
Abstract
Degeneration of retinal ganglion cells (RGCs) – an important cause of visual impairment – is often modeled by optic nerve transection, which leads to apoptotic death of these central nervous system neurons. With this model, we show that specific voltage-gated K+ channels (Kv1 family) contribute to the degeneration of rat RGCs and expression of apoptosis-related molecules in vivo. Retinal expression of Kv1.1, Kv1.2, Kv1.3 and Kv1.5 was examined by quantitative real-time reverse transcriptase-PCR and immunohistochemistry. Kv channel blockers and channel-specific short-interfering RNAs (siRNAs) were used to assess their roles in RGC degeneration. We found that (i) rat RGCs express Kv1.1, Kv1.2 and Kv1.3 (but not Kv1.5); (ii) intraocular injection of agitoxin-2 or margatoxin, potent blockers of Kv1.1, Kv1.2 and Kv1.3 channels, dose-dependently reduced the RGC degeneration; (iii) siRNAs applied to the cut optic nerve were rapidly transported throughout RGCs only, in which they reduced the expression of the cognate channel only. Our results show differential roles of the channels; siRNAs directed against Kv1.1 or Kv1.3 channels greatly reduced RGC death, whereas Kv1.2-targeted siRNAs had only a small effect, and siRNAs against Kv1.5 were without effect. (iv) Kv1.1 and Kv1.3 channels apparently contribute to cell-autonomous death of RGCs through different components of the apoptotic machinery. Kv1.1 depletion increased the antiapoptotic gene, Bcl-XL, whereas Kv1.3 depletion reduced the proapoptotic genes, caspase-3, caspase-9 and Bad.
Similar content being viewed by others

Main
Retinal ganglion cells (RGCs) consolidate visual processing and constitute the last step before signals are transmitted to higher brain centers. RGC death is a major cause of visual impairment in optic neuropathies, including glaucoma, age-related macular degeneration, diabetic retinopathy, uveoretinitis and vitreo-retinopathy. Optic nerve transection is a popular model used to assess potential neuroprotective strategies in vivo. The highly coordinated signaling response in axotomized RGCs results in activation of cell death programs and loss of ∼90% of RGCs within 14 days1, 2, 3, 4 (reviewed by Watanabe et al.,5 Levin,6 Isenmann et al.,7 Weishaupt and Bahr8). RGC death occurs by apoptosis, as shown by the presence of fragmented nuclei and DNA, caspase 3 activation and apoptotic bodies.3, 9, 10
Apoptosis in several cell types is accompanied by increased K+ currents, depletion of cytoplasmic K+ and by cell shrinkage (reviewed by Lang et al.,11 Bortner and Cidlowski,12 Burg et al.,13 Yu14). This ‘apoptotic volume decrease’ (AVD) precedes mitochondrial depolarization, apoptosome formation and cell fragmentation, and is considered a triggering event.11, 12, 13, 14 The concomitant decrease in cytoplasmic K+ concentration can activate molecules in the apoptotic cascade. Consequently, one experimental strategy has been to target K+ channels; an approach that is beginning to be investigated for RGCs (Diem et al.,15 this study). The link between specific K+ channels, AVD and apoptotic pathways is poorly understood, and K+ channel blockers have produced contradictory outcomes; that is, promoting16 or inhibiting14, 17, 18 apoptosis. Contributing to the confusion is that most of the blockers used were membrane permeant and not very selective, and could thus act on multiple channels at the plasma membrane or internally (e.g., on mitochondria). Even when a convincing link was shown between K+ efflux and apoptosis, the very few cell types in which the K+ channel was identified suggest that different cells use different channels, including voltage-gated K+ channel (Kv)1.3, Kv1.5, Kv2.1, Kir1.1 or TASK-3.18, 19, 20, 21, 22, 23
For K+ channels to be considered as targets to inhibit RGC degeneration, it is crucial to identify which channels are involved. In this study, we determined the distribution of four Shaker (Kv1) family K+ channels in the adult rat retina. Then, we analyzed their involvement in RGC degeneration after optic nerve transection using intraocular injection of potent channel blockers and short-interfering RNA (siRNA)-mediated knockdown of each channel in vivo. Our results support targeting specific Kv1 channels to reduce degeneration of these adult central nervous system (CNS) neurons.
Results
Retinal expression of Kv1. 1, Kv1.1, Kv1.3 and Kv1.5 mRNA
The healthy adult rat retina expressed mRNA for Kv1.1, Kv1.2, Kv1.3 and Kv1.5 at levels comparable with the housekeeping gene, hypoxanthine guanine phosphoribosyl transferase (HPRT-1). Expression of Kv1.1, Kv1.2 and Kv1.3 (but not Kv1.5) decreased 7 days after optic nerve transection, which is consistent with the observed immunolocalization of these channels (below). As expected, mRNA changes were relatively small because most RGCs are still present at 7 days after axotomy.2, 3, 6, 7, 8, 24 It was not feasible to monitor the time course of channel protein loss by immunoblotting, because RGCs comprise ∼7% of cells in the healthy adult retina.25
Kv1-selective blockers reduce RGC degeneration after axotomy
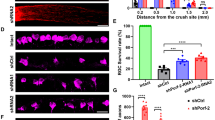
First, K+ channel blockers were tested. Agitoxin-2 (AgTx-2) (blocks Kv1.1 and Kv1.3) or margatoxin (MgTx) (blocks Kv1.2 and Kv1.3) were administered by intraocular injection at the time of axotomy and again at 4 days, when RGC apoptosis had begun.2, 3, 6, 7, 8, 24 To quantify normal RGC density, Fluorogold was injected into the superior colliculus 7 days before axotomy and allowed to undergo retrograde transport to the RGC somata. As commonly carried out, the density of surviving RGCs was counted in flat-mounted retinas at inner, middle and outer retinal eccentricities (see cartoon in Figure 1f). In naive retinas (no axotomy), RGC densities (cells/mm2; n=6) were as follows: 2 446±68 (inner eccentricity), 2 501±42 (middle) and 2 208±109 (outer eccentricity). At 1 day after axotomy (image in Figure 1b), RGC density (2 481±121 cells/mm2; n=4) was the same as in naïve retinas. In control axotomized retinas injected with saline (image in Figure 1c), there were few remaining RGCs at 14 days (cells/mm2; n=8); that is, 198±12 (inner eccentricity), 206±14 (middle) and 178±12 (outer). In contrast, following intraocular injection of AgTx-2 (see image in Figure 1d) or MgTx (Figure 1e), RGC survival at 14 days was dose-dependently increased (Figure 1f).

Kv1.3 blockers inhibit degeneration of retinal ganglion cells (RGCs) after axotomy. (a) Retinal transcript expression of selected Shaker-family (Kv1) K+ channels. Quantitative real-time RT-PCR (see Table 2 for primer sequences) was used to compare adult retinas from naive rats (n=11) with retinas 7 days after optic nerve transection (n=8). For each gene, the y axis shows relative expression, normalized to a housekeeping gene, HPRT-1 (see Materials and Methods section). Values are mean±S.E.M.; significant differences were determined by ANOVA, followed by Fisher's test: *P<0.05. (b–e) Representative confocal micrographs of RGCs in flat-mounted retinas from adult rats, which had been retrograde-labeled with Fluorogold applied to the cut optic nerve stump (see Materials and Methods section). Scale bar, 50 μm; applies to all panels. One day after optic nerve transection (b), a high density of Fluorogold-labeled RGCs and their axon bundles is seen at the nerve fiber layer. At 14 days after axotomy, Fluorogold-labeling shows that almost all RGCs have degenerated in control retinas (c) that were injected with saline only at the time of axotomy and 4 days later. In contrast, after intraocular injections of 50 μM agitoxin-2 (d) or 50 μM margatoxin (e), many more surviving RGCs are seen. (f) Summary of RGC densities 14 days after axotomy. Fluorogold-labeled RGCs in flat-mounted retinas were counted in a 70 000 μm2 area at three retinal eccentricities (inner, middle, outer; see cartoon inset) from each quadrant of the retina, for a total of 12 fields of cells per retina. Values shown are mean±S.E.M. for eight control retinas (axotomized, saline injected) and four retinas in each experimental group; that is, after intraocular injection of three concentrations of agitoxin-2 or margatoxin. At 50 μM, AgTx-2 and MgTx increased RGC survival by ∼2.8-fold and 3.3-fold, respectively. Significant differences between axotomized and treated retinas were determined by ANOVA, followed by Fisher's test: *P<0.05; **P<0.01; ***P<0.001
siRNA-mediated knockdown of specific Kv1 channels only in RGCs
Because no Kv1 channel blocker is perfectly selective, and drugs injected into the vitreous will reach several retinal cell types, we next exploited siRNA-mediated channel knockdown to target individual Kv1 channel subtypes in RGCs alone. The efficiency of retrograde siRNA transport was examined by injecting a Cy3-labeled control siRNA (directed against firefly luciferase) into the freshly cut nerve stump. One day later, almost every RGC cell body was co-labeled with siRNA and Fluorogold (Figure 2a). Thereafter, the same experimental design was used for each Kv channel type, as follows.

Retrograde labeling and siRNA uptake in retinal ganglion cells (RGCs). (a) Confocal micrographs of flat-mounted retinas show that RGCs selectively and rapidly take up siRNA after optic nerve transection. For this figure only, RGCs were retrograde-labeled with Fluorogold (false-colored green) by injecting it into the superior colliculus 7 days before axotomy. Then, immediately after axotomy, a Cy3-labeled control siRNA directed against firefly luciferase was applied to the cut nerve stump. By 24 h later, essentially all RGCs were co-labeled with the siRNA and Fluorogold. Scale bar, 50 μm. The inset in the merged image is at higher magnification. (b) RGC degeneration at 14 days after axotomy (representative fluorescence micrograph of a flat-mounted retina). Immediately after axotomy, the nerve stump was soaked in Fluorogold. The lack of fluorescent-labeled cell bodies indicates that most RGCs had degenerated by 14 days. Scale bar, 50 μm
Immunohistochemistry was used to examine the retinal expression of each channel. Then, to assess each channel's contribution to RGC death after axotomy, a Kv channel-specific siRNA (and Fluorogold) was applied to the freshly cut nerve stump. The control siRNA was compared with two or three siRNAs (10 ng of each) targeted against each channel. (None of the siRNAs was toxic at 1 or 100 pg, 10 or 100 ng.) In addition to the lack of staining with secondary antibody alone, several tests were carried out for antibody specificity. Loss of staining in the siRNA-treated RGCs with no other changes throughout the retina is itself an important test. Additional control experiments (not illustrated) were conducted for each channel found in RGCs (Kv1.1, Kv1.2, Kv1.3; see below). That is, the pattern of immunoreactivity for each channel in the retina was not affected by siRNAs directed against the other two channels. Specifically, Kv1.1 immunoreactivity was not affected by siRNAs against Kv1.2 or Kv1.3; Kv1.2 immunoreactivity was not affected by Kv1.1 or Kv1.3 siRNAs; and Kv1.3 immunoreactivity was not affected by Kv1.1 or Kv1.2 siRNAs. For each treatment, RGC density was quantified after 14 days. In a separate set of retinas, to determine whether the targeted channel was depleted from RGCs, immunohistochemistry was conducted at 5 days after axotomy, a time chosen to precede RGC loss by apoptosis.2, 3, 6, 7, 8, 24 Finally, to determine the efficacy of the selected siRNA, the density of surviving Fluorogold-labeled RGCs was determined at 14 days after axotomy in flat-mounted retinas. When control siRNA was used, we found that very few RGC somata remained (cells/mm2; n=8); that is, 200±23 (inner eccentricity), 203±21 (middle) and 165±18 (outer). At this time, most Fluorogold had been transported to the cell bodies, and few axons were stained.
Kv1.1 localization and increased RGC survival following its depletion from RGCs
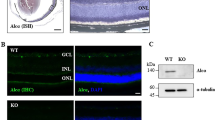
In the healthy adult rat retina, Kv1.1 immunoreactivity was widespread (Figure 3ai), with intense staining in the proximal and distal parts of the inner nuclear layer (INL), where somata of amacrine and horizontal cells are located. In the ganglion cell layer (GCL), there was a clear colocalization of Kv1.1 immunoreactivity with Fluorogold-labeled RGC somata (Figure 3aii–iv). Staining was more diffuse in the outer nuclear layer (ONL), where somata of photoreceptors are found. The Kv1.1 staining pattern was not affected by control siRNA against firefly luciferase (Figure 3aii and iv), showing no response or neurotoxicity from a nonspecific siRNA. By 5 days after axotomy, almost all Fluorogold was transported to RGC somata and no longer labeled their axons. Most importantly, both Kv1.1-specific siRNAs reduced Kv1.1 immunostaining in the Fluorogold-labeled RGCs (Figure 3a v,vi). As expected, Kv1.1 staining was unaffected in the displaced amacrine cells and in the INL and ONL. RGC survival after axotomy (Figure 3b) was significantly and similarly increased by either Kv1.1-specific siRNA, by ∼2.5-fold (to >500 cells/mm2) compared with control siRNA treatment. These results show that Kv1.1 protein is present in RGCs (and in amacrine, horizontal and photoreceptor cells), and that Kv1.1 in RGCs contributes to their degeneration after axotomy.

Expression pattern of the Shaker-family Kv1.1 channel and neuroprotection following siRNA-mediated Kv1.1 depletion from retinal ganglion cells (RGCs). (a) Kv1.1 immunoreactivity in the retina. Representative micrographs of transverse sections taken at the inner eccentricity (as illustrated in Figure 1f, inset) and labeled with a rabbit polyclonal anti-Kv1.1 antibody and a goat anti-rabbit secondary antibody, conjugated to either Alexa 488 (green) or Cy3 (red). When used, Fluorogold (false-colored green) was retrograde transported to the somata of RGCs by applying it to the freshly cut optic nerve stump. Scale bar, 50 μm in all main panels. (i) In the normal retina, Kv1.1 immunoreactivity (green) is seen in the inner nuclear layer (INL); outer nuclear layer (ONL); and ganglion cell layer (GCL). Both RGCs and displaced amacrine cells were labeled with Kv1.1 antibody (arrows show examples). (ii–iv) Control firefly luciferase-specific siRNA had no effect. Five days after Fluorogold (green), the control siRNA was applied to the freshly cut nerve stump, the Kv1.1 (red) staining pattern was similar to the naive retina, and most Fluorogold-labeled RGCs also stained for Kv1.3 (yellow in merged image). Below the remaining micrographs, the boxed areas in the main images are color separated and shown at higher magnification (scale bar, 20 μm applies to all expanded images). The fluorescence intensity was intentionally saturated to reveal any staining left after siRNA treatment. (v and vi) Kv1.1-specific siRNAs deplete Kv1.1 in RGCs. Five days after axotomy and administration of Fluorogold and either of two siRNAs directed against Kv1.1, the channel staining (red) in RGCs decreased. (b) Kv1.1 depletion reduces the death of RGCs. Summary of RGC densities in the inner, middle and outer eccentricities (see Figure 1f inset for locations) at 14 days after axotomy. For each retina, 12 fields of cells were counted, and values shown are mean±S.E.M. for four retinas from each experimental group, and eight axotomized retinas treated with the control siRNA (directed against firefly luciferase). Significant differences between control and treated retinas were determined by ANOVA, followed by Fisher's test (***P<0.001)
Kv1.3 localization and increased RGC survival following its depletion from RGCs
Kv1.3 immunoreactivity was widespread in the healthy retina (Figure 4ai). It was most intense in the nerve fiber layer (NFL), where axons of RGCs are found; the ganglion cell layer, the inner plexiform layer (IPL), where microglia and RGC dendrites are found; and the INL, which contains axons of amacrine and bipolar cells. Little or no Kv1.3 was detected in the ONL, where somata of photoreceptors are found. The cell bodies and axons of RGCs showed considerable Kv1.3 staining and colocalization with Fluorogold. The control siRNA did not affect the Kv1.3 staining, which remained extensively colocalized with Fluorogold in the somata of RGCs (panel a, ii–iv). In contrast, Kv1.3 immunoreactivity in axotomized RGCs was decreased after administering either Kv1.3-specific siRNA; Kv1.3-1169 siRNA was more effective. The summarized RGC density plots (Figure 4b) show that each of the three Kv1.3-specific siRNAs tested significantly increased RGC survival after axotomy. Kv1.3-1169 siRNA was more effective, increasing survival by as much as 3.5-fold compared with control siRNA. Importantly, siRNA-mediated depletion of Kv1.1 and Kv1.3 were not additive. A combination of the two most effective siRNAs elicited only marginally more RGC survival than either siRNA alone.

Expression pattern of Kv1.3 and neuroprotection following siRNA-mediated Kv1.3 depletion from RGCs. (a) Representative confocal micrographs of transverse sections taken at the inner eccentricity (illustrated in Figure 1f, inset). Kv1.3 was labeled with a rabbit polyclonal anti-Kv1.3 antibody and a Cy3-conjugated (red) goat anti-rabbit secondary antibody. Scale bar, 50 μm, applies to all main panels. (i) Kv1.3 immunoreactivity (red) in the healthy rat retina is seen throughout the inner nuclear (INL) and ganglion cell layers (GCL), and specifically, in RGCs (examples are shown by arrows). (ii–iv) Five days after applying the control siRNA, almost all Fluorogold-labeled RGCs (green) expressed Kv1.3 (red). As in Figure 3, boxed areas in the main images are shown at higher magnification, below (scale bar, 20 μm) after color separation and intentional saturation. (v and vi) Five days after applying either of two Kv1.3-specific siRNAs, Kv1.3 labeling of RGCs was decreased. (b) Kv1.3 depletion substantially reduces RGC death, as judged by their increased densities at 14 days after axotomy. Combined Kv1.1 and Kv1.3 depletion is not more effective. Values are mean±S.E.M. for four retinas from each experimental group, and eight axotomized retinas treated with the control siRNA against firefly luciferase. Significant differences between axotomized and treated retinas (***P<0.001) were determined by ANOVA, followed by Fisher's test
Kv1.2 and Kv1.5 localization and roles: Kv1.2 depletion marginally improves RGC survival, but Kv1.5 depletion does not
Kv1.2 immunoreactivity (Figure 5a) was much more restricted than Kv1.1 or Kv1.3 in both the healthy and damaged rat retina. There was intense Kv1.2 staining only in the NFL and specifically, in RGC axon fascicles; and weak staining in the IPL. Again, control siRNA treatment did not affect the Kv1.2 staining pattern after axotomy. Both Kv1.2-specific siRNAs greatly decreased Kv1.2 immunoreactivity in the axon fascicles, but RGC survival (Figure 5b) was only slightly increased (by ∼1.6-fold).

Expression pattern of Kv1.2 and marginal neuroprotection following siRNA-mediated Kv1.2 depletion from RGCs. (a) Representative confocal micrographs of transverse sections taken at the inner eccentricity (see Figure 1f inset). Kv1.2 immunoreactivity was monitored with a mouse monoclonal-antibody and a Cy3-conjugated (red) goat anti-mouse secondary antibody, and somata of retinal ganglion cells (RGCs) were Fluorogold-labeled (false-colored green). Scale bar, 50 μm; applies to all main panels. (i) In the normal retina, Kv1.2 staining (red) is seen in cross-sections of axon fascicles cut perpendicularly (arrows show examples). (ii–iv) After injection of the control siRNA against firefly luciferase, Kv1.2 staining (red) remained in RGC axon fascicles, which are seen parallel to the axon bundles. None of the Fluorogold-labeled RGC somata (green) were stained for Kv1.2. As in Figures 3 and 4, boxed areas in the main images are shown at higher magnification, below (scale bar, 20 μm) after color separation and intentional saturation. (v and vi) Kv1.2-specific siRNAs reduce axonal expression of Kv1.2 in RGCs. Five days after axotomy and injection of either siRNA, Kv1.2 immunostaining in RGC axons was less intense, but not abolished. (b) RGC densities at 14 days after axotomy are shown as mean±S.E.M. for four retinas from each experimental group, and eight control siRNA-treated retinas. Significant differences from axotomized retinas (*P<0.05; **P<0.01) were determined by ANOVA, followed by Fisher's test
Strong Kv1.5 immunoreactivity (Figure 6a) was seen in the ganglion cell layer and throughout the INL, where cell bodies of amacrine, bipolar and horizontal cells are found. In the ganglion cell layer, most Kv1.5 appeared to be in the end-feet of Muller glial cells, which form the inner limiting membrane, and in the INL, where their cell bodies are located. Similar staining was seen in undamaged retinas (not shown). Consistent with the lack of Kv1.5 staining in RGCs, RGC survival (Figure 6b) was not improved by either Kv1.5-specific siRNA. This result provides further evidence against nonspecific responses to siRNA treatment.

Expression pattern of Kv1.5; no protection by Kv1.5-targeted siRNAs. (a) A representative confocal micrograph of a transverse section taken at the inner eccentricity (as in Figure 1f, inset). Kv1.5 immunoreactivity was monitored with a rabbit polyclonal antibody, and a Cy3-conjugated (red) goat anti-rabbit secondary antibody. RGC somata were labeled with Fluorogold (false-colored green). Scale bar, 50 μm. Color-separated images of the ganglion cell layer (GCL) are shown below at the same magnification. Five days after axotomy and injection of control siRNA, strong Kv1.5 staining (red) was seen in the inner nuclear (INL) and GCL, but little colocalized with Fluorogold-labeled RGCs. (b) Kv1.5 depletion did not affect RGC densities at 14 days after axotomy. Data represent mean±S.E.M. for four retinas from each experimental group, and eight control retinas treated with siRNA against firefly luciferase
Kv1.1 and Kv1.3 depletion differentially affect apoptosis-related genes
Agitoxin-2, margatoxin or siRNA-mediated knockdown of Kv1.1 or Kv1.3 substantially increased RGC survival. Thus, we next examined possible neuroprotective mechanisms in response to Kv1.1 or Kv1.3 knockdown (Figure 7). Transcript expression was monitored for several genes known to have important roles in RGC death after axotomy.5, 6, 7, 8, 26, 27 Quantitative real-time reverse transcriptase-PCR (qRT-PCR) on whole retinas was conducted at 7 days after axotomy, a time when RGC apoptosis is well underway but before these cells are lost.3, 8 First, mRNA expression in naive healthy retinas was compared with axotomized control retinas that had been treated with control siRNA against firefly luciferase. After axotomy, Bcl-2, Bcl-XL, caspase 3 and caspase 9 were significantly upregulated in extracts from whole retina, while Bad decreased. Of the genes tested, the most highly expressed were Bad and caspase 3, which were ∼8 and 12%, respectively, of the level of the housekeeping gene, HPRT-1. Depletion of Kv1.1 from RGCs significantly increased the antiapoptotic gene, Bcl-XL. Kv1.3 depletion from RGCs significantly reduced the proapoptotic genes, Bad, caspase-3 and caspase-9. Thus, it appears that Kv1.1 and Kv1.3 channels differentially modulate gene expression in the injured adult CNS. Neither channel knockdown affected Bcl-2 mRNA levels.

siRNA-mediated depletion of Kv1.1 or Kv1.3 from RGCs differentially affects retinal expression of selected genes involved in apoptosis. Quantitative real-time reverse transcriptase-PCR (see Table 2 for primer sequences) was used to examine changes in mRNA expression. For each gene, the y axis shows relative expression, normalized to a housekeeping gene, HPRT-1 (see Materials and Methods section). Values are mean±S.E.M. for naive retinas (n=11), axotomized control retinas treated with an siRNA-directed against firefly luciferase (n=8), and Kv1.1 siRNA- or Kv1.3 siRNA-treated retinas (n=6 each). Significant differences were determined by ANOVA, followed by Fisher's test (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001)
Discussion
The optic nerve transection model was used to investigate roles of Kv1.1, Kv1.2, Kv1.3 and Kv1.5 channels in neurodegeneration in vivo. In this model, axotomy deprives RGCs of their efferent synapses, leading to apoptotic death through cell-autonomous (loss of neurotrophic support) and non-autonomous (e.g., glial-mediated) mechanisms. First, we assessed the mRNA expression and pattern of immunoreactivity for each channel before and after axotomy. Most importantly, Kv1.1, Kv1.2 and Kv1.3 (but not Kv1.5) were detected in RGCs. Next, the potential involvement of these channels in neurodegeneration was determined by quantifying RGC loss at 14 days after axotomy, with and without intraocular injection of two potent channel blockers. Both AgTx-2 and MgTx dose-dependently increased RGC survival. Then, to selectively target each Kv channel in RGCs only, channel-specific siRNAs were applied to the freshly cut optic nerve stump (axons of RGCs) and allowed to undergo retrograde transport to RGC somata. siRNA-mediated depletion of either Kv1.1 or Kv1.3 significantly increased RGC survival by several fold compared with axotomized control retinas. Kv1.2 depletion was less neuroprotective and Kv1.5 siRNAs had no effect. The highest RGC survival at 14 days after axotomy was ∼800 cells/mm2 after intraocular MgTx injection (∼750 cells/mm2 after siRNA-mediated Kv1.3 depletion), compared with 2300–2400 RGCs/mm2 in the healthy rat retina. Although incomplete, this rescue is comparable with that afforded by the growth factors, brain-derived neurotrophic factor, IGF-1, glial-derived neurotrophic factor and neurturin,10, 28 which are potent antiapoptotic proteins. The Kv channel blockers were more effective than our earlier results using TRAM-34, an inhibitor of KCNN4 (KCa3.1/SK4) channels.29 Finally, by quantifying changes in the expression of several apoptosis-related genes with and without siRNA-mediated channel depletion, we found that Kv1.1 and Kv1.3 are functionally linked to different apoptotic molecules. When monitored in the extracts of the whole retina, Kv1.1 depletion increased expression of the antiapoptotic gene, Bcl-XL, whereas Kv1.3 depletion reduced the proapoptotic genes, caspase-3, caspase-9 and Bad. This study provides new information about Kv1.1, Kv1.2, Kv1.3 and Kv1.5 distribution and roles in the rat retina. By using siRNA-mediated knockdown of specific K+ channels in identified CNS cells in vivo, we provide direct evidence that Kv1.1, Kv1.2 and Kv1.3 channels contribute to cell-autonomous neurodegeneration and to apoptosis-related molecules in vivo.
Little was previously known about retinal expression of Kv1.1, Kv1.2, Kv1.3 and Kv1.5 in mammals. The murine retina30 shows immunoreactivity for Kv1.1, Kv1.2 and Kv1.3, with strong Kv1.1 staining throughout the GCL and IPL, Kv1.2 staining in the NFL and GCL (with more diffuse labeling in the INL) and Kv1.3 staining in the GCL, IPL and INL. In this study on the adult rat retina, we observed intense staining for Kv1.1 in the GCL and INL, more diffuse staining in the ONL, and no detectable staining in the IPL. Kv1.3 immunoreactivity was detected in the NFL, GCL, IPL and INL of the rat (similar to the mouse).30 Most significantly, intense Kv1.1 and Kv1.3 staining was present in RGC somata (co-labeled with Fluorogold). The Kv1.2 distribution was more restricted, with intense staining in RGC axon fascicles (but not somata) and weaker staining in the IPL. This is consistent with an earlier observation that most Kv1.2 immunoreactivity is in axons and terminal fields in the hippocampus.31 We observed intense Kv1.5 staining only in the INL and on Muller glial cells in the GCL. Note that the distribution of channel immunoreactivity did not change at 5 days after axotomy, except when siRNA-mediated channel depletion was used.
To discuss our results in light of Kv channels previously described in neonatal mammalian RGCs, it is useful to address properties of the cloned channels. Kv1.3 channels produce a slowly inactivating current, whereas Kv1.1 and Kv1.2 produce currents with either fast or slow inactivation, depending on accessory molecules. Kv1.1 and Kv1.2 are blocked by sub-millimolar concentrations of tetraethylammonium (TEA), whereas all three channels are blocked by sub-millimolar concentrations of 4-aminopyridine and nanomolar concentrations of α-dendrotoxin (for properties and pharmacology, see Grissmer et al.32). Hence, these blockers were not useful for discerning the roles of the Kv channels in this study. Instead, we used AgTx-2, which is a potent blocker of Kv1.1 and Kv1.3; and MgTx, which blocks Kv1.2 and Kv1.3.32, 33 Recognizing that no K+ channel blocker is perfectly selective, we then exploited siRNA-mediated knockdown and identified substantial roles for Kv1.1 and Kv1.3, and a small contribution of Kv1.2. Electrophysiological studies of isolated mammalian RGCs have shown several voltage-gated K+ (Kv) currents. A fast transient Kv (usually called IA) is inhibited by 4-aminopyridine, a membrane-permeant nonselective blocker of Shaker Kv channels; and one or more delayed rectifier Kv currents (usually called IK) are inhibited by TEA,34, 35, 36 a membrane-permeant non-selective K+ channel blocker. Only one study15 has implicated K+ channels in RGC degeneration after optic nerve transection, but it is premature to ascribe the findings to a particular channel type. In that study, death of adult RGCs was reduced by intraocular injection of an extremely high concentration of TEA (300 mM), which probably acted on multiple channels and cell types. Their parallel in vitro studies of isolated RGCs from rat pups (6–16 days old) showed a delayed rectifier current that was reduced by TNF-α and increased by minoxidil sulfate, which abrogated the neuroprotective effect of TNF-α. Interestingly, minoxidil sulfate is not known as a Kv channel activator.
In future, it would be interesting to determine if a current carried by any of these channels is increased in RGCs after axotomy, but this will be difficult to assess for several reasons, as follows. Previous patch-clamp studies on RGCs have relied on acutely isolated or cultured cells prepared from neonatal rodents, and none have succeeded in identifying which channel gene underlies a particular current. Because there are dramatic developmental changes in RGC electrical activity and currents,34, 35 the analysis should be carried out in adult RGC's, which are difficult to isolate and purify. When RGCs are isolated, there is heterogeneity in the cells and possible changes in channel activity resulting from tissue ‘damage’ during isolation. The literature relating K+ currents to apoptosis in other cell types has relied on isolated cells (see below), and the increase in current thought to underlie induction of apoptosis is transient. After optic nerve transection in vivo, RGC apoptosis occurs over a period of 2 weeks and there is no way of knowing at what time point a change in K+ current might occur. It would also be interesting to determine if neuroprotection owing to Kv channel knockdown is long lasting. Several other neuroprotective strategies delay, rather than prevent degeneration of axotomized RGCs, whether treatment was by intraocular injections or a continuous supply of the neuroprotective agent using viral gene transfer.1, 4, 10
What are the potential links between specific K+ channels, RGC apoptosis and genes affected by siRNA-mediated knockdown? Neurons can express multiple K+ channel types and their normal activity is clearly not toxic. However, apoptosis in several cell types is accompanied by an ‘AVD’, increased K+ currents and a decrease in cytoplasmic K+ concentration11, 12, 13 (recently reviewed by Yu14), which is considered crucial for both triggering (e.g., caspase 8) and effector (e.g., caspase 3) molecules for apoptosis.12, 13, 23 The limited information on channel identity points to involvement of different K+ channels in different cell types. Kv1.3 is apparently responsible for the increased K+ efflux underlying lymphocyte apoptosis.11, 12, 19, 20, 37 Other K+ channels implicated in apoptosis are Kir1.1 in hippocampal neurons,22 Kv2.1 in cortical neurons,23 TASK-3 in cerebellar granule cells18 and Kv1.5 in smooth muscle cells.21 It is intriguing that we found contributions of both Kv1.1 and Kv1.3 to RGC degeneration. Our results suggest that Kv contributions depend on their location; that is, Kv1.1 and Kv1.3 were highly expressed in RGC somata and had the greatest influence on cell survival, whereas the predominantly axonal Kv1.2 channel contributed little. On the basis of association of RGC death with activation of caspases and Bcl-2 family molecules (reviewed by Levin,6 Isenmann et al.,7 Weishaupt and Bahr8), we monitored expression of several molecules thought to lie downstream of the AVD. In whole retinas analyzed 7 days after axotomy (when the rate of apoptosis is maximal), the two Kv channels modulated different components of the apoptotic pathway. Kv1.3 depletion decreased expression of caspase-3, caspase-9, and Bad; molecules that exacerbate apoptotic RGC death after axotomy. Kv1.1 depletion increased mRNA expression of Bcl-XL, a molecule that protects the mitochondrial membrane potential and is neuroprotective for injured RGCs.27 Note that RGCs comprise only ∼7% of retinal cells and 25–50% of RGCs degenerate by 7 days;2, 3, 24 thus, transcript changes in individual RGCs are likely under-represented.
These results have broader implications because RGC death is a major cause of visual impairment in glaucoma, age-related macular degeneration, diabetic retinopathy, uveoretinitis and vitreo-retinopathy. This study provides the first evidence that targeting specific Kv1 channels might have therapeutic potential for treating CNS diseases or insults that induce neuronal apoptosis. Moreover, because Kv1.3 is important for activation of T lymphocytes, it is already considered a therapeutic target for immune-mediated disorders (e.g., arthritis, multiple sclerosis), and for transplant rejection of organs, tissues and cells.33 Several potent peptide blockers of Kv1.3 have been identified, and some have been tested in vivo in animal models to assess immune suppression.
Materials and Methods
Optic nerve transection and RGC labeling
Experiments were carried out in accordance with guidelines from the Canadian Council on Animal Care. The left optic nerve of adult female Sprague–Dawley rats (225–250 g; Charles River, Quebec, Canada) was transected, as previously described.2, 3, 15, 24 Briefly, rats were anesthetized by intraperitoneal injection of 7% chloral hydrate (420 mg/kg), and body temperature was maintained with a regulated heating pad. Access to the optic nerve was through a lesion in the superior rim of the orbit of the eye, being careful not to damage the lacrimal gland or retinal blood supply. A 1–2 mm longitudinal incision was made in the dural sheath within 2 mm of the eye to avoid severing arteries and veins. The optic nerve was pulled through the dura, a 1 mm section removed, and the nerve stump returned to the dura. RGCs were retrograde-labeled with Fluorogold in either of two ways, depending on the experiment. (i) To count normal RGC density and characterize the efficacy of retrograde siRNA transport, Fluorogold was injected into the superior colliculus 7 days before axotomy,3, 24 and a Cy3-labeled control siRNA, directed against firefly luciferase, was injected into the freshly cut nerve stump with a Hamilton syringe. Retinas were fixed 24 h later. Retrograde transport to the RGC somata from the cut nerve stump has been well characterized8, 24, 26, 38, 39 and, as expected, essentially all RGCs were labeled with siRNA or Fluorogold (Figure 3a). (ii) For the remaining experiments, RGCs were retrograde labeled with Fluorogold by briefly placing a 2-mm3 piece of Gelfoam soaked in 5 μl of 3% Fluorogold (false-colored green) against the freshly cut optic nerve stump.
Intraocular administration of Kv channel blockers
We used two small-molecular weight peptidyl inhibitors of specific Shaker (Kv1) channels. AgTx-2 blocks Kv1.1 and Kv1.3, and MgTx blocks Kv1.2 and Kv1.3.32, 33 When applied to cells transfected with Kv1.1, Kv1.2 or Kv1.3, the blockers are effective at sub-nanomolar concentrations. Much higher concentrations are necessary in vivo, because compounds injected into the vitreous chamber of the eye slowly diffuse out of the orbit and into the retina and the peptide blockers can degrade with time. Two intraocular injections of 500 nM, 5 μM or 50 μM of AgTx-2 or MgTx were given; one at the time of optic nerve transection and the second at 4 days. Each drug was dissolved in saline (100 mM NaCl, 0.1% bovine serum albumin, 1 mM EDTA, 10 mM TRIS buffer, pH 7.5) and 3 μl was injected at each time point. Control injections consisted of the saline solution alone. Injections were administered posterior to the limbus of the eye using a pulled glass micropipette to ensure that anterior ocular structures were not damaged and care was also taken not to puncture the lens. No attempt was made to block Kv1.5 because no potent, selective inhibitor is available.
RNA interference to reduce specific Kv channels in RGCs only
RNA interference in vivo was used to assess the role of four Shaker-family channels (Kv1.1, Kv1.2, Kv1.3, Kv1.5) in the degeneration of RGCs after axotomy. Immediately after optic nerve transection, RGCs were labeled using Fluorogold-soaked Gelfoam (as above), and then 5 μl of a solution containing 10 ng of a selected siRNA was injected into the transected optic nerve stump using a Hamilton syringe. A similar siRNA injection technique has been used to knockdown apoptosis-related proteins and transcription factors.26 As a control treatment throughout these studies, an irrelevant, Cy3-labeled siRNA directed against a non-mammalian protein, firefly luciferase (Upstate, Charlottesville, VA, USA) was injected. The Kv-channel-specific siRNAs (Table 1) were synthesized using in vitro transcription procedures (Silencer siRNA kit, Ambion, Austin, TX, USA) according to the manufacturer's protocol. For simplicity, we named each siRNA construct for the first nucleotide in the targeted channel sequence.
Immunohistochemistry
The normal retinal expression of each Kv channel type was determined by immunohistochemistry, as was the reduction of each channel protein following siRNA treatment. Retinas were fixed and labeled with a Kv channel antibody; that is, rabbit polyclonal-anti-Kv1.1 (APC-009; Alomone Labs, Jerusalem, Israel), mouse monoclonal-anti-Kv1.2 (05-408; United Biomedical Inc., Hauppauge, NY, USA), rabbit polyclonal-anti-Kv1.3 (gift from Dr. Debra Fadool, Florida State University), and rabbit polyclonal-anti-Kv1.5 (Alomone Labs, APC-004). All primary antisera were diluted (1 : 200 dilution in phosphate-buffered saline (PBS), pH 7.6) containing 3% normal goat serum and 0.3% Triton X-100. Transverse frozen sections of retina were incubated in a primary antiserum overnight at room temperature. Sections were then rinsed (3 × , 15 min each) in PBS, and incubated with the appropriate secondary antibody for 4 h at room temperature. The secondary antisera, affinity-purified Cy3- or Alexa 488-conjugated goat-anti-rabbit or goat-anti-mouse (Jackson ImmunoResearch, West Grove, PA, USA) were diluted in a similar buffer to primary antisera. After incubation, sections were rinsed (3 × , 15 min each) and mounted with 1 : 1 glycerol/PBS. For each immunohistochemistry assay, the concentration of antibodies, fluorescence intensity gain, and pinhole and neutral density filters were kept constant. There was no staining when each primary antibody was omitted, and the black regions within each image (Figures 2, 3, 4, 5 and 6) show the lack of nonspecific staining. The specificity of these antibodies has been previously characterized by others and by us; for example, in our earlier study40 each channel was heterologously expressed in a rat microglia cell line (MLS-9) that does not express these channels. The lack of cross-reactivity is also shown by the very different staining patterns for each antibody (Figures 2, 3, 4, 5 and 6).
Quantifying RGC survival after axotomy
Retinal ganglion cell survival after optic nerve transection was quantified 14 days after axotomy, by which time most RGCs normally die.3, 6, 7, 8, 24 Comparisons were made between retinas treated with a Kv-channel-specific siRNA or the control siRNA directed against firefly luciferase. Rats were killed by an overdose of anesthesia (chloral hydrate), their eyes were enucleated, the cornea and lens removed, and the remaining eye cups fixed (1.5 h, room temperature) in 4% paraformaldehyde made in PBS containing 2% sucrose. Then, the eye cup was washed with PBS for 15 min, and the neural retina was dissected away from the sclera and flat-mounted in 1 : 1 glycerol/PBS. The surviving RGCs (labeled with Fluorogold) were counted from confocal micrographs (Zeiss LSM 510). Because RGC density varies somewhat across the retina, we used a standard procedure.2, 3, 24 That is, cells in flat-mounted retinas were counted in a 70 000 μm2 area at three retinal eccentricities (inner, middle, outer; see cartoon in Figure 1f) from each quadrant of the retina, for a total of 12 fields of cells per retina.
Quantitative real-time reverse transcriptase–PCR
Whole retinas were harvested 7 days after axotomy, and changes in gene transcript expression were monitored by qRT-PCR, as previously described.29 Gene-specific primers (Table 2) were designed using the ‘Primer3Output’ program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). RNeasy mini kits (Qiagen Inc. Mississauga, ON, Canada) were used to isolate RNA after degrading any contaminating DNA with DNaseI (0.1 U/ml, 15 min, 37 °C; Amersham Biosciences, Baie d′Urfe, Qc, Canada). A two-step reaction was performed according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA); that is, total RNA (2 μg) was reverse transcribed in 20 μl volume using 200 U of SuperScriptII RNase H-reverse transcriptase, with 0.5 mM dNTPs (Invitrogen) and 0.5 μM oligo-dT (Sigma-aldrich, St. Louis, MO, USA). Amplification was performed on an ABI PRISM 7900 Sequence Detection System (PE Biosystems, Foster City, CA, USA) at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s, 55 °C for 15 s and 72 °C for 30 s. ‘No-template’ and ‘no-amplification’ controls were included for each gene, and melt curves showed a single peak, confirming specific amplification. The threshold cycle (Ct) for each gene was determined and normalized against the housekeeping gene, HPRT-1. To monitor retinal responses to optic nerve transection, normal retinas (naive) were compared with axotomized retinas. To assess responses to siRNA-mediated Kv channel knockdown, the comparison was between a control siRNA (directed against firefly luciferase) and a channel-specific siRNA delivered to the cut optic nerve stump, as described above. Significant differences were identified by ANOVA, followed by Fisher's mean interval comparisons.
Abbreviations
- AgTx-2:
-
agitoxin-2
- AVD:
-
apoptotic volume decrease
- CNS:
-
central nervous system
- GCL:
-
ganglion cell layer
- INL:
-
inner nuclear layer
- IPL:
-
inner plexiform layer
- Kv:
-
voltage-gated K+ channel
- MgTx:
-
margatoxin
- NFL:
-
nerve fiber layer
- ONL:
-
outer nuclear layer
- RGC:
-
retinal ganglion cell
- siRNA:
-
short-interfering RNA
References
Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ . Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci USA 1994; 91: 1632–1636.
Villegas-Perez MP, Vidal-Sanz M, Rasminsky M, Bray GM, Aguayo AJ . Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol 1993; 24: 23–36.
Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ . Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J Neurosci 1994; 14: 4368–4374.
Di Polo A, Aigner LJ, Dunn RJ, Bray GM, Aguayo AJ . Prolonged delivery of brain-derived neurotrophic factor by adenovirus-infected Muller cells temporarily rescues injured retinal ganglion cells. Proc Natl Acad Sci USA 1998; 95: 3978–3983.
Watanabe M, Sawai H, Fukuda Y . Survival of axotomized retinal ganglion cells in adult mammals. Clin Neurosci 1997; 4: 233–239.
Levin LA . Intrinsic survival mechanisms for retinal ganglion cells. Eur J Ophthalmol 1999; 9 (Suppl 1): S12–S16.
Isenmann S, Kretz A, Cellerino A . Molecular determinants of retinal ganglion cell development, survival, and regeneration. Prog Retin Eye Res 2003; 22: 483–543.
Weishaupt JH, Bahr M . Degeneration of axotomized retinal ganglion cells as a model for neuronal apoptosis in the central nervous system - molecular death and survival pathways. Restor Neurol Neurosci 2001; 19: 19–27.
Quigley HA, Nickells RW, Kerrigan LA, Pease ME, Thibault DJ, Zack DJ . Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci 1995; 36: 774–786.
Kermer P, Klocker N, Labes M, Bahr M . Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 in vivo. J Neurosci 2000; 20: 2–8.
Lang F, Foller M, Lang KS, Lang PA, Ritter M, Gulbins E et al. Ion channels in cell proliferation and apoptotic cell death. J Membr Biol 2005; 205: 147–157.
Bortner CD, Cidlowski JA . Cell shrinkage and monovalent cation fluxes: role in apoptosis. Arch Biochem Biophys 2007; 462: 176–188.
Burg ED, Remillard CV, Yuan JX . Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Br J Pharmacol 2008; 153 (Suppl 1): S99–S111.
Yu SP . Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol 2003; 70: 363–386.
Diem R, Meyer R, Weishaupt JH, Bahr M . Reduction of potassium currents and phosphatidylinositol 3-kinase-dependent AKT phosphorylation by tumor necrosis factor-(alpha) rescues axotomized retinal ganglion cells from retrograde cell death in vivo. J Neurosci 2001; 21: 2058–2066.
Takaba H, Nagao T, Yao H, Kitazono T, Ibayashi S, Fujishima M . An ATP-sensitive potassium channel activator reduces infarct volume in focal cerebral ischemia in rats. Am J Physiol 1997; 273 (2 Part 2): R583–R586.
Wei L, Yu SP, Gottron F, Snider BJ, Zipfel GJ, Choi DW . Potassium channel blockers attenuate hypoxia- and ischemia-induced neuronal death in vitro and in vivo. Stroke 2003; 34: 1281–1286.
Lauritzen I, Zanzouri M, Honore E, Duprat F, Ehrengruber MU, Lazdunski M et al. K+-dependent cerebellar granule neuron apoptosis: role of TASK leak K+ channels. J Biol Chem 2003; 278: 32068–32076.
Bock J, Szabo I, Jekle A, Gulbins E . Actinomycin D-induced apoptosis involves the potassium channel Kv1.3. Biochem Biophys Res Commun 2002; 295: 526–531.
Storey NM, Gomez-Angelats M, Bortner CD, Armstrong DL, Cidlowski JA . Stimulation of Kv1.3 potassium channels by death receptors during apoptosis in Jurkat T lymphocytes. J Biol Chem 2003; 278: 33319–33326.
McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 2004; 95: 830–840.
Nadeau H, McKinney S, Anderson DJ, Lester HA . ROMK1 (Kir1.1) causes apoptosis and chronic silencing of hippocampal neurons. J Neurophysiol 2000; 84: 1062–1075.
Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E . Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci 2003; 23: 4798–4802.
Koeberle PD, Ball AK . Effects of GDNF on retinal ganglion cell survival following axotomy. Vision Res 1998; 38: 1505–1515.
Yang J, Tezel G, Patil RV, Wax MB . Flow cytometry for quantification of retrogradely labeled retinal ganglion cells by Fluoro-Gold. Curr Eye Res 2000; 21: 981–985.
Lingor P, Koeberle P, Kugler S, Bahr M . Down-regulation of apoptosis mediators by RNAi inhibits axotomy-induced retinal ganglion cell death in vivo. Brain 2005; 128 (Part 3): 550–558.
Malik JM, Shevtsova Z, Bahr M, Kugler S . Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL gene transfer. Mol Ther 2005; 11: 373–381.
Koeberle PD, Ball AK . Neurturin enhances the survival of axotomized retinal ganglion cells in vivo: combined effects with glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor. Neuroscience 2002; 110: 555–567.
Kaushal V, Koeberle PD, Wang Y, Schlichter LC . The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci 2007; 27: 234–244.
Pinto LH, Klumpp DJ . Localization of potassium channels in the retina. Prog Retin Eye Res 1998; 17: 207–230.
Monaghan MM, Trimmer JS, Rhodes KJ . Experimental localization of Kv1 family voltage-gated K+ channel alpha and beta subunits in rat hippocampal formation. J Neurosci 2001; 21: 5973–5983.
Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA et al. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol 1994; 45: 1227–1234.
Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD . K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci 2004; 25: 280–289.
Reiff DF, Guenther E . Developmental changes in voltage-activated potassium currents of rat retinal ganglion cells. Neuroscience 1999; 92: 1103–1117.
Robinson DW, Wang GY . Development of intrinsic membrane properties in mammalian retinal ganglion cells. Semin Cell Dev Biol 1998; 9: 301–310.
Lipton SA, Tauck DL . Voltage-dependent conductances of solitary ganglion cells dissociated from the rat retina. J Physiol 1987; 385: 361–391.
Hughes Jr FM, Cidlowski JA . Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo. Adv Enzyme Regul 1999; 39: 157–171.
Kugler S, Klocker N, Kermer P, Isenmann S, Bahr M . Transduction of axotomized retinal ganglion cells by adenoviral vector administration at the optic nerve stump: an in vivo model system for the inhibition of neuronal apoptotic cell death. Gene Therapy 1999; 6: 1759–1767.
Sarthy PV, Curtis BM, Catterall WA . Retrograde labeling, enrichment, and characterization of retinal ganglion cells from the neonatal rat. J Neurosci 1983; 3: 2532–2544.
Kotecha SA, Schlichter LC . A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci 1999; 19: 10680–10693.
Acknowledgements
We are grateful to Xiaoping Zhu for expert technical assistance. This work was supported by operating grants to LCS from the Canadian Institutes for Health Research (MT-13657), the Heart and Stroke Foundation, Ontario chapter (HSFO; T4670; T5782) and the Krembil Scientific Development Seed Fund. PDK was supported by a postdoctoral fellowship from HSF Canada. Preliminary data from this paper have been published as abstracts in conference proceedings: Society for Neuroscience Annual Meeting (2005), Federation of European Neuroscience Societies (2006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by L Greene
Rights and permissions
About this article
Cite this article
Koeberle, P., Wang, Y. & Schlichter, L. Kv1.1 and Kv1.3 channels contribute to the degeneration of retinal ganglion cells after optic nerve transection in vivo. Cell Death Differ 17, 134–144 (2010). https://doi.org/10.1038/cdd.2009.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.113
Keywords
This article is cited by
-
Peripheral Administration of the Kv1.3-Blocking Peptide HsTX1[R14A] Improves Cognitive Performance in Senescence Accelerated SAMP8 Mice
Neurotherapeutics (2023)
-
Novel use of a chemically modified siRNA for robust and sustainable in vivo gene silencing in the retina
Scientific Reports (2020)
-
Dopamine D2 Receptor-Mediated Modulation of Rat Retinal Ganglion Cell Excitability
Neuroscience Bulletin (2020)
-
The versatile Kv channels in the nervous system: actions beyond action potentials
Cellular and Molecular Life Sciences (2020)
-
Combined transcriptomic and proteomic analysis reveals a diversity of venom-related and toxin-like peptides expressed in the mat anemone Zoanthus natalensis (Cnidaria, Hexacorallia)
Archives of Toxicology (2019)
