
Abstract
Autophagy is a cell biological process, enabling cells to autodigest their own cytosol when starved, remove cytoplasmic protein aggregates too large for proteasomal degradation, eliminate aberrant or over-proliferated organelles, and sanitize the cytoplasm by killing intracellular microbes. The role of autophagy has been expanded in recent years to include diverse immunological effector and regulatory functions. In this review, we summarize the multiple immunological roles of autophagy uncovered to date and focus primarily on details of induction of autophagy by pattern recognition receptors, as a newly established Toll-like receptor output. Taken together with other links between autophagy and innate and adaptive immunity processes, this cell-autonomous antimicrobial defense may be evolutionarily positioned at the root of immunity with the multiple innate and adaptive immunity connections uncovered to date reflecting a co-evolution of this ancient cell-defense mechanism and more advanced immunological systems in metazoans.
Similar content being viewed by others

Main
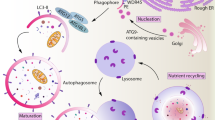
Autophagy is an evolutionarily ancient, highly conserved cytoplasmic homeostasis process, operational in all eukaryotes.1 In one of its classical presentation, autophagy is a regulator of biomass quantity, quality, and distribution, targeting degradation of many cytoplasmic constituents. Autophagosomes can capture and digest protein aggregates exceeding proteasomal substrate size cut-off, portions of the cytosol, and a variety of cytoplasmic organelles. Autophagy is heralded by the formation within the cytosol of crescent-shaped nascent autophagic isolation membranes (phagophores) that corral cytoplasmic targets into double membrane autophagosomes, which are relatively short-lived and undergo flux by maturation into autolysosomes (Figure 1a). Autolysosomes lose their inner membrane and the captured cargo is exposed to the degradative lysosomal enzymes.1

Autophagy – morphological stages and regulation. (a) Initiation: formation of phagophore crescents poised to capture cytoplasmic targets (cytosol, organelles such as mitochondria, pathogens). Elongation: phagophore enlarges assisted by two systems (i) Atg5–Atg12/Atg16 protein complex and (ii) LC3-II, which is a lipidated protein (Atg8) with phosphatidylethanolamine added to its C-terminus. It warps around its cytoplasmic target and closes to form double membrane autophagosome. Maturation: Autophagosome received lysosomal components by vesicular trafficking or fusion with lysosomes (Lys) and converts into an acidified, hydrolytic organelle termed autolysosome. Growth factor and nutritional signals through Tor and Atg1 control the downstream Atg factors. (b) Autophagy is additionally controlled by the Class III PI3 kinase, hVPS34 in a protein complex with the key autophagy regulator Beclin 1 (Atg6). When Beclin 1 is associated through its BH3-like domain with Bcl-2 family members, autophagy is inhibited. When Bcl-2–Beclin 1 complex is disrupted, this is compatible with autophagy activation. Different factors (above the arrow) can effect dissociation of Bcl-2–Beclin 1 complexes, and assist or lead to autophagy activation
Autophagy participates in a broad spectrum of biological processes including aging, development, degenerative diseases, and cancer.1, 2 Another major function of autophagy is in immunity, an area of major growth in autophagy research.3, 4 We now know that autophagy can eliminate some intracellular microbes,5, 6, 7, 8 contribute to MHC II-restricted endogenous antigen presentation,9, 10, 11, 12 act as an effector of Th1/Th2 polarization,13 control B- and T-cell development and homeostasis,14, 15, 16, 17 shape central tolerance,18 and contribute to chronic inflammatory diseases.19, 20 Several of the above immunological functions of autophagy have been reviewed recently.3, 4, 21 In this review, we will focus on the relationship between autophagy and pattern recognition receptors (PRR) with specific emphasis on Toll-like receptors (TLR) (Figure 2). Why is this topic of particular importance? Firstly, the relationship between TLRs and autophagy, which is both regulatory and effector in nature, clearly links two evolutionarily old innate immunity mechanisms – signaling through PRRs and autophagy. Secondly, the PRR–autophagy and autophagy–PRR connections elevate autophagy from a seemingly isolated, standalone cell-autonomous defense mechanism to a fully integrated immunity process. In this review, we will describe and discuss the reported experimental findings regarding PRRs and autophagy and their broader implications.

Pattern recognition receptor (PRR) agonists, signaling modules, and immunological outputs. Red, input microbial products (PAMPs) acting as PRR agonists: DAP, diaminopimelic acid, LipoProt, lipoprotein; MDP, muramyl dipeptide, PG, peptidoglycan, β-gluc, β-glucan; ss, single stranded; ds, double stranded. PRRs (blue): CLR, C-type lectin receptors; NLR, NOD-like receptors; TLR, toll-like receptors; Cytosolic, endosomal or plasma membrane (PM) localization is indicated; RLR, RIG-I-like receptors; CARD, caspase recruitment domain; PYR, pyrin domain; BIR, baculovirus inhibitor repeat domain. Green, adapters interacting with PRRs: ASC, apoptosis-associated speck-like protein containing a CARD. Gray (boxed, center) signaling pathways engaged by PRR and downstream adapters. Gray (bottom), proinflammatory cytokines output. Autophagy is shown as a new, earlier unappreciated output of PRR signaling that can (i) be directly microbicidal, (ii) fuel further PRR activation, or (iii) participate in adaptive immunity processes. A colour version of this figure is available online
Autophagy Pathway and Its Regulation
In its simplest rendition, autophagy is a starvation survival mechanism endowing cells with the ability to autodigest, and thus sacrifice portions of their cytoplasm for reuse in support of vital functions.1, 22 This is evident when cells are starved for amino acids or when growth factors are withdrawn, with a related signaling cascade feeding into the control of the Ser/Thr kinase Tor, which is the main growth rheostat controlling biomass increase or decrease (Figure 1a). When Tor is active, cells add biomass; when Tor is inactive (e.g. when cells are starved), autophagy is induced through the factor termed Atg1 (Figure 1a). In a sequence of events downstream of Atg1, two protein–protein (Atg5–Atg12, noncovalently complexed with Atg16) and protein–lipid conjugation ((light chain 3) LC3-II, which is C-terminally lipidated with phosphatidylethanolamine) systems are induced to help form phagophore crescents that elongate and close to form double membrane autophagosomes. These receive lysosomal contents and eventually mature into hybrid organelles termed autolysosomes in which degradation of the captured material takes place (Figure 1a).
Another level of regulation of the autophagy induction and execution rests on a protein complex between Beclin 1 (Atg6) and the evolutionarily ancient phosphatidylinositol 3-kinase hVPS34 (Figure 1b). Both Beclin 1 and hVPS34 are positive regulators of autophagy. A key regulatory mechanism is that Beclin 1 is complexed through its BH3-like domain23 with Bcl-2 family proteins.24 Interaction of Beclin 1 with the Bcl-2 family proteins (Bcl, Bclxl, Bcl-w) is subject to regulation by Jun-N-terminal kinase 1 (JNK-1),25 death-associated protein kinase (DAPK),26 myeloid differentiation primary response gene (88) (MyD88),27 alternate reading frame of the INK4a/ARF locus ARF tumor suppressor (human p14ARF; murine p19ARF),28 and certain BH3 proteins such as Bnip3.29 JNK-1 phosphorylates Bcl-2 and dissociates it from the Beclin 1-hVPS34 complex, thus activating it and promoting autophagy25 (Figure 1b). DAPK phosphorylates Beclin 1, also leading to dissociation of the Bcl-2–Beclin 1 complex.26 ARF and MyD88 affect the amount of Bcl-2 in relevant complexes with Beclin 127, 28 (Figure 1b). Thus, Bcl-2–Beclin 1 interaction is a key brake in the autophagy system, and is regulated by kinases, tumor suppressors, and, perhaps somewhat unexpectedly, by the key adapter proteins in innate imunity signaling downstream of TLRs, MyD88 and possibly another TLR adapter, TRIF.27 Given that MyD88 seems to act in this manner, it is perhaps not surprising that TLRs can induce autophagy at least under some conditions.
PRR and TLR Signaling in Conventional Innate Immunity Processes
PRR responses, downstream signaling, and effector outputs are collectively needed for early and effective clearance of pathogens. Ideally, they are tightly controlled to avoid excessive response that could be deleterious to the host. In most cases, several PRRs can recognize a panel of products released from a given pathogen and activate shared signaling pathways (Figure 2). Ultimately, an interplay between several signaling pathways in the same cell or in multiple cell types activated through cognate pathways, determines the specific immune response directed at clearing the pathogen and resolving infection.30 A recent series of studies has added autophagy to the list of known effector outputs of PRR signaling.27, 31, 32, 33, 34
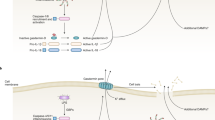
Four major classes of cell-associated PRRs have been characterized in metazoans: (i) TLR; (ii) retinoic acid-inducible gene I (RIG-I)-like receptors (RLR); (iii) nucleotide-binding and oligomerization domain (NOD)-like receptors (NLR);35 and (iv) C-type lectin receptors (CLR) (Figure 2). This review is limited to these sensu stricto PRR categories, and does not address the non-conventional PRRs, which include scavenger receptors, integrins, complement receptors, interferon-inducible proteins, GPI-anchored proteins, collectins, pentraxins, and lipid transferases classified as PRRs,30 simply because at present there is no information whether these affect autophagy. TLRs are the best-characterized receptors among the PRR. All known TLRs in mammals are type I integral membrane glycoproteins containing an extracellular domain with leucine-rich repeats responsible for ligand recognition and a cytoplasmic Toll/Interleukin-I receptor homology (TIR) domain required for initiating signaling.30 Working as homo- or heterodimers, they recognize diverse microbial components in bacteria, fungi, parasites, and viruses.30 TLR1–9 are conserved between the humans and the mouse, TLR10 is expressed in humans, but not in the mouse, whereas TLR11 is present in the mouse, but not in humans. TLRs 1, 2, 4, 5, and 6 are located mainly on the cell surface (Figures 1 and 2), and primarily recognize bacterial components. TLRs 3, 7, 8, and 9 are mostly in the endocytic compartments and mainly recognize viral products.30 TLR1 and TLR2 heterodimerize with the dimer sensing bacterial triacylated lipopeptides (represented frequently in experiments by Pam3CSK4). TLR2 can also heterodimerize with TLR6 to recognize bacterial diacylated lipopeptides (represented by Pam2CSK4). TLR4 and TLR9 homodimerize, and sense the gram negative bacterial lipopolysaccharide (LPS) and unmethylated CpG-containing DNA motifs (CpG), respectively. TLR3 and TLR5 are presumed to be homodimers, and sense double-stranded RNA (dsRNA) and bacterial flagellin, respectively. TLR7 and TLR8 are believed to form homodimers that can sense guanosine- or uridine-rich single-stranded RNA (ssRNA) and synthetic imidazoquinoline compounds (imiquimod or R837, resiquimod or R848).36, 37 TLRs alone27, 31, 32, 33, 34 and other PRRs alone38 can activate autophagy (Figure 2). Furthermore, TLRs can cooperate with other PRRs, for example, TLR2 may act in combination with CLRs, for example, Dectin 1 (Figure 2) that reacts to fungal cell wall product β-glucan to orchestrate responses (including autophagy) to complex stimuli.31, 34
After recognition of the pathogen-derived components, individual TLRs trigger distinctive responses (Figure 2) by recruiting different combinations of four TIR domain-containing adapter molecules that act as activators of downstream signaling (Figure 3): MyD88, used by all TLRs except TLR3; MyD88 adapter-like (MAL) also known as TIR domain-containing adapter protein, employed by TLR2 and TLR4 as a bridge to recruit MyD88; TIR domain-containing adapter-inducing interferon-β (TRIF) also known as TIR domain-containing adapter molecule 1 (TICAM-1), employed by TLR3 and TLR4; and TRIF-related adapter molecule (TRAM) or TICAM-2, used only by TLR4 to bridge interactions with TRIF.30, 37, 39 The fifth member of this family of adapters, Sterile α- and HEAT-armadillo-motif-containing protein (SARM), acts as a negative regulator by interacting with TRIF to inhibit its signaling pathways.37 Some TLRs signal exclusively in a MyD88-dependent manner (TLR1, 2, 5, 6, 7, 8, and 9), TLR3 signals exclusively in a MyD88-independent manner by using TRIF only. TLR4 signals through both MyD88 (assisted by MAL) and TRIF (assisted by TRAM) adapters. These signaling pathways activate the transcription nuclear factor kappa enhancer-binding protein (NF-κB) and activator protein-1 (AP-1), which are common to all TLRs, leading to the production of inflammatory cytokines and chemokines.30 TLR 3, 4, 7, 8, and 9 also activate interferon regulatory factor 3 (IRF3) and/or IRF7, leading to the production of type I interferons (IFNs) such as IFN-α family and IFN-β in a cell-type-specific manner.30 Subsequently, the cytokines and chemokines initiate and amplify inflammatory responses by recruiting and activating appropriate cells such as monocytes, neutrophils, and natural killer cells.30 Type I IFNs can induce antiviral state in most cells.30

Signaling and regulation of PRR-induced autophagy. 1. PAMP agonists stimulate TLRs (TLR4 and TLR7/TLR8 are depicted) leading to signaling through adaptors (TRAM–TRIF or MAL–MyD88) and downstream kinases (not shown – see text). 2. One molecular mechanism linking TLR signaling and autophagy induction is the association of Beclin 1 (a key regulator of autophagy) and MyD88-containing protein complexes, affecting Bcl-2–Beclin 1 interactions: when Bcl-2 is in a complex with Beclin-1 this inhibits autophagy; when Bcl-2 dissociates from Beclin 1 (as shown to be the case downstream of TLR4 signaling), Beclin-1 (along with other Atg factors and type III PI3K hVPS34, not shown) is free to initiate autophagy. 3. Autophagy can act as a topological inversion device delivering PAMP molecules to endosomal TLRs. Note that the topological inversion occurs by sequestration of cytosolic PAMPs (e.g. from a replicating virus) into the autophagosome, in which they now are in organellar lumen, which puts them topologically on the same side of the membrane as the receptor domain of endosomal TLRs. 4. PGRP-LE, a Drosophila PRR, reacts to bacterial PAMPs and induces autophagy as an innate immunity output protecting the fly from infection in vivo. 5. Inhibitory action of PAMP through NF-κB on autophagy. Inhibition of autophagy by NF-κB has been earlier described in the context of TNF-α signaling. A balance between activating/amplifying pathways 1,2, and 3, and inhibitory signaling through pathway 5 may determine the net outcome in terms of induction or inhibition of autophagy. These relationships have not been explored, but need to be delineated. 6–8, immunological outputs of PAMP–PRR–autophagy cascade: 6. Autophagy induced by PAMPs may result in direct elimination of offending microbes. 7. Autophagy assists cytosolic antigen delivery to MHC II processing and loading compartments, akin to the delivery of cytosolic PAMPs to the lumenal domains of endosomal TLRs. It is not known whether PRR-induced autophagy assists endogenous antigen MHC II presentation, but this can be predicted from the depicted circuitry. 8. Autophagy may inhibit IL-1β activation or secretion; it is not known whether autophagy acts on inflammasome, an apparatus that processes inactive pro-IL-1β and secretes it as active IL-1β, normally activated by PAMPs or danger-associated molecular patterns (DAPMs) – body's own molecules also known as ‘alarmins’ capable of inducing inflammation and defenses. 9. When individual PAMP–PRR pairs do not activate autophagy, it is possible that a combinatorial engagement of multiple PRRs may be needed to activate autophagy (as shown for TLR2/6 plus Dectin). Green arrows and boxes, positive regulatory pathways and molecules; Red arrows and letters, negative regulatory pathways; Gray arrows, immunological outputs. A colour version of this figure is available online
In general, when a TLR is activated (except TLR3), IL-1R-associated kinase 4 (IRAK4) seems to be recruited immediately downstream of MyD88. IRAK4 recruits IRAK1, and phosphorylation of IRAK1 releases it from the complex to bind tumor necrosis-factor (TNF)-receptor-associated factor 6 (TRAF6).40 TRAF6 forms oligomers and this oligomerization activates its ubiquitin E3 ligase activity, leading to Lys 63 polyubiquitination of target proteins including TRAF6 itself.41 Ubiquitinated TRAF6 recruits transforming-growth-factor-β-activated protein kinase 1 (TAK1)-binding protein 2, and activates TAK1, which then phosphorylates and activates the inhibitor of NF-κB kinase (IKK) complex, composed of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (NEMO, also known as IKKγ).41 Active IKKβ phosphorylates inhibitor of NF-κB (IκB) proteins and this phosphorylation targets IκB for polyubiquitination and subsequent degradation of IκB by the proteasome, thereby releasing the NF-κB dimer (which consists of p50 and REL-A, or p65) to let it enter the nucleus in which it regulates pro-inflammatory genes.41 Ubiquitin-activated TAK1 also phosphorylates and activates mitogen-activated protein-kinase (MAPK) kinases (MKKs), which in turn activate the JNK, p38, and probably extracellular signal-regulated kinases ERK, leading to the activation of AP-1, thus augmenting inflammatory cytokine expression.30, 41
In the case of TLR7, TLR8, and TLR9, in addition to NF-κB and MAPK activation, MyD88 activates IRF7, which leads in a cell type-dependent manner to type I IFN production. A complex comprising MyD88, IRAK1, IRAK4, TRAF6, and IRF7 has been detected, with IRF7 being phosphorylated by IRAK1 at multiple serine clusters in the C-terminus, and this phosphorylation triggers its dimerization and nuclear translocation to induce type I IFN expression, activating preferentially IFN-α promoters.37, 39, 42 The activation of TLR3-induced pathway and TLR4-induced MyD88-independent pathway relies on TRIF as an adapter.37, 43 TRIF associates to TANK-binding kinase-1 (TBK1) through NAK-associated protein 1 and possibly TRAF3.37 TBK1 and IκB kinase-ɛ (IKKɛ) are the crucial kinases for IRF3 activation39 and subsequent induction of the IFN-β gene. After TRIF engagement, there are two pathways to activate NF-κB: N-terminus of TRIF binds TRAF6 to activate NF-κB, but not IRF3; and C-terminus of TRIF recruits receptor-interacting protein-1 (RIP1) to activate NF-κB.37 In the TLR4 signaling, the MyD88-dependent pathway leads to an early-phase activation of NF-κB and MAPKs, whereas the TRIF-dependent pathway induces a late-phase activation of NF-κB and MAPKs.43 It also seems that the TRIF- and MyD88-dependent activation downstream of TLR4 is sequential and compartment-specific, with the MAL–MyD88 pathway being engaged by TLR4 on the plasma membrane, whereas the TRAM–TRIF pathway being engaged from early endosomes.44 TRIF also mediates induction of apoptosis, and the pathway seems to involve RIP1, Fas-associated death domain, and caspase-8. This induction is likely only when the antiapoptotic NF-κB signaling pathway is inhibited.45
Autophagy as a Newly Recognized Immunological Effector of TLR Signaling: TLR4 and TLR7/8
Recently, the repertoire of the known outputs after PRR activation has been broadened to include induction of autophagy downstream of TLR stimulation (Figures 1 and 2).27, 31, 33, 34 The quintessential PRR TLR4 (Figure 3) can induce autophagy in the mouse macrophage cell line RAW264.7 on stimulation with the bacterial PAMP LPS. This has been reported by four different groups27, 31, 33, 34 showing a redistribution of microtubule-associated protein 1 LC3, one of the several mammalian paralogs of the yeast Atg8, from diffuse cytosolic to punctuate pattern. The puncta formation by GFP-LC3 is applied as one of the most reliable autophagy markers. LPS also increased the amount of the lipidated form of LC3 (LC3-II), detected by western blots, in RAW264.7 cells and in murine primary bone marrow macrophages (BMMs).31, 33 An increase in numbers of double-membrane vacuoles after LPS stimulation was observed by electron microscopy in RAW264.7 macrophages.33 The punctate distribution of LC3 was also observed by immunofluorescence in human alveolar macrophages on LPS stimulation.33 The above induction of autophagy with LPS was dependent on TLR4, TRIF, RIP1, and p38 MAPK, but reportedly independent of MyD88.33 Xu et al.,33 have furthermore reported that LPS increases class III PI3 kinase hVPS34 association with membranes and augments the expression of the murine immunity-related GTPase LRG47, both known to be involved in autophagy induction.6, 46, 47, 48 The same group, reported that LPS caused colocalization of intracellular Mycobacterium tuberculosis with autophagosomes.
Two different agonists of mouse TLR7 (Figure 3) have been shown to induce autophagy in RAW264.7 cells, and weakly in BMMs. Single stranded RNA (ssRNA) induced puncta formation, LC3-II conversion, and formation of the typical autophagosomal profiles at later stages of maturation observed by electron microscopy.31 LC3-II conversion was detectable as early as 30 min on ssRNA stimulation in the presence of Bafilomycin A (to inhibit LC3-II degradation by blocking autophagosomal maturation).31 The same PAMP, ssRNA, induced LC3-II conversion in another murine macrophage cell line, J774, and in BMMs.31 Imiquimod, a different mTLR7 ligand, induced GFP-LC3 puncta formation31, 34 and increased proteolysis of long-lived proteins in RAW264.7 macrophages.31 Both TLR7 ligands were able to induce GFP-LC3 puncta formation in murine primary macrophages.31 The autophagy induction by TLR7 ligands was Beclin 1-, TLR7-, and MyD88-dependent.31 The autophagic activation by TLR7 ligands had the same effect on intracellular mycobacteria as starvation-, rapamycin-, or LRG47 expression-induced autophagy:6, 48 stimulation of BCG infected macrophages with ssRNA or imiquimod killed between 20 and 40% of the intracellular mycobacteria.31 This killing was dependent on autophagy and on TLR7-MyD88 signaling, as siRNAs for Beclin 1, Atg5, TLR7, or MyD88 decreased the bactericidal effects.31 An induction of autophagy in human cells after TLR7/8 activation was also apparent by LC3-II conversion assays in HeLa cells infected with HIV-1, a pathogen known to present a natural ligand to hTLR7 and hTLR8. The LC3-II conversion was blocked when hTLR8 was knocked down with siRNA.31
Other TLRs and Autophagy
Delgado et al.31 screened a full panel of standard PAMP ligands for all the major TLRs using RAW264.7 cells, at standard concentrations and for a specific time, and observed induction of autophagy on poly(I:C) (TLR3 or MDA5 ligand), LPS (TLR4 ligand), ssRNA, and imiquimod (TLR7/8 ligands). Flagellin, a ligand for TLR5 did not induce autophagy in RAW264.7 cells and in murine primary BMMs, as judged by the absence of LC3-II conversion in western blots, after stimulating cells for 2 h with bacterial flagellin.31 In the comprehensive screen by Delgado et al.,31 the ligand for TLR9, CpG, was not able to induce autophagy in murine RAW264.7 macrophages: there was no increase in GFP-LC3 puncta formation (in the presence or absence of the autophagic protease inhibitors, E-64d and PepstatinA), and there was no LC3-II conversion by immunoblots (unpublished observations), there was no increased proteolysis of stable proteins, and also there was no killing of intracellular BCG. Sanjuan et al.34 reported an opposite result, showing GFP-LC3 puncta formation in RAW264.7 macrophages on 3 h of incubation with CpG. This apparently contradictory result may be explained by the fact that the CpG oligonucleotides used by the two groups were different, although both of them were type B.49 Both groups also stimulated cells with a different concentration of the respective CpG, with the commonly used 3 μM giving a negative result31 and half of that concentration giving a positive induction of autophagy.34 Independent of these studies, Atg factors are involved in signaling pathways emanating from TLR9, by an unknown mechanism, as secretion of type I IFN by plasmocytoid dendritic cells (pDCs) on stimulation with type A CpG required Atg5.32 It has also been recently published that in B cells, the internalized B-cell receptor (BCR) signaling leads to delivery of TLR9-containing endosomes to the autophagosome, presumably activating TLR9 response if CpG is present within the antigen cargo of the BCR-containing compartment.50 Regardless of the variable reports on CpG signaling in induction of autophagy, in pDCs and in B cells, autophagy at the very least helps TLR ligands to meet their cognate receptor or vice-versa.32, 50
TLR3 can be activated by dsRNA or its synthetic analog poly(I:C). Delgado et al.31, have reported that poly(I:C) was able to induce autophagy by GFP-LC3 puncta formation and increased proteolysis of long-lived proteins in murine RAW264.7 macrophages, and by LC3-II conversion in BMMs. As TLR4 activates autophagy by using the TRIF-dependent pathway, and TLR3 also activates the TRIF pathway, this supports the possibility that a TLR3 ligand induces autophagy on TLR3 activation. However, Delgado et al., did not prove that this autophagic induction was because of TLR3 activation, as poly(I:C) can also activate the RLR termed MDA5.35 Thus, it is not known whether TLR3, MDA5, or both lead to autophagy induction on stimulation with dsRNA.
Autophagy Delivers Cytosolic Ligands to Lumenal TLRs
In addition to serving as an immunological effector output of TLR stimulation, autophagy also works in TLR activation by physically bringing cytosolic PAMPs to their cognate endosomal TLRs. This was best documented in the case in which autophagy was found needed for recognition of vesicular stomatitis virus by TLR732 (Figure 3). It seems that autophagy in this role delivers cytosolic PAMPs to their cognate endosomal TLRs, so that TLR signaling and innate immune response can be initiated.32 The concept of autophagic capture of ligands in the cytosol and their delivery to immune receptors facing the lumen of endosomal compartments is in principle very similar to the process of endogenous antigen presentation through MHC II molecules.11, 12 During endogenous antigen presentation, cytosolic self or foreign (e.g. viral) proteins are captured by autophagosomes and are delivered to MHC II-processing and -loading compartments. It turns out that antigen-loading compartments for MHC II presentation continuously receive input from autophagosomes, as shown by Munz and colleagues.11 It is assumed that a similar process may occur with cytosolic PAMPs and endosomal TLR7, initially separated by a membrane fence, but brought together by autophagy within the lumen of TLR-containing organelles. This is an important flip side of relationships between TLRs and autophagy, indicating that in addition to autophagy being an effector function downstream of PRR activation, it is just as important upstream of TLR action.
Autophagy is not Always Detectable after Stimulation of Individual TLRs
In contrast to the studies documenting induction of autophagy after TLR stimulation, several groups have reported inability to detect induction of autophagy on PRR stimulation in primary cells. For example, in pDCs, direct stimulation of TLR7 does not lead to induction of autophagy.32 Dendritic cells also show high level of basal autophagy engaged in MHC II presentation, and afford very little or no induction of autophagy on stimulation with other types of immunological agonists.12 In a recent study by Saitoh et al.,19 it was reported that they could not elicit autophagy induction with TLR agonists. In keeping with this, an ostensibly lower responsiveness of BMMs has been noted.31 It is very likely that some cells either have high basal level of autophagy, as in the case of dendritic cells, or perhaps induce competing signaling pathways that may act to inhibit autophagy induction. This is not a surprise, given that there are multiple regulatory circuits controlling autophagy. Some of the candidate molecules that may be involved in inhibiting autophagy induction on exposure of cells to PRR ligands may be the known antagonists of pro-inflammatory signaling such as SARM and IRAK-M, but there are no studies at present on this topic. Most importantly, it is well known that TLRs induce NF-κB signaling (Figure 3, pathway rendered in red). NF-κB is a known potent inhibitor of autophagy.51 This positive and negative regulation of autophagy is akin to the classical case of a balance between two types of opposing signals downstream of TNF-α stimulation: NF-κB and other signals compete in determining the final outcome of cell death or survival. In keeping with this notion, the studies regarding CpG PAMP and TLR9 (discussed above) showed no induction of autophagy when higher (standard) concentrations of CpG were used31 (with a documented strong induction of NF-κB), whereas induction of autophagy was detected using lower concentrations of CpG that might have not stimulated as much NF-κB.34 Thus, negative regulation of PRR-induced autophagy may be just as important as the positive signals, and may provide a tighter control in primary cells in which it has been ostensibly observed. These issues may be of higher biological significance (i.e. when do cells undergo autophagy on stimulation with microbial ligands) than just the simple fact that PAMPs can induce autophagy, and need more study.
Combinatorial Stimulation of PRRs: Is There a Code for Activating Autophagy by PRRs?
Even in those cases, in which a single PAMP–PRR fails to induce autophagy, a more complex PAMP combinations have been found to induce autophagy.31, 34 One such case involves failure to induce autophagy by Pam3CSK4 or Pam2CSK4 (ligands for TLR1/TLR2 and TLR2/TLR6, respectively), contrasting with a strong induction on exposure to zymosan, yeast cell wall particles that engage both TLR2/TLR6 and Dectin-1.31, 34 This was shown by GFP-LC3 puncta formation using fluorescence microscopy31 and by LC3-II conversion in immunoblots.34 The GFP-LC3 translocation in response to zymosan was shown to be TLR2-dependent, but MyD88-independent, in primary macrophages.34 These observations suggest that there may be a combinatorial code in some cells or under certain conditions required to unlock the cascade-linking PRR stimulation and autophagy induction.
TLRs Can Result in Phagosomes Acquiring LC3
Sanjuan et al.,34 have reported that Pam3CSK4 coating of latex beads (used as model particles to study phagosomes) induced a rapid recruitment of LC3 to the phagosome in RAW264.7 macrophages.34 The LC3 recruitment to the phagosome of engulfed zymosan was Atg5- and Atg7-dependent.34 Zymosan also induced a rapid association of GFP-Beclin 1 with the phagosome.34 The killing of live Saccharomyces cerevisiae engulfed by macrophages was dependent on Atg7.34 These experiments suggest that phagosomal and autophagosomal pathways may intersect or that LC3-II can be recruited to phagosomal membranes. Two (or more) models are possible: (i) Sanjuan et al.34 proposed that LC3-II is directly recruited to the phagosomal membrane, thus expanding the domain of LC3 action to conventional phagosomes. In an artificial experimental set up designed to establish the principles of LC3-II targeting, Yoshimori and colleagues52 have shown that LC3-II can be coerced to appear on non-autophagic membranes bypassing Atg16, a factor that seems to earmark membranes that will become LC3-II-positive. Whether the phenomenon observed by Sanjuan et al.34 is Atg16 driven, or some other factor may target LC3-II remains to be explored. (ii) Given that the LC3-II ortholog in yeast, Atg8, is fusogenic – that is, mediates membrane tethering and hemifusion between adjacent vesicles,53 it is possible that LC3-II in small quantities on phagosomes may promote interactions between autophagosomes and phagosomes, leading to outright LC3-positive phagosomal structures. In any event, it is not surprising that phagosomes and autophagosomes have a special relationship, given that phagosome formation can be viewed as autophagy of the external space, topologically equivalent to the autophagosomes in the cytosol only capturing the external space (representing a quasi-lumen) delimited by the plasma membrane. It will be of interest to explore whether the autophagosome and the phagosome represent two extremes of a phagocytic continuum, as recently proposed.54
Autophagy, Other PRRs, and Inflammasome
There are at present no reports on Nod1, Nod2, or the members of the large NLR family in relationship to autophagy, although NLRs seem ideally positioned for detection of microbial products in the cytosol, in which autophagic capture of microbes would be the most relevant. A recent study in Drosophila38 has shown that a cytosolic PRR, PGRP-LE, recognizing diaminopimelic acid-type peptidoglycan (a PAMP for Nod in mammalian cells), induces autophagy and that autophagy protects the fly from Listeria monocytogenes infection. This study is important for another reason, as it is one of the two recent reports showing in vivo role of autophagy in animal models.38, 55 With regards to RLRs, no studies have been reported linking RLR activation and autophagy induction. However, it seems that Atg5–Atg12 complex plays a negative regulatory function in RIG-I interactions with its downstream adapter molecule IPS-1 (also known as VISA or MAVS), blocking RIG-I signaling and type I IFN production.56
Motivated by reports that autophagy plays a role in PRR responses and the reports57 that Atg16L is a risk locus in Crohn's disease (a form of inflammatory bowel disease), Saitoh et al.19 investigated the role of autophagy in inflammation with the emphasis on IL-1β, a cytokine generated by the action of inflammasome in response to various PAMPs and danger-associated signals. Saitoh et al., generated Atg16L ΔCCD mice (which die within 1 day of birth just like the Atg5−/− knockout mice), and showed that macrophages form these mice show elevated IL-1β production. Comparing mouse chimeras repopulated with Atg16L1 ΔCCD or wild-type stem cells, Saitoh et al. observed 100% mortality of the Atg16L1 ΔCCD mouse chimeras in experimentally induced colitis. This was reversible by IL-β-neutralizing antibodies. Thus, it seems that another function of autophagy in the context of pro-inflammatory signaling is to temper IL-1β secretion, and possibly inflammasome activation or availability, although the latter possibility has not been investigated.
Is there a Connection Between Innate Immunity Roles of Autophagy and Cell Survival/Cell Death Pathways?
In the above sections, we have analyzed in details the relationship between PRR and autophagy induction. However, when we introduced the topic at the beginning of this review, we saw that one of the key regulatory relationships in control of autophagy is the Bcl-2–Beclin 1 interaction (Figure 1). As Bcl-2 is best known for its antiapoptotic/anti-cell death role, it seems potentially counterintuitive that this same mechanism would fit with innate immunity defenses. In Figure 4 we propose a model (Figure 4), in which we attempt to bring together these seemingly disparate functions (i) autophagy as a process affecting cell survival or death and (ii) autophagy as an innate immunity mechanism. Autophagy is known to interact with cell death (apoptosis and necrosis) pathways and possibly when excessive may contribute to cell death,58 although the presently emerging view is that autophagy is primarily a cell survival pathway and that cell death, when it happens, does not happen ‘by autophagy’, but ‘with autophagy’, as noted by Kroemer and Levine.59 In the model shown in Figure 4, autophagy is considered a cell survival process in the context of defense against intracellular pathogens. We surmise that this was one of the very early innate defense mechanisms, and that autophagy has been used to eliminate microbes that manage to erode into the cytoplasm of eukaryotic cells. In this model, when pathogens outgrew autophagic defense, evolution led to a second stage process resulting in cell death (e.g. apoptosis), again with the net effect of limiting the spread of the pathogen within the multicellular organism or species. We propose that this set of relationships may have led to the parallels in wiring of the regulatory circuitries controlling autophagy (MyD88/TRIF–Bcl-2–Beclin 1) and apoptosis (Bcl-2–Bax/Bak) (Figure 4a). Of interest, is to point out that mitochondria neatly fit into this model (Figure 4b), as they are believed to have originated from a Rickettisia-like α-protobacterium that established a symbiotic relationship with pre-eukaryotic cells. It is thus not surprising that both apoptosis and many innate immunity-signaling transactions are associated with mitochondria.

Proposed evolutionary relationships between autophagy, apoptosis, intracellular pathogens, and mitochondria. See text section ‘Is there a connection between innate immunity roles of autophagy and cell survival/cell death pathways?’ for details.
Conclusions
Autophagy has multiple roles in immunity and inflammation. This is now clear from a number of studies: (i) aberrant autophagy shows genetic association in human populations with the chronic inflammatory syndrome known as Crohn's disease;19, 20, 57, 60, 61 (ii) autophagy is critical for central immunological tolerance (preventing multiorgan inflammation) in which it works in thymic selection;18 (iii) it plays in vivo defense against infection in a PRR-driven38 or other innate immunity systems-dependent55 manner; and (iv) a molecular signaling connection between PRR (specifically TLR) signaling and autophagy has been established.27, 31, 32, 33, 34 Many questions remain: (i) When and how positive and negative regulation of autophagy by PRR signaling leads to autophagy activation? (ii) Is there a ‘code’ for a combination of PAMP–PRR pairs that need to be engaged to activate antimicrobial autophagy? (iii) To which extent autophagy contributes to other inflammatory conditions apart form Crohn's disease? (iv) Is the PRR–autophagy axis limited only to the autophagic role in immunity? (v) Could it be linked with other conditions such as cancer (where inflammation has been implicated but mechanistically remains purely understood), neurodegeneration, and perhaps aging? and (vi) Can we glean a better understanding of cancer and innate immunity by looking at them through a unified theory, such as the one in Figure 4?
Abbreviations
- TLR:
-
toll-like receptor
- PRR:
-
pattern recognition receptors
- JNK-1:
-
Jun-N-terminal kinase 1
- DAPK:
-
death-associated protein kinase
- MyD88:
-
myeloid differentiation primary response gene
- RLR:
-
retinoic acid-inducible gene I (RIG-I)-like receptors
- NLR:
-
nucleotide-binding and oligomerization domain (NOD)-like receptors
- CLR:
-
C-type lectin receptors
References
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451: 1069–1075.
Levine B, Kroemer G . Autophagy in the pathogenesis of disease. Cell 2008; 132: 27–42.
Levine B, Deretic V . Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7: 767–777.
Lunemann JD, Munz C . Autophagy in CD4+ T-cell immunity and tolerance. Cell Death Differ 2009; 16: 79–86.
Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T et al. Autophagy defends cells against invading group A Streptococcus. Science 2004; 306: 1037–1040.
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V . Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119: 753–766.
Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C . Escape of intracellular Shigella from autophagy. Science 2005; 307: 727–731.
Deretic V . Autophagy in innate and adaptive immunity. Trends Immunol 2005; 26: 523–528.
Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005; 307: 593–596.
Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 2005; 102: 7922–7927.
Schmid D, Munz C . Innate and adaptive immunity through autophagy. Immunity 2007; 27: 11–21.
Schmid D, Pypaert M, Munz C . Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007; 26: 79–92.
Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity 2007; 27: 505–517.
Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol 2006; 177: 5163–5168.
Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW . A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204: 25–31.
Pua HH, He YW . Maintaining T lymphocyte homeostasis: another duty of autophagy. Autophagy 2007; 3: 266–267.
Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008; 4: 309–314.
Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L . Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008; 455: 396–400.
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456: 264–268.
Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456: 259–263.
Orvedahl A, Levine B . Eating the enemy within: autophagy in infectious diseases. Cell Death Differ 2009; 16: 57–69.
Kundu M, Thompson CB . Autophagy: basic principles and relevance to disease. Annu Rev Pathol 2008; 3: 427–455.
Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007; 26: 2527–2539.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122: 927–939.
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B . JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30: 678–688.
Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-X(L) and induction of autophagy. EMBO Rep 2009; 10: 285–292.
Shi CS, Kehrl JH . MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 2008; 283: 33175–33182.
Pimkina J, Humbey O, Zilfou JT, Jarnik M, Murphy ME . ARF induces autophagy by virtue of interaction with Bcl-xl. J Biol Chem 2009; 284: 2803–2810.
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6: 458–471.
Lee MS, Kim YJ . Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem 2007; 76: 447–480.
Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V . Toll-like receptors control autophagy. EMBO J 2008; 27: 1110–1121.
Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A . Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007; 315: 1398–1401.
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT . Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27: 135–144.
Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450: 1253–1257.
Takeuchi O, Akira S . MDA5/RIG-I and virus recognition. Curr Opin Immunol 2008; 20: 17–22.
Takeda K, Akira S . Toll-like receptors in innate immunity. Int Immunol 2005; 17: 1–14.
O'Neill LA, Bowie AG . The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007; 7: 353–364.
Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R et al. Autophagic control of Listeria through intracellular innate immune recognition in Drosophila. Nat Immunol 2008; 9: 908–916.
Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 2004; 5: 1061–1068.
Kobayashi K, Hernandez LD, Galán JE, Janeway Jr CA, Medzhitov R, Flavell RA . IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 2002; 110: 191–202.
Chen ZJ . Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol 2005; 7: 758–765.
Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci USA 2004; 101: 15416–15421.
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003; 301: 640–643.
Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R . TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol 2008; 9: 361–368.
Ruckdeschel K, Pfaffinger G, Haase R, Sing A, Weighardt H, Häcker G et al. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-beta, but not MyD88, in bacteria-infected murine macrophages. J Immunol 2004; 173: 3320–3328.
Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T . Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2001; 2: 330–335.
Stein MP, Feng Y, Cooper KL, Welford AM, Wandinger-Ness A . Human VPS34 and p150 are Rab7 interacting partners. Traffic 2003; 4: 754–771.
Singh SB, Davis AS, Taylor GA, Deretic V . Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006; 313: 1438–1441.
Krieg AM . Now I know my CpGs. Trends Microbiol 2001; 9: 249–252.
Chaturvedi A, Dorward D, Pierce SK . The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 2008; 28: 799–809.
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Souquère S et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 2006; 281: 30373–30382.
Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T . The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19: 2092–2100.
Nakatogawa H, Ichimura Y, Ohsumi Y . Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130: 165–178.
Deretic V . Autophagosome and phagosome. Methods Mol Biol 2008; 445: 1–10.
Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 2008; 4: 458–469.
Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 2007; 104: 14050–14055.
Xavier RJ, Podolsky DK . Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007; 448: 427–434.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Kroemer G, Levine B . Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9: 1004–1010.
Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet 2007; 39: 830–832.
McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet 2008; 40: 1107–1112.
Acknowledgements
This work was supported by grants AI069345, AI45148, AI42999 from the National Institutes of Health, 107160-44-RGRL from amfAR, a Bill and Meilinda Gates Grand Challenge Explorations grant and a grant from the Crohn's and Colitis Foundation of America.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Piacentini
Rights and permissions
About this article
Cite this article
Delgado, M., Deretic, V. Toll-like receptors in control of immunological autophagy. Cell Death Differ 16, 976–983 (2009). https://doi.org/10.1038/cdd.2009.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.40
Keywords
This article is cited by
-
Quinolinate promotes macrophage-induced immune tolerance in glioblastoma through the NMDAR/PPARγ signaling axis
Nature Communications (2023)
-
Bi-Directional Relationship Between Autophagy and Inflammasomes in Neurodegenerative Disorders
Cellular and Molecular Neurobiology (2023)
-
The dependency of autophagy and ubiquitin proteasome system during skeletal muscle atrophy
Biophysical Reviews (2021)
-
Does the Global Outbreak of COVID-19 or Other Viral Diseases Threaten the Stem Cell Reservoir Inside the Body?
Stem Cell Reviews and Reports (2021)
-
Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients
Journal of Neuroinflammation (2019)
