
Abstract
Most cells undergo apoptosis through the intrinsic pathway. This is dependent on mitochondrial outer membrane permeabilisation (MOMP), which is mediated by the pro-apoptotic Bcl-2 family proteins, Bax and Bak. During apoptosis, Bax translocates from the cytosol to the outer mitochondrial membrane (OMM), wherein it contributes to the formation of pores to release cytochrome-c. However, it remains unclear whether Bax translocation is sufficient to bring about MOMP or whether Bax requires further signals on the OMM to be fully activated. We have previously shown that during mammary epithelial cell anoikis, Bax translocation does not commit cells to MOMP and detached cells are rescued if survival signals from the extracellular matrix (ECM) are restored. These findings implied that a second signal is required for mitochondrial Bax to fully activate and cause MOMP. We now identify p38MAPK (mitogen-activated protein kinase) as this necessary signal to activate Bax after its translocation to mitochondria. The inhibition of p38MAPK did not prevent Bax translocation, but its activity was required for mitochondrial Bax to bring about MOMP. p38MAPK was activated and recruited to a high molecular weight mitochondrial complex after loss of ECM attachment. Artificially targeting p38MAPK to the OMM increased the kinetics of anoikis, supporting a requirement for its mitochondrial localisation to regulate Bax activation and drive commitment to apoptosis.
Similar content being viewed by others


Main
Metazoan cells are pre-programmed to undergo apoptosis in response to becoming damaged or losing essential survival signals. Apoptosis in response to these cellular stresses is initiated through the intrinsic, or mitochondrial, pathway.1 During intrinsic apoptosis, mitochondrial outer membrane permeabilisation (MOMP) results in the release of proteins, such as cytochrome-c and second mitochondrial activator of caspases (Smac)/Diablo, which activate the caspases to drive cell death. The release of cytochrome-c and Smac/Diablo is an extremely rapid event, with mitochondria throughout the cell releasing their contents within a few minutes of MOMP first commencing.2, 3 The resulting activation of caspases occurs immediately and cell death is complete within a few minutes. After MOMP, cells rapidly lose viability, even if caspase activation is inhibited. Thus, MOMP represents a point at which cells cannot be rescued from an apoptotic fate.
The Bcl-2 family of proteins regulates MOMP.4 These are characterised by a number of shared regions of sequence homology, the Bcl-2 Homology (BH) domains. Pro-survival members, such as Bcl-2 and Bcl-XL, share BH-domains 1–4. In contrast, the pro-apoptotic proteins, Bax and Bak, only posses BH-domains 1, 2 and 3. A third group of Bcl-2 proteins are the BH3-only proteins, which function by sensing survival and stress signals, and transmit these to the pro- and anti-apoptotic multi-domain proteins.5 The multi-domain pro-apoptotic proteins, Bax and Bak, are expressed in virtually all tissues, and are absolutely required for MOMP to occur.6 Cells derived from mice in which Bax and Bak are genetically deleted are resistant to a wide range of apoptotic stimuli. Mitochondrial Bax and Bak are thought to mediate cytochrome-c release by forming pores in the outer mitochondrial membrane (OMM).
Many cells require attachment to the correct extracellular matrix (ECM) to suppress apoptosis.7 Apoptosis caused by loss of ECM-dependent survival signals is known as anoikis. Loss of sensitivity to anoikis has been shown to directly contribute to the ability of tumour cells to metastasise.8 We have previously shown that Bax associates with mitochondria rapidly after detachment of mammary epithelial cells (MECs) from ECM.9, 10 However, translocation of Bax can precede MOMP by a considerable amount of time, and can even be reversed if cells are able to reattach to ECM before becoming committed to apoptosis. MOMP requires further conformational changes in Bax that occur concomitantly with cytochrome-c release. This suggests that distinct signals regulate both the initial association of Bax with the OMM and its subsequent activation to induce MOMP.11 Here, we identify a role for p38 mitogen-activated protein kinase (p38MAPK) in regulating Bax-dependent MOMP during anoikis. We show that after detachment of MECs from ECM, p38MAPK becomes activated and associated with the mitochondrial fraction. This p38MAPK activity is not required for Bax to translocate to mitochondria. Instead, p38MAPK regulates subsequent Bax activation, controlling MOMP and commitment to apoptosis.
Results
p38MAPK is required for MECs to undergo anoikis
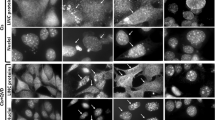
We have previously shown that, after detachment of MECs from ECM, Bax rapidly redistributes to mitochondria.10 In adherent MECs, GFP-Bax shows a cytosolic distribution (Figure 1a). In contrast, detachment for just 15 min results in GFP-Bax associating with mitochondria, indicated by co-localisation with cytochrome-c. Identical results are obtained if the distribution of endogenous Bax is examined by subcellular fractionation (Figure 1b). Prolonged detachment from ECM is required before loss of mitochondrial cytochrome-c and apoptotic nuclear morphology is observed (Figure 1a and b; Figure 6a). We found that if MECs that had been detached for 30 min were allowed to reattach to ECM, endogenous Bax was lost from the mitochondrial fraction (Figure 1c). These data, together with our previously published observations, suggest that after Bax translocation, a second signal is required before it activates and forms the MOMP-inducing pore.

Bax translocation to mitochondria precedes cytochrome-c release and cell death. (a) MECs transiently expressing YFP-Bax, either adherent or detached from ECM for 15 min or 4 h, were fixed and immunostained for YFP or cytochrome-c. Nuclei were stained using Hoechst dye. (b) MECs were left adherent or were detached from ECM for the indicated time periods before fixing, and apoptosis was quantified by examining nuclear morphology (n=3, error bars indicate standard error of the mean). Data were analysed by ANOVA, using Bonferroni's post-test. A significant increase in apoptosis was seen at 4 h (*). There was no significant increase at 1 h (n/s). Adherent cells and those detached for 15 min and 4 h were separated into cytosolic and mitochondrial membrane fractions. Fractions were immunoblotted for Bax. (c) MECs were left adherent, detached for 30 min, or detached and replated onto ECM for the indicated time periods. Cells were separated into cytosolic and membrane fractions, and immunoblotted for the indicated proteins. APAF-1 is a cytosolic marker. mtHsp70 is a mitochondrial marker
To identify signalling pathways regulating this second event, we examined known survival pathways previously linked to adhesion signalling. MECs were either left adherent or detached from ECM for various time periods, and phosphorylation of several signalling enzymes was examined by immunoblotting (Figure 2a). We have previously shown that focal adhesion kinase (FAK) regulates anoikis in MECs,14 and as expected its activation, judged by phosphorylation on tyrosine 397, is turned off within 15 min of cells detaching from ECM. Paxillin, a downstream substrate of FAK, becomes dephosphorylated within the same time period. When we looked at MAPK signalling, we observed that although ERK remained activated throughout, both JNK and p38MAPK became activated after detachment. Furthermore, their activation, as judged by phosphorylation on the threonine and tyrosine residues within the activation loop, increased significantly with prolonged detachment from ECM.

p38MAPK and JNK are activated during anoikis. (a) MECs were left adherent or were detached from ECM for the indicated time periods. Whole cell lysates were prepared and immunoblotted using the indicated phospho and total antibodies. Note that phosphorylation of FAK and paxillin are only seen in adherent cells, whereas that of JNK and p38MAPK increases with time of detachment. Phospho-ERK levels do not alter and active caspase 3 is observed at later time points. (b) MECs pre-treated with either a JNK inhibitor (SP600125) or a p38MAPK inhibitor (SB203580) were detached from ECM for the indicated time periods. Untreated cells were given DMSO alone. Apoptosis was quantified by nuclear morphology. The data represent the mean of three experiments. Bars indicate standard error of the mean. (c) Whole cell lysates of adherent or detached MECs, treated with either SB203580 (+) or DMSO (−), were immunoblotted for active caspase 3 and actin. Detached cells were harvested after 5 h. Cells treated with staurosporine for 5 h were used as a control for active caspase 3. (d) MECs pre-treated with either DMSO (−) or SB203580 (+) were treated with anisomycin to activate p38MAPK. Phosphorylation of Hsp27 was determined by immunoblotting. SB203580 completely abolished anisomycin-induced Hsp27 phosphorylation
To test the requirement for p38MAPK and JNK activity for MEC anoikis, we pre-incubated cells with either JNK (SP600125) or p38MAPK (SB203580) inhibitors before detaching them from ECM for the indicated time periods (Figure 2b). Apoptosis, as judged by nuclear morphology, was unaffected by inhibition of JNK compared with untreated cells. In contrast, SB203580 inhibited anoikis significantly over the time course investigated. To confirm this, we immunoblotted cell lysates from adherent or detached cells treated with SB203580 or DMSO for active caspase 3 (Figure 2c). No active caspase 3 was detected in adherent cells. In detached cells, the amount of active caspase 3 was noticeably reduced when p38MAPK was inhibited. To confirm the effectiveness of SB203580, MECs were treated with anisomycin, a potent activator of p38MAPK with or without pre-incubation with SB203580. Lysates were then prepared and immunoblotted for phospho-Hsp27 (Figure 2d). SB203580 completely inhibited anisomycin-induced Hsp27 phosphorylation. We asked whether p38MAPK activity was required for MECs’ apoptosis in response to other stimuli. MECs were treated with anisomycin, the EGF receptor inhibitor, Iressa or UV irradiation, in the absence or presence of SB203580. Apoptosis was observed by immunoblotting for activated caspase 3 (Supplemental Figure 1). SB203580 inhibited caspase activation in response to all three stimuli.
We tested two siRNA duplexes targeted to p38MAPKα. MECs were transfected with each p38MAPKα siRNA or a control duplex, and immunoblotted using either a total p38MAPK antibody or the one specific for p38MAPKα (Figure 3a). One duplex (siRNA p38α-13) reduced expression of p38MAPKα by 70%. The reduction in total p38MAPK was around 50%, suggesting that another isoform, probably p38MAPKβ, was also expressed. We were unable to directly test expression of p38MAPKβ, as we could not find an antibody that detected the endogenous protein (data not shown). When we examined detachment-induced p38MAPK phosphorylation, this was almost completely abolished in MECs transfected with siRNA p38α-13, but not with the control siRNA (Figure 3b). Detachment-induced phosphorylation of Hsp27 was also reduced. We examined apoptosis by immunoblotting siRNA-transfected cells, detached for various time periods, using anti-active caspase 3 (Figure 3c). siRNA p38α-13 inhibited detachment-induced activation of caspase 3, confirming the requirement for p38MAPK in MEC anoikis. siRNA knockdown of p38MAPK did not affect the adhesion-dependent phosphorylation of FAK. However, it did reduce the small amount of active caspase 3 seen in adherent cells, suggesting that activation of p38MAPK is also controlling the basal level of apoptosis in these cells.

p38MAPK siRNA inhibits anoikis. (a) MECs were transfected with siRNA duplexes (either control or one of two p38MAPK-targeted duplexes). After 72 h, lysates were prepared and quantitative immunoblotting performed using antibodies against either total p38MAPK or p38MAPKα and IR-dye-conjugated secondary antibodies. Triplicate results are shown, along with the loading control (calnexin) against which p38MAPK readings were normalised. Mean for each duplex are quantified and shown in the histograms. Error bars indicate standard error of the mean. (b) MECs transfected with either p38α-13 or a control duplex were grown for 72 h. MECs were then either left adherent or detached for the indicated time periods. WCL were prepared and immunoblotted for phospho-p38MAPK, phospho-Hsp27 and total Hsp27.(c) MECs transfected as in (b) were detached for up to 8 h and WCL immunoblotted for phospho-p38MAPK, active caspase 3 and phospho-FAK Y397
p38MAPK associates with mitochondria during anoikis
We compared the phosphorylation of p38MAPK and known downstream effectors after detachment from ECM or treatment with anisomycin (Figure 4a). Anisomycin resulted in more robust phosphorylation of p38MAPK than that seen after detachment, but phosphorylation of Hsp27 was observed after either treatment. Interestingly, phosphorylation of the transcription factor ATF2 occurred in response to anisomycin, but not after detachment from ECM. Phosphorylation of a restricted set of substrates suggested that p38MAPK might be recruited to a specific subcellular compartment after loss of ECM attachment. To determine whether this was the case, we isolated cytosolic and mitochondrial fractions from MECs that were either left adherent, detached from ECM for 60 min, or detached for 60 min and then replated onto ECM (Figure 4b). As previously shown, detachment from ECM resulted in Bax associating with the mitochondrial fraction. Reattachment to ECM resulted in loss of Bax from the mitochondria. Detachment also brought about an accumulation of p38MAPK in the mitochondrial fraction, and this was also reversed if MECs were allowed to reattach to ECM. Interestingly, phosphorylated p38MAPK was exclusively present in the membrane fraction. To confirm mitochondrial localisation, we coexpressed in MECs a V5-epitope-tagged p38MAPK along with a mitochondrial marker (Supplemental Figure 2). After detachment from ECM, we observed co-localisation of V5-tagged p38MAPK with mitochondria.

p38MAPK is recruited to the mitochondrial membrane fraction during anoikis. (a) MECs were either left adherent and untreated, detached from ECM or treated with anisomycin. Whole cell lysates were prepared and immunoblotted using the indicated antibodies. p38MAPK and Hsp27 are phosphorylated by both detachment and anisomycin, whereas phospho-ATF2 is only seen after anisomycin treatment. (b) MECs were left adherent, detached for 60 min, or detached and replated. Cytosolic and membrane fractions were prepared and immunoblotted for phospho- and total p38MAPK, Bax, and cytosolic and membrane markers (APAF-1 and mtHsp70, respectively). (c) MECs transiently transfected with expression vectors for MKK6Ala or MKK6Glu, or with empty vector, were left adherent or detached for 60 min. Cytosolic and membrane fractions were prepared and immunoblotted for endogenous phospho-p38MAPK. APAF-1 and mtHsp70 were used as cytosolic and membrane fraction markers. (d) Mitochondrial membrane fractions were prepared from MECs detached for 60 min. These were extracted in BN-PAGE buffer, and separated in the first dimension by BN-PAGE and in the second dimension by conventional SDS-PAGE. Fractions were then immunoblotted for Bax and phospho-p38MAPK. The positions of BN-PAGE molecular weight markers (KLH, β-Amylase and BSA) are shown.
We asked whether activation of p38MAPK by upstream MKKs was sufficient to induce its translocation to mitochondria. Cells were transiently transfected with expression vectors for either an active MKK6 (MKK6Glu) or an inactive form (MKK6Ala). Cells were left adherent or detached for 30 min, and cytosolic and membrane fractions were prepared. Immunoblotting indicated that, although transient expression of MKK6Glu resulted in phosphorylation of endogenous p38MAPK, this was present in the cytosol of the adherent cells and was not recruited to the membrane fraction (Figure 4c; compare lanes 5 and 11). Even in the presence of MKK6Glu, detachment from ECM was required for the association of phosphorylated p38MAPK with the mitochondrial fraction (Figure 4c; compare lanes 6 and 12). Identical results were observed with an active MKK3 (data not shown). Thus, ECM-withdrawal was necessary for translocation of p38MAPK to mitochondria.
As MAPKs can be recruited to specific subcellular compartments through scaffold proteins, we asked whether p38MAPK was associated with other molecules in the mitochondrial fraction in detached cells. We have recently shown, using BN-PAGE, that a number of Bcl-2 family proteins are recruited into high molecular weight complexes on mitochondria.13 We used BN-PAGE to examine the native molecular weight of activated p38MAPK in detached cells. Mitochondrial fractions were prepared from MECs after detachment from ECM for 1 h. These were then separated by 2D BN-PAGE and immunoblotted for phospho-p38MAPK (Figure 4d). Interestingly, phosphorylated p38MAPK was exclusively present in a high molecular weight complex of approximately 200kDa, similar to that previously shown for Bax. These data indicate that detachment from ECM induces p38MAPK to associate with protein complexes on mitochondria.
p38MAPK activity is required for MOMP
As p38MAPK was recruited to the mitochondrial fraction concomitant with Bax, we asked whether it regulated Bax translocation. MECs transiently expressing YFP-Bax were left adherent or detached from ECM for 15 min, either with or without pre-treatment with SB203580. Inhibiting p38MAPK had no effect on the detachment-induced co-localisation of YFP-Bax and on the mitochondrial heat shock protein 70 (mtHsp70) (Figure 5a). We examined translocation of endogenous Bax in detached MECs after p38MAPK inhibition by subcellular fractionation (Figure 5b). Treatment with SB203580 resulted in a small amount of phospho-p38MAPK being detected in the cytosol. However, there was no effect on the recruitment of Bax to the membrane fraction. We also examined the distribution of Bid and Bcl-XL (Figure 5c). Inhibiting p38MAPK had no effect on the redistribution of Bid after detachment. Similarly, there was no alteration in the membrane association of Bcl-XL.

p38MAPK activity during anoikis is not required for translocation of Bax to mitochondria. (a) MECs transiently expressing YFP-Bax were pre-treated with either DMSO (−) or SB203580 (+). Cells were either left adherent or detached for 15 min, fixed in 4% formaldehyde and immunostained for YFP and mtHsp70. Nuclei were stained using Hoechst dye. Note that YFP-Bax is cytosolic in the adherent cells, but has a punctate distribution identical to mtHsp70 in the detached cells. This is not altered by treatment with SB203580. (b) MECs pre-treated with either DMSO (−) or SB203580 (+) were left adherent or detached for 60 min. Cytosolic and membrane fractions were prepared and immunoblotted for phospho-p38MAPK, total p38MAPK, Bax, cytosolic (APAF-1) and mitochondrial (mtHsp70) markers. Note that there is no difference in Bax redistribution in SB203580-treated cells. (c) MECs pre-treated with either DMSO (−) or SB203580 (+) were left adherent or detached for 60 min. Cytosolic and membrane fractions were prepared and immunoblotted for Bid, Bcl-XL, APAF-1 and mtHsp70
We have recently shown that the Bax activation step associated with MOMP is a distinct regulatory step from its translocation.11 As p38MAPK did not regulate Bax translocation, we asked whether it had a role in its subsequent activation. MECs treated with either DMSO or SB203580 were detached from ECM for various time periods. Cells were fixed and immunostained for cytochrome-c, and the number of cells having undergone MOMP was quantified by counting the percentage of cells without mitochondrial cytochrome-c (Figure 6a). As previously shown, MECs required prolonged detachment from ECM before undergoing MOMP. However, the increase in the number of cells showing release of cytochrome-c after 6 h detachment was inhibited by SB203580.

p38MAPK activity is required for Bax-induced MOMP during anoikis. (a) Untransfected MECs pre-treated with either DMSO (−) or SB203580 (+) were left adherent or detached for various time periods. Cells were fixed in 4% formaldehyde and immunostained for cytochrome-c. The percentage of cells showing diffuse cytochrome-c was quantified. (b) As in (a), except that cells were transiently expressing YFP-Bax. Only YFP-Bax-positive cells were counted. (c) As in (b), except that cells were transiently expressing YFP-BaxP13A. For (a–c), the data represent the mean of three independent experiments. Error bars show standard error of the mean. Data were analysed by two-way ANOVA using Bonferroni's post-test. * Indicates a significant difference between DMSO and SB203580-treated cells at the same time point. n/s indicates not significant. (d) MECs transiently expressing YFP-Bax were left adherent or detached from ECM for 4 h, in the presence of either DMSO or SB203580, as indicated. Cells were fixed and immunostained with anti-Bax N20. Nuclei were stained using Hoechst dye. All images were taken at the same exposure and subjected to identical processing. (e) Mitochondrial membrane fractions were prepared from MECs detached for 60 min in the presence of either DMSO or SB203580. These were extracted in BN-PAGE buffer, and separated in the first dimension by BN-PAGE and in second dimension by conventional SDS-PAGE. Fractions were immunoblotted for Bax. The position of BN-PAGE molecular weight markers are shown
The kinetics of progression to MOMP can be increased by overexpressing YFP-Bax in MECs (Figure 6b). In cells overexpressing YFP-Bax, a significant increase in MOMP was observed after 4 h detachment. However, MOMP in YFP-Bax overexpressing cells was also inhibited by SB203580. We have recently shown that mutating proline 13 in Bax to alanine results in a protein that seems to drive cells through MOMP without any delay after loss of adhesion. We expressed YFP-BaxP13A in MECs and treated them with SB203580. Surprisingly, we found that even this more readily activated form of Bax required p38MAPK activity to induce MOMP (Figure 6c).
Bax activation is associated with the exposure of cryptic N-terminal epitopes.15 We have shown that, during anoikis, exposure of these epitopes occurs not when Bax initially translocates to the OMM but during MOMP.11 MECs transiently expressing YFP-Bax were left adherent or detached from ECM in the presence or absence of SB203580. Cells were then immunostained for exposure of the Bax N-terminal epitope (Figure 6d). In adherent cells, YFP-Bax was diffuse and did not show epitope exposure. In agreement with our previous results, after detachment in the absence of SB203580, cells that showed apoptotic nuclei stained positively for anti-Bax N20. However, cells that did not show apoptotic morphology did not stain with anti-Bax N20, even though Bax had a punctate, mitochondrial distribution. Cells detached in the presence of SB203580 showed mitochondrial Bax, but did not show N-terminal epitope exposure or apoptotic nuclei. We also asked whether p38MAPK was required for the formation of the high molecular weight Bax complexes. Detached cells, either untreated or treated with SB203580, were analysed by 2D BN-PAGE and immunoblotted for Bax (Figure 6e). Interestingly, the membrane-associated 200-kDa complexes of Bax were unaffected by inhibition of p38MAPK, suggesting that these form before Bax activation and N-terminal epitope exposure, in agreement with our previous data.13
Together, these results suggest that Bax activation after its translocation to the OMM requires p38MAPK activity.
p38MAPK association with mitochondria drives MEC commitment to apoptosis
We wanted to determine whether p38MAPK recruitment to mitochondria drove MECs through to the commitment point of apoptosis. Overexpression of p38MAPK or activated MKK6 did not result in apoptosis (data not shown). As activation of p38MAPK was insufficient to drive its association with the mitochondrial fraction, we decided to target it constitutively to the OMM. We generated YFP-tagged forms of p38MAPK α and β isoforms onto which we had attached the tail anchor sequence of Bcl-XL (YFP-p38αXT and YFP-p38βXT). YFP-p38αXT and YFP-p38βXT colocalised with mtHsp70, indicating mitochondrial targeting (Figure7a and data not shown). Immuoblotting indicated that YFP-XT, YFP-p38αXT and YFP-p38βXT were expressed at similar levels, as was a non-targeted YFP-p38α (Figure 7b). In adherent cells, overexpression of YFP-p38αXT, YFP-p38βXT or YFP-p38α with monomeric red fluorescent protein (mRFP)-XT did not result in phosphorylation on ser180/tyr182. However, coexpression with mRFP-MKK6GluXT resulted in a significant increase in the phosphorylation of all three YFP-p38 constructs.

Mitochondrial targeting of p38MAPK increases the rate of anoikis and commitment to apoptosis. (a) MECs were transfected with the indicated YFP expression vectors for p38MAPKαXT or p38MAPKα. Cells were fixed and immunostained for mtHsp70. Nuclei were stained using Hoechst dye. (b) Cells were transfected with the indicated expression vectors for YFP-XT, YFP-p38αXT, YFP-p38βXT and YFP-p38α, along with either mRFP-MKK6GluXT or control mRFP-xT. Whole cell lysates were prepared and immunoblotted as indicated. (c) MECs transiently expressing YFP-p38MAPKαXT, YFP-p38MAPKβXT or YFP-XT were left adherent or detached from ECM for the indicated time periods. Cells were fixed and immunostained for active caspase 3. The number of YFP-expressing cells that were positive for active caspase 3 was quantified. Data are the mean of three independent experiments. Error bars show standard error of the mean. Data were analysed by two-way ANOVA using Bonferroni's post-test. Significant P-values are indicated at the 8-h time point. (d) Cell as in (c), but apoptosis was quantified based on nuclear morphology. Data are the mean of three independent experiments and were analysed as in (c). (e) MECs transiently expressing the indicated proteins were left adherent or detached from the ECM for 6 h. Cells were fixed and immunostained for active caspase 3. Apoptosis was scored by counting the percentage of YFP-expressing cells with active caspase-3. Data were analysed by two-way ANOVA using Bonferroni's post-test. Significant P-values are indicated at the 6-h time point. (f) MECs transiently expressing the indicated proteins were detached from ECM for 15 min before replating. Cells were allowed to reattach for 5 h, after which the ratio of transfected cells in the supernatant (det) to those that had reattached (att) was calculated. The higher ratio of det:att indicates that more number of cells had committed to apoptosis and could not be rescued by reattachment to ECM. *Indicates significance by ANOVA. n/s indicates not significant
We next asked whether expression of mitochondrial-targeted p38MAPK altered the kinetics of apoptosis after detachment from ECM. MECs expressing YFP-p38αXT, YFP-p38βXT or YFP-XT were either left adherent or detached for various time periods. Apoptosis was quantified by immunostaining for active caspase 3 or by examining nuclear morphology (Figure 7 c and d). There was no significant increase in the amount of apoptosis in adherent cells expressing YFP-p38αXT or YFP-p38βXT compared with that in YFP-XT. However, after detachment from ECM, both YFP-p38αXT and YFP-p38βXT increased the rate of apoptosis, the α isoform having the most significant effect. We asked whether the membrane targeting was important for this effect on anoikis by expressing either YFP-p38αXT or the non-targeted YFP-p38α. There was no increase in apoptosis seen with expression of either protein in adherent cells (Figure 7e). After detachment from ECM for 6 h expression, both YFP-p38XT and YFP-p38α increased the amount of apoptosis. However, this was significantly greater (P<0.001) in the OMM-targeted YFP-p38XT.
Finally, we asked whether p38MAPK activation on mitochondria regulated MEC commitment to apoptosis. We have previously described an assay to measure apoptosis commitment after MEC detachment from ECM, which uses the ability of cells to be rescued from anoikis if adhesion signals are restored before they commit to apoptosis.11 MECs were detached from ECM for 15 min and then allowed to reattach for 5 h, after which those cells that had committed to apoptosis would be found in the supernatant and those rescued would remain adherent. We transiently expressed in MECs YFP-XT, YFP-p38αXT or YFP-p38βXT, along with either mRFP-XT or mRFP-MKK6GluXT. After 5 h of reattachment, the ratio of transfected cells in the supernatant to those still adherent was determined (Figure 7f). Expression of either of the YFP-p38XT isoforms alone did not increase the number of cells that had committed to apoptosis after 15 min detachment, with the same ratio of detached to attached cells seen with expression of YFP-XT. Thus, although overexpressing mitochondrial-targeted p38MAPK increased the rate of anoikis in MECs, it alone was insufficient to cause commitment to apoptosis after just 15 min detachment from ECM. We had seen relocalisation of phosphorylated MKK3 or MKK6 to mitochondria (data not shown). We, therefore, speculated that to accelerate commitment, we would need to force the recruitment of not only p38MAPK but also the activating MKK as well. In support of this, coexpressing mRFP-MKK6GluXT along with either YFP-p38αXT or YFP-p38βXT significantly increased the number of MECs that were committed to apoptosis after just 15 min detachment. These data indicate that activation of p38MAPK on mitochondria can alter the kinetics of MECs’ commitment to apoptosis.
Discussion
The activation of the multi-domain pro-apoptotic Bcl-2 protein, Bax, is a critical event in the intrinsic pathway for cell death. The biochemical pathways that regulate Bax on mitochondria were recently described as ‘the Holy Grail’.4 In this paper, we show a role for p38MAPK in such a pathway during epithelial cell anoikis. In particular, we show that p38MAPK activity is essential for the Bax-dependent commitment of MECs to apoptosis. Loss of epithelial/ECM adhesion resulted in the recruitment of activated p38MAPK to the mitochondrial fraction. Detachment-induced p38MAPK activation was not required for Bax translocation to the OMM or its association with high molecular weight complexes. However, inhibiting p38MAPK activity protected cells from anoikis by preventing Bax-dependent MOMP. Mitochondrial p38MAPK was found to be associated with a high molecular weight complex, suggesting that it forms a regulatory scaffold during anoikis.
Bax activation involves a series of conformational changes, including reorientation of the C-terminal tail anchor, exposure of otherwise cryptic N-terminal antibody epitopes and its association with high molecular weight complexes.10, 15, 16, 17, 18, 19, 20 During this process, Bax translocates from the cytosol to mitochondria, wherein it ultimately forms pores in the OMM. We have previously shown that during anoikis, Bax activation occurs in distinct stages before MOMP; an initial, reversible translocation to the mitochondria is followed by irreversible activation, concomitant with cytochrome-c release.10, 11, 13 The second stage of activation, which occurs as the cryptic N-terminal epitopes become exposed, was found to equate with epithelial cell commitment to apoptosis. A number of other studies have also shown that Bax recruitment to the OMM does not necessarily mean that MOMP and cell death are inevitable.21, 22 Together, these data indicate that separate signalling events control Bax translocation and its subsequent activation into a cytochrome-c-releasing pore. Our present data indicate that the second step of this process involves p38MAPK.
p38MAPKs are activated in response to a wide variety of cellular stresses and can regulate apoptosis through phosphorylation of downstream targets.23 Many of the well-characterised p38MAPK targets are transcription factors. For example, activation of p38MAPK in response to DNA damage can induce p53-mediated upregulation of the pro-apoptotic BH3-only protein, Noxa.24 In intestinal epithelial cells, p38MAPK-dependent upregulation of Fas ligand occurs after detachment from ECM to promote anoikis.25 However, MEC anoikis does not require new gene expression.26 In the present study, we showed that ATF2, a transcription factor substrate of p38MAPK, was not activated in response to ECM-withdrawal, but was after treatment with anisomycin. Although we saw p38MAPK-dependent phosphorylation of Hsp27 after detachment from ECM, we do not yet know what are the relevant substrates for the regulation of anoikis. A number of studies have implicated p38MAPK as a direct regulator of Bax. In cardiomyocytes, p38MAPK activity has been shown to be required for mitochondrial translocation of Bax in response to ischaemia.27 Another study has suggested that p38MAPK and JNK can directly phosphorylate Bax in HepG2 cells following a variety of stresses, including UV irradiation and etoposide.28 This phosphorylation by p38MAPK was suggested to regulate Bax mitochondrial translocation. However, our data indicate that p38MAPK activity was not required for mitochondrial association of Bax after loss of ECM attachment, a step that is regulated by adhesion-dependent tyrosine kinase signalling (J Keeble and AP Gilmore, unpublished data). Rather, p38MAPK was required for Bax that is already associated with the OMM, and present in a high molecular weight complex, to activate into a cytochrome-c-releasing pore. We found that inhibiting p38MAPK prevented apoptosis in response to a number of stimuli and also reduced the small amount of caspase activation seen in untreated cells. It is noteworthy that mitochondrial localisation of p38MAPK was observed in a small proportion of adherent cells that were not treated with any apoptotic stimulus (supplemental Figure 2b). These data imply that p38MAPK may have a general role in apoptosis during normal cell turnover, as well as in response to specific stress stimuli. To date, phosphorylation of Bax by p38MAPK has not been detected in our system.
The members of the MAPK family respond to a wide range of stimuli, and yet it is well established that specific stimuli result in a restricted set of substrates and effectors becoming activated.29 Our data suggest that p38MAPK activation during anoikis results in the activation of distinct set of substrates compared with anisomycin treatment, and this may be because of a restricted subcellular localisation. Specificity of MAP kinase signalling can be regulated by scaffold proteins, not only through recruiting both substrates and upstream activators but also by restricting MAPK localisation to particular subcellular compartments. Multiple scaffolds have been identified for ERK (for example, β-Arrestins, kinase suppressor of Ras, MEKK 1) and JNK (for example, filamin and JNK-interacting proteins (JIPs) 1–4).30, 31 However, there are relatively few known p38MAPK scaffolds, with JIP2 and JIP4 reported to bind to and possibly regulate localisation of p38MAPK activity.32, 33, 34 Osmosensing scaffold for MEKK3 (OSM) has also been shown to act as a p38MAPK scaffold.35 OSM responds to osmotic stress by recruiting p38MAPK and Rac to membrane ruffles, wherein they co-operate to reorganise the actin cytoskeleton. In a similar way, our data showed that p38MAPK activated during anoikis was recruited into a mitochondrial membrane-associated, high molecular weight complex. This recruitment has a role in the commitment of MECs to apoptosis, as shown when we bypassed the normal recruitment of p38MAPK by targeting it to mitochondria using the tail anchor sequence of Bcl-XL. Similar use of artificial scaffolds has been previously shown to direct MAPK signalling in yeast.36, 37 To date, no MAPK scaffold proteins have been characterised that direct signalling to mitochondria.30 Thus, the membrane-associated complex that we observe in detached cells may contain novel p38MAPK-interacting proteins that direct a specific stress response to control apoptosis commitment.
Resistance to anoikis is an important factor in cancer metastasis.8 We have identified a novel pathway of Bax regulation involving mitochondrial recruitment of p38MAPK. Together, our data suggest that p38MAPK acts as part of a novel mitochondrial complex to regulate Bax activation during anoikis. It will now be important to identify components of this complex.
Materials and Methods
Reagents and antibodies
Acrylamide and sodium dodecyl sulphate were obtained from Bio-Rad. β-mercaptoethanol, dithiothreitol, Tricine, Bis/Tris, aminocaproic acid, CHAPS, n-octylglucoside, Coomassie brilliant blue G-250 and poly-2-hydroxyethyl methacrylate (poly-HEMA) were obtained from Sigma. Triton X-100 was purchased from BDH. Protease Inhibitor Cocktail (539131), staurosporine, anisomycin, p38 inhibitor (SB203580) and JNK inhibitor (SP600125) were obtained from Calbiochem.
The following antibodies were used: Bax monoclonal 5B7 (B9054) and Actin purchased from Sigma; anti-BaxN20 (06-499) obtained from Upstate Biotechnology; polyclonal BaxΔ21 (sc-6236), p38β (E20) and ERK (C-14) purchased from Santa Cruz; Bid (AF860) and cleaved caspase-3 (AF835) obtained from R&D Systems; Apaf-I (AAP-300) purchased from Bioquote; mitochondrial Hsp70 (MA3-028) obtained from Affinity Bioreagents; phospho-JNK (T183Y185; 44-682G) and phospho-FAK (Y397; 44-624G) purchased from Biosource; phospho-paxillin (Y31; 44-720G) obtained from Invitrogen; phospho-ERK (T202Y204; 9101), phospho-p38 (T180Y182; 9913), phospho-HSP27 (S82; 2401), phospho-ATF2 (T71; 9221), total p38 (9212), p38α (9218), JNK (9252) and active caspase 3 (9661) purchased from Cell Signaling Technology; and paxillin (clone 349) and cytochrome-c (clone 6H2.B4) obtained from BD Biosciences. The total-FAK antibody was a gift from Andy Ziemiecki, University of Berne, Switzerland and the Tubulin antibody was a gift from Keith Gull, University of Manchester, UK.
The following vectors and cDNAs were used: the yellow fluorescent protein (YFP) and YFP-Bax constructs have been described previously (Upton et al 11). Expression constructs encoding p38α, p38β, MAP kinase kinase 3(MKK3) and MKK6 were kindly provided by Dr Alan Whitmarsh, University of Manchester, UK. Mitochondrial targeting of proteins was achieved by creating fusion constructs using the Bcl-XL C-terminal membrane anchor sequence (XT). siRNA oligos against p38MAPKα were obtained from Ambion.
Cell culture and transfection
Fsk-7 cells, a mouse MEC line, were cultured as previously described.12 Expression vectors were transfected into FSK-7 using Lipofectamine and Plus reagent, as previously described.10 Cells were treated with 10 μM staurosporine, 10 μM anisomycin, 10 μM JNK inhibitor (SP600125) or 1.0 μM p38 inhibitor (SB203580); control cells were treated with vehicle (DMSO) alone. For anoikis studies, cells were plated onto the non-adherent substratum poly-HEMA; control cells were plated onto glass coverslips. siRNA transfections were performed using Lipofectamine 2000. Quantitative immunoblotting was performed using the Li-Cor Odyssey, as previously described.13
Cell Fractionation and blue native polyacrylamide gel electrophoresis (BN-PAGE)
Adherent and detached Fsk-7 cells were subfractionated into a cytosolic and membrane fractions, as described.10 Membrane pellets were extracted in 1% CHAPS, 10% glycerol, 0.5 M aminocaproic acid in 50 mM Bis/Tris, pH 7.0, containing protease inhibitors and left on ice for 10 min before centrifuging at 1 00 000 × g for 30 min. Two-dimensional (2D) blue native electrophoresis was performed, as previously described.13 Immunodetection was performed by SuperSignal chemiluminesence (Pierce).
Immunofluorescence and imaging
For imaging, cells on coverslips were fixed using 4% formaldehyde in PBS and permeabilised using 0.5% Triton X-100/PBS. Non-adherent cells were cytospun onto polysine-coated slides before fixation and permeabilisation, as described above. Cells were immunostained using primary antibodies followed by fluorophore-conjugated goat anti-IgG secondary antibodies (Molecular Probes). Nuclei were stained using 1 μg/ml Hoechst 33258 dye. Images were collected on an Olympus IX70 microscope, equipped with a Deltavision imaging system, using a 100 × PLAN-APO 1.4NA objective. Images were processed by constrained iterative deconvolution on softWoRxTM software (Applied Precision).
For quantification of cell phenotypes, cells were visualised on an Axioplan 2 imaging microscope using a 63 × 1.4NA Plan Apochromat objective (Zeiss MicroImaging). For transfection experiments, only the transfected cells were counted. A minimum of 300 cells were counted per condition.
Abbreviations
- BN-PAGE:
-
blue native polyacrylamide gel electrophoresis
- ECM:
-
extracellular matrix
- FAK:
-
focal adhesion kinase
- MAPK:
-
mitogen-activated protein kinase
- MEC:
-
mammary epithelial cell
- MKK:
-
MAP kinase kinase
- MOMP:
-
mitochondrial outer membrane permeabilisation
- mRFP:
-
monomeric red fluorescent protein
- mtHsp70:
-
mitochondrial heat shock protein 70
- OMM:
-
outer mitochondrial membrane
- poly-HEMA:
-
poly-2-hydroxyethyl methacrylate
- SMAC:
-
second mitochondrial activator of caspases
- YFP:
-
yellow fluorescent protein
References
Martinou JC, Green DR . Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2001; 2: 63–67.
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR . The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2000; 2: 156–162.
Rehm M, Dussmann H, Prehn JH . Real-time single cell analysis of Smac/DIABLO release during apoptosis. J Cell Biol 2003; 162: 1031–1043.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Bouillet P, Strasser A . BH3-only proteins—evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J Cell Sci 2002; 115: 1567–1574.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Gilmore AP . Anoikis. Cell Death Differ 2005; 12: 1473–1477.
Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS . Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 2004; 430: 1034–1039.
Gilmore AP, Metcalfe AM, Romer LH, Streuli CH . Integrin-mediated survival signals regulate the apoptotic function of Bax through its conformation and subcellular localization. J Cell Biol 2000; 149: 431–445.
Valentijn AJ, Metcalfe AD, Kott J, Streuli CH, Gilmore AP . Spatial and temporal changes in Bax subcellular localization during anoikis. J Cell Biol 2003; 162: 599–612.
Upton JP, Valentijn AJ, Zhang L, Gilmore AP . The N-terminal conformation of Bax regulates cell commitment to apoptosis. Cell Death Differ 2007; 14: 932–942.
Kittrell FS, Oborn CJ, Medina D . Development of mammary preneoplasias invivo from mouse mammary epithelial-cell lines invitro. Cancer Res 1992; 52: 1924–1932.
Valentijn AJ, Upton JP, Gilmore AP . Analysis of endogenous Bax complexes during apoptosis using blue native PAGE: implications for Bax activation and oligomerization. Biochem J 2008; 412: 347–357.
Zouq NK, Keeble JA, Lindsay J, Valentijn AJ, Zhang L, Mills D et al. FAK engages multiple pathways to maintain survival of fibroblasts and epithelia—differential roles for paxillin and p130Cas. J Cell Sci 2009; 122: 357–367 Epub 2009 Jan 6.
Hsu YT, Youle RJ . Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem 1998; 273: 10777–10783.
Antonsson B, Montessuit S, Sanchez B, Martinou JC . Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem 2001; 276: 11615–11623.
Desagher S, Osen Sand A, Nichols A, Eskes R, Montessuit S, Lauper S et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 1999; 144: 891–901.
Eskes R, Desagher S, Antonsson B, Martinou JC . Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 2000; 20: 929–935.
Hsu YT, Wolter KG, Youle RJ . Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA 1997; 94: 3668–3672.
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ . Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 1997; 139: 1281–1292.
Nechushtan A, Smith CL, Hsu YT, Youle RJ . Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 1999; 18: 2330–2341.
Soucie EL, Annis MG, Sedivy J, Filmus J, Leber B, Andrews DW et al. Myc potentiates apoptosis by stimulating Bax activity at the mitochondria. Mol Cell Biol 2001; 21: 4725–4736.
Wada T, Penninger JM . Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004; 23: 2838–2849.
Cuadrado A, Lafarga V, Cheung PC, Dolado I, Llanos S, Cohen P et al. A new p38 MAP kinase-regulated transcriptional coactivator that stimulates p53-dependent apoptosis. EMBO J 2007; 26: 2115–2126.
Rosen K, Shi W, Calabretta B, Filmus J . Cell detachment triggers p38 mitogen-activated protein kinase-dependent overexpression of Fas ligand. A novel mechanism of Anoikis of intestinal epithelial cells. J Biol Chem 2002; 277: 46123–46130.
Wang P, Valentijn AJ, Gilmore AP, Streuli CH . Early events in the anoikis program occur in the absence of caspase activation. J Biol Chem 2003; 278: 19917–19925.
Capano M, Crompton M . Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J 2006; 395: 57–64.
Kim BJ, Ryu SW, Song BJ . JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem 2006; 281: 21256–21265.
Raman M, Chen W, Cobb MH . Differential regulation and properties of MAPKs. Oncogene 2007; 26: 3100–3112.
Morrison DK, Davis RJ . Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol 2003; 19: 91–118.
Whitmarsh AJ . The JIP family of MAPK scaffold proteins. Biochem Soc Trans 2006; 34: 828–832.
Buchsbaum RJ, Connolly BA, Feig LA . Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol Cell Biol 2002; 22: 4073–4085.
Kelkar N, Standen CL, Davis RJ . Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol Cell Biol 2005; 25: 2733–2743.
Schoorlemmer J, Goldfarb M . Fibroblast growth factor homologous factors and the islet brain-2 scaffold protein regulate activation of a stress-activated protein kinase. J Biol Chem 2002; 277: 49111–49119.
Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE et al. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol 2003; 5: 1104–1110.
Harris K, Lamson RE, Nelson B, Hughes TR, Marton MJ, Roberts CJ et al. Role of scaffolds in MAP kinase pathway specificity revealed by custom design of pathway-dedicated signaling proteins. Curr Biol 2001; 11: 1815–1824.
Park SH, Zarrinpar A, Lim WA . Rewiring MAP kinase pathways using alternative scaffold assembly mechanisms. Science 2003; 299: 1061–1064.
Acknowledgements
We thank Alan Whitmarsh and Cathy Tournier for their comments on the manuscript. This work was supported by Cancer Research UK, The Wellcome Trust and Breast Cancer Campaign.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by L Scorrano
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Rights and permissions
About this article
Cite this article
Owens, T., Valentijn, A., Upton, JP. et al. Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ 16, 1551–1562 (2009). https://doi.org/10.1038/cdd.2009.102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.102
Keywords
This article is cited by
-
BAX activation in mouse retinal ganglion cells occurs in two temporally and mechanistically distinct steps
Molecular Neurodegeneration (2023)
-
Anoikis resistance––protagonists of breast cancer cells survive and metastasize after ECM detachment
Cell Communication and Signaling (2023)
-
Osmoregulation, physiological metabolism, and oxidative stress responses to water salinity in adult males of Chinese mitten crabs (Eriocheir sinensis)
Aquaculture International (2023)
-
Non-canonical phosphorylation of Bmf by p38 MAPK promotes its apoptotic activity in anoikis
Cell Death & Differentiation (2022)
-
Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression
Nature Reviews Cancer (2014)
