, 81% at 6 h and 86% at 24 h (Figure 5a and b). Interestingly, the intracellular ROS level in unstressed cells treated with OTR4120 was similar to the level measured in non-stressed control cells. These results indicate that the GAG–mimetic can directly or indirectly reduce intracellular ROS production induced by H2O2 treatment.

ROS generation in mouse skin fibroblast induced by H2O2. (a) Representative images of intracellular ROS fluorescence merging with transmission by laser confocal scanning system. At 1, 3, 6 and 24 h after H2O2 withdrawal, ROS generation was visualized by H2DCF-DA fluorescence dye for each different experimental group. (b) Quantification of the ROS production was achieved and analyzed with Olympus Fluoview 5.0 software. Results represent at least 100 cells in three independent experiments
Early lysosomal membrane permeabilization
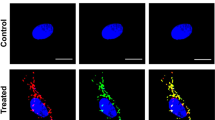
LMP was analyzed by the acridine orange (AO) relocation method using confocal microscopy. Figure 6a illustrates typical appearances and subcellular localizations of the AO-probe in control and OTR4120-treated cells. AO was exclusively found as red granules localized in the cytoplasm with a perinuclear distribution consistent with that of lysosomes, together with some faint green staining of the nucleoli and cytoplasm. In cells stressed with H2O2 for 30 min and observed every 5 min after oxidant withdrawal, a more diffuse green staining in the cytoplasm was observed, together with time-dependent red granules disappearing. When fibroblasts were treated with OTR4120 after the stress period, the red granules, monitored continuously for 15 min, slowly decreased. Simultaneously, we observed a decrease of the green fluorescence smearing into the cytoplasm compared to the stressed cells. This clearly demonstrates the protective effect of the GAG–mimetic against lysosomal membrane disruption induced by oxidative stress.

(a) Lysosome disruption induced by oxidative stress and recovered by OTR4120. Representative images of lysosomes disruption over the time (0, 5 and 15 min after treatments were shown) through laser confocal microscope. Lysosome membrane stability was visualized using the acridine orange dye for each different treatment: control cells, treated with OTR4120 (1 μg/ml) alone, stress with H2O2 (5 mM, 30 min), or H2O2+OTR4120. Results represent three independent experiments. (b) Cathepsin D activity in cytosolic extracts of normal, stress (5 mM, 30 min) and stress (5 mM, 30 min)+OTR4120 (1 μg/ml) skin fibroblasts. Results are expressed in percentage of normal cell extract levels and presented as mean±S.E.M. of three independent experiments in duplicate. *P<0.01 and **P<0.05 versus control unstressed cell; δδδP<0.001 versus stressed cells. Western blot analysis of cathepsin D release in cytosolic extract at the end of the stress and 1 h after. Non-stressed cells and/or untreated cells by OTR4120 (1 μg/ml) were represented by ‘−’. This immunoblot was representative of two independent experiments
Cathepsin D activity
Cathepsin D has recently been involved in the regulation of apoptosis in skin models associating oxidative stress. Classically, oxidative stress induced the lysosome membrane disruption and the cytosolic release of cathepsins, in particular cathepsin D, in skin fibroblasts.19 In our model, cathepsin D activity was increased in cytosolic extracts by 24% at the end of the stress and by 60% 1 h later. OTR4120 (1 μg/ml) treatment restores the cathepsin D activity level close to the control unstressed cells (Figure 6b). Western blot analysis of cytosolic extracts showed a little increase of cathepsin D at the end of the stress whereas a high level was detected after 1 h. Remarkably, OTR4120 cell treatment inhibited this response to stress (Figure 6d). As the inhibition of the lysosomal enzymes by GAGs has been previously reported,10 we tested the effects of OTR4120 on cathepsin D activity and showed that this protease was inhibited by the GAG–mimetic in a concentration–dependent manner (Figure 7c). The IC50 of the GAG–mimetic was 19.5±2.7 nM and total inhibition was obtained at 100 nM.

Cathepsin D inhibition and activity assay. Pepstatin A, a cathepsin inhibitor offered similar protection to inhibition of caspase-9 (a) and caspase-3 (b) to OTR4120. Error bars indicate S.E.M. (c) Cathepsin D activity was inhibited by OTR4120 with a concentration–dependent process. The IC50 of the GAG–mimetic was 19.5±2.7 nM and the total inhibition was obtained at 100 nM. Results represent three independent experiments in duplicate. *P<0.01 and **P<0.05 versus control unstressed cell
Effect of pepstatin A on caspases activation
To investigate if cathepsin D plays a role in our model of skin fibroblasts cell death and in the activation of caspases, the activities of caspase-9 and-3 were analyzed with and without treatment by pepstatin A, a specific cathepsin D inhibitor. As seen in Figure 7a and b, treatment of cells with pepstatin A, significantly prevented caspase-9 and -3 activations and thus offered similar protection to that afforded by OTR4120.
Protective effect of natural GAGs
The action HS, CS and Hep was considered in this model of oxidative stress-induced cell death by assessing cytochrome c release and caspase-9 and 3 activities in the same experimental conditions than the GAG–mimetic. As expected, HS and CS decrease the cytochrome c release and caspases activations whereas surprisingly no effect was observed for Hep treatment (Figure 8).

Effects of HS, CS and Hep on skin fibroblasts submitted to an oxidative stress. Cells were submitted to H2O2 (0.5 mM) during 30 min and treated or not by HS, CS or Hep at 1 μg/ml. (a) Western blot analysis of cytochrome c release in cytosolic extracts 1 h after the end of stress. Non-stressed cells and/or HS, CS or Hep untreated cells (1 μg/ml) were represented by ‘−’. This immunoblot represents 3 independent experiments. (b) Caspases-9 activity modifications by HS, CS or Hep treatments. Caspase-9 activation was measured 6 h after the end of the stress using specific fluorescent substrate Ac-LEHD-AFC. Results are expressed in percentage of unstressed control group and represent three independent experiments in triplicate. Results are presented as the mean±S.E.M. **P<0.01 versus control unstressed cells. (c) Caspase-3 activity was measured at 24 h after H2O2 withdrawal in each group using Ac-DEVD-AFC as substrate. Results are expressed in percentage of unstressed control group
Discussion
The structural complexity of GAGs and the growing evidences indicating their multifunctional roles in the regulation of complex biological processes points out their potential involvement in the regulation of another major process: apoptosis. During the last few years, the use of GAGs at high doses have clearly shown their antioxidant properties both in vitro and in vivo experimental models; they reduced hydroxyl radical generation, inhibited lipid peroxidation and improved antioxidant defences.20, 21, 22 Although their mechanisms of actions have not been well defined, some of these activities have been associated to a ROS scavenger activity observed only when GAGs were used at high concentrations (mg g/kg or mg g/ml).20, 21, 22 The ROS scavenger hypothesis is not consistent when GAGs or their mimetics are used at low doses (μg/kg or μg/ml). Here, based in the intracellular GAG fate, we consider an additional hypothesis related to an antiapoptotic effect at low GAGs concentrations. We used a cellular model of acute oxidative stress in primary culture of skin fibroblasts. This model is characterized by the induction of H2O2 stress during 30 min followed by elimination of the oxidants from the cell environment before natural GAGs or GAG–mimetic treatment. This circumvents the direct interaction of the polysaccharides with extracellular soluble molecules and especially with extracellular ROS avoiding, in this way, ROS scavenger activities in the ECM. As shown in Figure 1, H2O2 treated cells were characterized by an increase of membrane lipid peroxidation, a decrease of aconitase and an increase in SOD activities.
In this system, we investigated the antiapoptotic effect of the GAG–mimetic OTR4120, member of the RGTAs family of glycanase-resistant polysaccharides that exert, at low concentrations, remarkable wound-healing effects in injured tissue models involving oxidative stress.11, 15, 16, 17 RGTAs are characterized by their capacity to surrogate natural GAGs destroyed by glycanase activities during stress. As expected, the GAG–mimetic decreased lipid peroxidation of cell membranes and restored the activity of aconitase close to control level (Figure 1) indicating that, at low concentrations, the polysaccharide can regulate oxidative stress, and in consequence apoptosis.
Thus, the effect of OTR4120 was investigated in some key steps of the extrinsic and intrinsic pathways of apoptosis. In the extrinsic pathway, also known as ‘death receptor pathway’, apoptosis is triggered by the ligand-induced activation of death receptors at the cell surface. Fas and the TNF-α receptor transmit a signal to the cytoplasm that leads to the activation of caspase-8, which initiates a cascade of caspases activation including caspase-3 and leading to cell death. To evaluate whether the GAG–mimetic OTR4120 affects H2O2-induced cell death through this pathway, we tested whether the sulfated polysaccharide could modulate the activity of TNF-α or bind to it. The measurement of TNF-α and caspase-8 activities (Figure 4a) in stressed and non-stressed cells treated or not with the mimetic showed no differences. Accordingly, a lack of affinity between TNF-α and the mimetic was also observed. This suggests that, under the working conditions, this extrinsic pathway of apoptosis is not activated in the H2O2-stressed cells and that the GAG–mimetic by itself it not able to protect the cell by this path.
The intrinsic pathway of apoptosis, also called ‘mitochondrial pathway’, is the result of an intracellular cascade of events linked to mitochondrial membrane permeabilization (MMP). MMP induces the release of mitochondrial pro-apoptotic proteins, which in turn triggers the formation of a caspase-9 homodimer that activates caspase-3. This caspase cleaves enzymes and activates endonucleases to finally induce apoptosis. To confirm the activation of this pathway in our oxidative stress model, we examined the integrity of the mitochondrial membrane in stressed and non-stressed cells and looked into the GAG–mimetic effect. One indicator of alterations on the mitochondrial membrane is the change in its mitochondrial membrane potential (Δψm). In our model, oxidative stress induced the Δψm collapse in cells 1 h after H2O2 removal, whereas in OTR4120-treated cells, Δψm was partly restored (Figure 2b and c) as previously described for other sulfated polysaccharides in a model of T-cell apoptosis.23 Our results also demonstrated that OTR4120 exerts not obvious effects on the physiological Δψm level in non-stressed cells indicating that the compound acts on the cascade of events allowing restoration of Δψm. Western blot analysis confirmed that cytochrome c release to the cytosol is increased 1 h after stress induction, which ascertains mitochondrial dysfunction (Figure 2d). OTR4120-treated stressed cells, compared to non-treated stressed cells, showed a lower level of cytosolic cytochrome c 1 h after stress. This level is not modified between time (24 h), which suggests that cytochrome c stays in mitochondria favoring its recovery. Interestingly, a known apoptosis ATP-dependent process is the intracellular migration of mitochondria characterized by decomposition of extended mitochondrial profiles to small roundish mitochondria known as ‘thread-grain transition’.18, 24, 25 In our model, the mitochondria assembling analysis showed that localization of the round shed mitochondria was pronounced near the cell nucleus in stressed cells compared to control cells. As expected, this effect was diminished in OTR4120-treated cells (Figure 2a). Moreover, analysis of caspase activities and immunoblots on H2O2-stressed cells showed a caspase-9 activation with a peak level ∼6 h after the stress induction and a caspase-3 maximum activation ∼24 h after. Importantly, in OTR4120-treated cells, the two caspases showed similar activity levels to those of the non-stressed control cells (Figure 3a and b). It demonstrates that OTR4120 is able to interfere with apoptosis induced through mitochondrial damage. Mitochondria is proven to be the most powerful intracellular source of ROS but they are also recognized as the main target of these species.26 In the mimetic untreated cells the initial H2O2 exposure increases the intracellular ROS levels observed 1 h after the stress. OTR4120 treatment reduced these ROS levels (Figure 5a and b) possibly due to protection of mitochondria.
In the other side, it is important to point out that in our model the GAG–mimetic affects important key points of the intrinsic pathway of apoptosis in a time-dependent process: MMP collapse was observed at least 1 h after stress, the mimetic restored MMP → intracellular ROS increased also in the first hour, the mimetic reduces these levels → caspase-9 was activated at 6 h → caspase-3 was activated at 24 h, the mimetic effect was transduced by restoring caspases to control levels. This indicates that the polysaccharide can interact somewhere between or before the mitochondrial step of the intrinsic pathway of apoptosis. Previous reports have demonstrated that several GAGs are efficient inhibitors of lysosomal enzymes, as cathepsins.10 Our results showed that the GAG–mimetic may also interact with cathepsin D and inhibit its activity (Figure 7c). Cathepsins B, L and D have shown to play important roles in the regulation of apoptosis.27, 28 Although cathepsin B induced cleavage of the protein Bid leading to its pro-apoptotic activation,29 it has appeared that cathepsin D can trigger apoptosis through multiple pathways that may overlap with traditional mediators. Importantly, in skin fibroblasts submitted to oxidative stress, only cathepsin D could mediate apoptosis through the activation of Bax and this by an unknown mechanism.19, 28, 30 This resulted in the collapse of mitochondrial membrane potential independently of cathepsin B activity and Bid cleavage.2, 19 Even if there are many controversies on the involvement of cathepsin D and pro-cathepsin D in Bax activation,31, 32 our results show that treatment with Pepstatin A, a specific inhibitor of cathepsin D, protects cells from oxidative stress injuries to the same extent as the GAG–mimetic. This can directly or indirectly concern the activation of Bax and Bak proteins, which proved to have a decisive role in the disruption of mitochondrial function.33 These proteins have recently been considered as key elements of a novel death cascade involving lysosomal communication with mitochondria.34 Lysosomal cathepsins release to the cytosol was associated to LMP and Bax activation leading to MMP before Cytochrome c release.2 In our model, the AO subcellular redistribution from lysosomes to cytosol showed a clear lysosomal membrane disruption 15 min after H2O2 withdrawal from cells (Figure 6a) and was confirmed by an increase of the cytosolic cathepsin D activity (Figure 6b). This lysosomal destabilization can then be associated to the cytosol release of cathepsin D followed by the observed definite signs of mitochondrial damage including collapse of Δψm and increases in caspases-9 and -3 activities. Although it is not so simple to establish which occurs first, MMP or LMP, ROS generation following mitochondrial damage could act as a feedback to the lysosomes resulting in lysosomal disruption with further release of lysosomal enzymes.29, 35 Thus, we suggest that lysosomal membrane disruption induced by oxidative stress can be inhibited by OTR4120 treatment, preventing the release of cathepsin D to the cytosol.
The results of cell death protection by natural GAGs assessed by cytochrome c release and caspases activities indicate that endogenous GAGs are effectively able to regulate apoptosis but to a lower extent than their mimetic. This could be explained by mimetic resistance to glycanases degradation. Importantly, the non-effect observed by Hep treatment, although surprising, agrees with the existence of particular GAG sequences bearing sulfate groups in optimal extents and/or positions for specific interaction with HBP.1, 36 This suggests the existence of a sulfation code for the regulation of apoptosis.
Thus, intracellular or lysosomal GAGs, their metabolites, or their mimetics, may interact with lysosomal enzymes and modify their fates. This supports the hypothesis pointing out lysosomes as targets for regulation of apoptosis7 and suggests that GAGs can be added to the molecular elements of the apoptosis cascade. It is important to remark that GAGs regulate other cellular processes as proliferation and differentiation, which involve signaling events that could also be assessed in this model, but later on. Moreover, the observed cascade of early antiapoptotic effects suggests rapid interactions of GAGs with direct mediators of cell death, as cathepsins. This process can then be pulled out from other time consuming events.
Taken together, our data suggest that lysosomal disruption and cathepsin D release may be key players of the H2O2-induced apoptosis in fibroblasts. As GAGs and GAGs metabolites are common residents in lysosome, they can readily be implicated in the regulation of the apoptosis mediated between lysosomes and mitochondria. Discriminating between intracellular and extracellular actions of GAGs or their mimetics will eventually be facilitated by further studies. Progress in this area will open a promising new therapeutic strategy in different pathologies where oxidative stress is implicated including lysosomal storage, skin and neurodegenerative disorders.
Materials and Methods
Chemicals and reagents
The GAG–mimetic OTR4120, or RGTA, used in this study was obtained from OTR3 (Paris, France). The structure characterization of this water-soluble dextran derivative has been described elsewhere.37 Briefly, the product is composed of about 200 glucosidic units bounded by β1-6 linkages. This compound contains carboxylic and sulfate substitutions in contents comparable to those found in heparin: the sulfate degree of substitution was 1.1 and that of carboxylate (dsC) was 0.5. Dulbecco's Minimal Essential medium (DME), RPMI 1640, fetal bovine serum (FBS), penicillin/streptomycin, trypsin-EDTA solution and phosphate buffered saline (PBS) were from GibcoBRL (France). 3-(4,5-Dimethyl-2-thiazolyl)-3,5-diphenyl-2H-tetrazolium bromide (MTT), bovine serum albumin (BSA), H2O2, cis- aconitate, CS, HS and Hep were from Sigma (France). 2′,7′-Dichlorofluorescein diacetate (H2DCFH-DA) was from Biotium (USA). 5,5′,6,6′-Tetrachloro-1,1′, 3,3′-tetraethyl-imidacarbocyanine iodide (JC-1) was from Fluka. Mitotracker Green FM was from Molecular Probes. Recombinant murine TNFα/TNFSF1A and anti-mouse mouse TNFα antibody were purchased from R&D Systems Europe (Abingdon, UK). Donkey anti-goat IgG HRP conjugate, caspase-3, -8, -9, cytochrome c, cathepsin D antibodies were from Santa Cruz. Caspase substrates Ac-DEVD-AFC, Ac-IETD-AFC and Ac-LEHD-AFC were purchased from Calbiochem.
Cell culture
Fibroblasts were derived from back skin biopsis of healthy young Wistar rats (3 months old). Biopsies were cut tangentially to the epidermis to separate superficial and deep fibroblasts in the dermis. Cells were cultured in medium DME containing 4500 mg/l Glucose, 20% FBS and 100 IU/ml penicillin/streptomycin, at 37°C in humidified 95% air with 5% CO2. For experiments involving caspase activity determination, cells were plated in six-well culture plates (TPP, Switzerland); for confocal imaging, cells were seeded in 35 mm glass bottom culture dishes (MatTek Corp, USA). Cells were used within fifth passage. The L929 cell line was purchased from ATCC, and cultivated in RPMI-1640 supplemented with 10% FBS and 100 IU/ml penicillin/streptomycin at 37°C in humidified 95% air with 5% CO2.
Induction of oxidative stress in fibroblasts
Skin fibroblasts were plated into six-well culture plates (TPP, Switzerland) at a density of 0.5 × 105 cells per well. After 24 h incubation, oxidative stress was induced in the cells by treatment with 0.5 mM H2O2 incubated at 37°C for 30 min. Then the medium was discarded and replaced by 2 ml of the same fresh medium supplemented or not with the GAG–mimetic. 1, 3, 6 and 24 h later, cells were subjected to morphological and biochemical evaluation.
Confocal microscope imaging
To study the inhibition of oxidative stress at the single cell level, a confocal laser-scanning system (Olympus, FV500, Tokyo, Japan) microscope was used. The confocal unit was interfaced with an Olympus IX-81 inverted fluorescence microscope. For the study of various intracellular components and functions the following vital fluorescent probes were used: H2DCFH-DA 5 μM, for the generation of ROS, MitoTracker Green FM 100 nM, a mitochondria-selective stain in living cell, operating at a λexc of 488 nm and at λem of 520 nm; JC-1, 5 μM, for Δψm; AO, for assessment of lysosomal membrane integrity, scanning at a λexc of 488 nm and at λem of 520 nm and 615 nm. The optimum time loading for each of the probes was determined experimentally. Images were recorded with a × 60 oil immersion objective using a zoom factor of 2.0 and sequential scanning with the 488 nm spectral line of an argon-ion laser, and/or the 543 nm line from a green helium-neon laser, and/or the 633 nm line of a red helium-neon laser. All images were acquired and analyzed with an Olympus Fluoview 5.0 software.
Cell status
Aconitase activity was assayed by ultraviolet absorbance as described by Drapier.38 Lipid peroxidation was assessed as the generation of thiobarbituric acid-reactive substances, that is, lipid peroxides, according to Zini et al.39 The xanthine and xanthine oxidase couple system is used as superoxide generator and nitroblue tetrazolium as a probe, a common assay for SOD activity. The reduction of nitroblue tetrazolium, reflecting the level of free O2•− anions, is measured spectrophotometrically at 560 nm.
Caspase-3, -8, -9 activations
The caspase assay is based on the ability of the active enzymes to cleave the fluorogenic substrates Ac-DEVD-AFC (caspase-3), Ac-IETD-AFC (caspase-8), or Ac-LEHD-AFC (caspase-9). Cells treated under various conditions were harvested through trypsinization and washed with PBS. The cell pellet was gently suspended in buffer containing HEPES 30 mM, EDTA 0.3 mM, NaCl 100 mM, Triton X-100 0.15% and DTT 10 mM and centrifuged; the supernatant was used for the assay. Caspase substrates were added to a final concentration of 100 μM. The plate was covered, mixed gently on a Titramax 100 plate shaker and incubated at 37°C for 2 h. Then incubated samples were measured at λex of 400 nm and λem of 505 nm in a fluorescent micro plate reader (Labsystems Fluoroskan II). For Western blotting experiments with caspases, cytochrome c, cytosolic and mitochondrial extracts were prepared by washing cells with PBS, then swelling buffer (10 mM HEPES, pH 7.0, 5 mM MgCl2, 0.67 mM dithiothreitol, complete protease inhibitors (Roche Applied Science, Indianapolis, IN, USA). Cells were swollen in an equal volume of swelling buffer for 15 min on ice. Cells were disrupted by passing through a 22-G needle 10 times, until 80% of nuclei-stained positive with trypan blue. The suspension was centrifuged two times at 15 000 × g for 30 min, 4°C. The supernatant was centrifuged at 100 000 × g, 1 h, 4°C to obtain the S100 fraction used for immunoblot experiments.
Binding of TNFα to OTR4120-BSA
OTR4120 was covalently bound to BSA in a potassium phosphate buffer (0.2 M, pH 8) medium as previously described.40 The protein concentration was determined by the kit BCA protein assay (Pierce). The surface of 96 flat-well were coated overnight at 4°C with 100 μl of OTR4120-BSA (20 μg/ml of equivalent BSA), prepared complexes in 50 mM Tris-HCl, pH 7.4, supplemented with 12.5 mM EDTA. After three washings with PBS containing 0.05% Tween 20 (washing buffer), the non-specific interactions were saturated with PBS containing 3% BSA for 1 h at room temperature. The plates were washed again and the various concentrations of rmTNFα were added at 37°C for 2 h, after washing 10 μg/ml goat anti-mouse TNF-α were added at 37°C for 1.5 h. After a new wash, horseradish peroxidase (HRP)-conjugated rabbit anti-goat IgG (1 : 10 000 in PBS) was added to each well and incubated for 1 h at room temperature. HRP label was visualized using tetramethylbenzidine substrate kit (TMB kit, Pierce). The reaction was terminated with 1 M H2SO4 and the absorbance measured at 450 nm. All experiments were carried out in duplicate and were repeated three times.
TNFα activity assay by L929
L929 were used for their sensitivity to TNF-α mediated cytotoxicity by using a modified method of Flick and Gifford.41 Briefly, 96 flat-well plates were seeded with 3.5 × 104 cells per well. Actinomycin D (50 μl of 4 μg/ml) was introduced and after 12 h incubation, 50 μl of various concentrations of rmTNFα and OTR4120 were added. The plates were incubated for 24 h. For the cell viability measurement, MTT (1 g/l) was added into each well and incubated for 4 h at 37°C. Thereafter, the MTT-solution was removed and the formed intracellular dye solubilized by DMSO. The absorbance of the solution was measured at 570 nm using a micro-ELISA plate reader. Mean percentage cytotoxicity was determined to allow normalization of data in each experiment.
Cathepsin D activity
The assay is based on the hydrolysis of a fluorimetric substrate, MCA-Gly-Lys-Pro-lle-Leu-Phe-Phe-Arg-Leu-Lys(DNP)-D-Arg-NH2 trifluoroacetate salt, by the enzyme. After cathepsin D cleavage of the peptide between the two phenylalanine residues, the quenching is relieved and the released MCA-peptide fluoresces at λem 393 nm after excitation at λex 328 nm. The enzyme was pre-incubated 10 min at 37°C with various concentration of OTR4120 before the specific substrate addition (20 μM). Then, cathepsin D activity was measured after 20 min of incubation at 37°C. Analyses performed in the presence of 0.3 mM pepstatin A were used to estimate contributions of cathepsin D to the monitored reaction. The activity of cathepsin D was measured in cytosolic extracts obtained by incubating cells 5 min at 4°C in a buffer (250 mM sucrose, 70 mM KCl, 137 mM NaCl, 4.3 mM NaH2PO4, 1.4 mM KH2PO4, pH 7.2) containing 100 μg/ml of digitonine. After a centrifugation of 10 min at 12 000 g, supernatant corresponding to the cytosolic extract was assayed by acid phosphatase test to verify the integrity of lysosomes. Then, cathepsin D activity was measured as previously described, or the presence of this protease detected by immunoblots.
Data analysis
Group data are expressed as mean±S.E.M. Comparisons among groups were made by ANOVA (F-test), Bonferroni adjusted t-tests were used for multiple group comparisons, and unpaired t-test was used for single comparisons. A two-tailed P<0.05 was selected to indicate a statistically significant difference. All values were calculated using GraphPad Prism 5.00 (GraphPad Software, San Diego, CA).
Abbreviations
- GAGs:
-
glycosaminoglycans
- HS:
-
heparan sulfate
- CS:
-
chondroitin sulfate
- HEP:
-
heparin
- ROS:
-
reactive oxygen species
- H2O2:
-
hydrogen peroxide
- Δψm:
-
mitochondrial membrane potential
- RGTA:
-
regenerating agent
- HBP:
-
heparin-binding proteins
- MDA:
-
malondialdehyde
- AO:
-
acridine orange
References
Kreuger J, Spillmann D, Li JP, Lindahl U . Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol 2006; 174: 323–327.
Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem 2003; 278: 31401–31411.
Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M et al. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol 2001; 153: 999–1010.
Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C et al. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest 2000; 106: 1127–1137.
Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L et al. Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci USA 2002; 99: 6286–6291.
Nilsson E, Ghassemifar R, Brunk UT . Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem J 1997; 29: 857–865.
Guicciardi ME, Leist M, Gores GJ . Lysosomes in cell death. Oncogene 2004; 23: 2881–2890.
Ishidoh K, Kominami E . Processing and activation of lysosomal proteinases. Biol Chem 2002; 383: 1827–1831.
Caglic D, Pungercar JR, Pejler G, Turk V, Turk B . Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J Biol Chem 2007; 282: 33076–33085.
Avila JL, Convit J . Inhibition of leucocytic lysosomal enzymes by glycosaminoglycans in vitro. Biochem J 1975; 152: 57–64.
Escartin Q, Lallam-Laroye C, Baroukh B, Morvan FO, Caruelle JP, Godeau G et al. A new approach to treat tissue destruction in periodontitis with chemically modified dextran polymers. FASEB J 2003; 17: 644–651.
Tardieu M, Gamby C, Avramoglou T, Jozefonvicz J, Barritault D . Derivatized dextrans mimic heparin as stabilizers, potentiators, and protectors of acidic or basic FGF. J Cell Physiol 1992; 150: 194–203.
Ledoux D, Papy-Garcia D, Escartin Q, Sagot MA, Cao Y, Barritault D et al. Human plasmin enzymatic activity is inhibited by chemically modified dextrans. J Biol Chem 2000; 275: 29383–29390.
Alexakis C, Mestries P, Garcia S, Petit E, Barbier V, Papy-Garcia D et al. Structurally different RGTAs modulate collagen-type expression by cultured aortic smooth muscle cells via different pathways involving fibroblast growth factor-2 or transforming growth factor-beta1. FASEB J 2004; 18: 1147–1149.
Garcia-Filipe S, Barbier-Chassefiere V, Alexakis C, Huet E, Ledoux D, Kerros ME et al. RGTA OTR4120, a heparan sulfate mimetic, is a possible long-term active agent to heal burned skin. J Biomed Mater Res A 2007; 80: 75–84.
Lallam-Laroye C, Escartin Q, Zlowodzki AS, Barritault D, Caruelle JP, Baroukh B et al. Periodontitis destructions are restored by synthetic glycosaminoglycan mimetic. J Biomed Mater Res A 2006; 79: 675–683.
Morvan FO, Baroukh B, Ledoux D, Caruelle JP, Barritault D, Godeau G et al. An engineered biopolymer prevents mucositis induced by 5-fluorouracil in hamsters. Am J Pathol 2004; 164: 739–746.
Youle RJ, Karbowski M . Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 2005; 6: 657–663.
Johansson AC, Steen H, Ollinger K, Roberg K . Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ 2003; 10: 1253–1259.
Campo GM, Avenoso A, Campo S, Ferlazzo AM, Micali C, Zanghi L et al. Hyaluronic acid and chondroitin-4-sulphate treatment reduces damage in carbon tetrachloride-induced acute rat liver injury. Life Sci 2004; 74: 1289–1305.
Campo GM, Avenoso A, Campo S, Ferlazzo AM, Calatroni A . Administration of hyaluronic acid and chondroitin-4-sulfate limits endogenous antioxidant depletion and reduces cell damage in experimental acute pancreatitis. Pancreas 2004; 28: E45–E53.
Campo GM, D'Ascola A, Avenoso A, Campo S, Ferlazzo AM, Micali C et al. Glycosaminoglycans reduce oxidative damage induced by copper (Cu+2), iron Fe+2) and hydrogen peroxide (H2O2) in human fibroblast cultures. Glycoconj J 2004; 20: 133–141.
Miao B, Li J, Fu X, Gan L, Xin X, Geng M . Sulfated polymannuroguluronate, a novel anti-AIDS drug candidate, inhibits T cell apoptosis by combating oxidative damage of mitochondria. Mol Pharmacol 2005; 68: 1716–1727.
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 2001; 1: 515–525.
Skulachev VP, Bakeeva LE, Chernyak BV, Domnina LV, Minin AA, Pletjushkina OY et al. Thread-grain transition of mitochondrial reticulum as a step of mitoptosis and apoptosis. Mol Cell Biochem 2004; 256–257: 341–358.
Morin C, Simon N . Mitochondria: a target for neuroprotective interventions in cerebral ischemia-reperfusion. Curr Pharm Des 2006; 12: 739–757.
Isahara K, Ohsawa Y, Kanamori S, Shibata M, Waguri S, Sato N et al. Regulation of a novel pathway for cell death by lysosomal aspartic and cysteine proteinases. Neuroscience 1999; 91: 233–249.
Ollinger K . Inhibition of cathepsin D prevents free-radical-induced apoptosis in rat cardiomyocytes. Arch Biochem Biophys 2000; 373: 346–351.
Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM et al. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 2004; 279: 3578–3587.
Kagedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS et al. Lysosomal membrane permeabilization during apoptosis – involvement of Bax? Int J Exp Pathol 2005; 86: 309–321.
Schestkowa O, Geisel D, Jacob R, Hasilik A . The catalytically inactive precursor of cathepsin D induces apoptosis in human fibroblasts and HeLa cells. J Cell Biochem 2007; 101: 1558–1566.
Green DR, Amarante-Mendes GP . The point of no return: mitochondria, caspases, and the commitment to cell death. Results Probl Cell Differ 1998; 24: 45–61.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Kroemer G, Jaattela M . Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5: 886–897.
Zhao M, Antunes F, Eaton JW, Brunk UT . Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem 2003; 270: 3778–3786.
Ouidja MO, Petit E, Kerros ME, Ikeda Y, Morin C, Carpentier G et al. Structure-activity studies of heparan mimetic polyanions for anti-prion therapies. Biochem Biophys Res Commun 2007; 363: 95–100.
Papy-Garcia D, Barbier-Chassefiere V, Rouet V, Kerros M, Klochendler C, Tournaire M et al. nondegradative sulfation of polysaccharides. Synthesis and structure characterization of biologically active heparan sulfate mimetics. Macromolecules 2005; 38: 4647–4654.
Drapier JC, Hibbs Jr JB . Aconitases: a class of metalloproteins highly sensitive to nitric oxide synthesis. Methods Enzymol 1996; 269: 26–36.
Zini R, Morin C, Bertelli A, Bertelli AA, Tillement JP . Effects of resveratrol on the rat brain respiratory chain. Drugs Exp Clin Res 1999; 25: 87–97.
Barbosa I, Morin C, Garcia S, Duchesnay A, Oudghir M, Jenniskens G et al. A synthetic glycosaminoglycan mimetic (RGTA) modifies natural glycosaminoglycan species during myogenesis. J Cell Sci 2005; 118 (Part 1): 253–264.
Denizot F, Lang R . Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986; 89: 271–277.
Acknowledgements
We thank Dr. Pierre Rustin (INSERM U676, Robert Debré, Paris France) for his helpful discussion.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by L Scorrano
Rights and permissions
About this article
Cite this article
Yue, XL., Lehri, S., Li, P. et al. Insights on a new path of pre-mitochondrial apoptosis regulation by a glycosaminoglycan mimetic. Cell Death Differ 16, 770–781 (2009). https://doi.org/10.1038/cdd.2009.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.9
Keywords
This article is cited by
-
Repair Effects of Astragalus Polysaccharides with Different Molecular Weights on Oxidatively Damaged HK-2 Cells
Scientific Reports (2019)
-
RGTA® or ReGeneraTing Agents mimic heparan sulfate in regenerative medicine: from concept to curing patients
Glycoconjugate Journal (2017)
-
In vitro and in vivo evaluation of cord blood hematopoietic stem and progenitor cells amplified with glycosaminoglycan mimetic
Stem Cell Research & Therapy (2016)
