
Abstract
Granzyme F (GzmF) belongs to a unique group of granzymes in mice. Murine GzmF is highly expressed in NK3.1 cells and in lymphokine-activated killer (LAK) cells. However, the manner in which GzmF works in granule-mediated cytolysis is unknown. In this study, we first demonstrated that GzmF causes a novel cell death pathway. The death is characterized by an externalization of phosphatidylserine, by nuclear condensation, mitochondrial damage, cytochrome c (cyt c) release, caspase inactivation and single-stranded DNA nicking. GzmF-induced chromatin was incompletely condensed and segmented at the nuclear periphery. Cellular organelles were damaged and the cytoplasm showed an extensive vacuolization that is reminiscent of necroptosis. GzmF can cause rapid mitochondrial swelling, depolarization and reactive oxygen species accumulation. GzmF-induced death does not involve caspase activation, Bid cleavage or activation of DNA nickase NM23H1. GzmF-silenced LAK cells showed reduced cytotoxicity against caspase-inhibited target tumor cells. Moreover, cyt c release is independent of Bid or Bax/Bak. We further showed that GzmF impairs mitochondrial electron transport to abolish ATP generation. ATP decline may contribute to a failure of apoptosome formation, leading to caspase inactivation.
Similar content being viewed by others


Main
Perforin (PFP)/Granzymes (Gzms) of cytotoxic lymphocytes have a major role in the clearance of tumor cells and virus-infected target cells.1, 2 A large number of Gzms have been identified in mice (A, B, C, D, E, F, G, K, M and N), but only GzmA and GzmB have been extensively studied.3 GzmB is an Aspase that cleaves after Asp residue of physiological substrates. It activates several caspases and directly cleaves some death substrates, such as the inhibitor of caspase-activated DNase (ICAD), to mediate classical apoptosis with double-stranded DNA fragmentation.4 GzmB can process Bid to truncated Bid (tBid) that translocates into the mitochondria to cause cytochrome c (cyt c) release, which leads to apoptosome assembly and caspase cascade activation.5 GzmA is a tryptase that cleaves its specific substrates after basic residues Arg or Lys. GzmA mediates single-stranded DNA nicking and caspase-independent death through the destruction of an endoplasmic reticulum-associated SET complex.6, 7 Murine GzmC was reported to trigger a caspase-independent death program that differs from GzmA or GzmB.8 Recently, several groups have demonstrated that human GzmK, GzmH and GzmM mediate non-redundant death pathways.9, 10, 11, 12, 13
Natural murine GzmF was first isolated from CTL granules by Masson and Tschopp in 1987 and the gene was cloned by Jenne et al.14, 15 Murine GzmF belongs to the third group (group 3) of Gzms, including GzmD, E, F, G, L and N.3, 16 Members of group 3 Gzms show high nucleotide similarity and are physically clustered with GzmB and GzmC genes at chromosome 14 of mice. However, the role of GzmF in granule-mediated cell death has not been defined.
Granzyme F is highly expressed in NK3.1 cells and in lymphokine-activated killer (LAK) cells derived from splenocytes.17 A recent study showed that GzmB locus knockout mice (GzmB−/−/+PGK-neo) with a significant reduction of GzmF and GzmC expression displayed a more severe defect in killing tumor cells than did mice deficient in GzmB only.18 These findings suggest that GzmF and GzmC may have important roles in the cytotoxic functions of killer lymphocytes. In this study, we demonstrated that GzmF causes a novel cell death pathway. The death is characterized by phosphatidylserine (PS) externalization, nuclear condensation, mitochondrial damage, cyt c release, caspase inactivation and single-stranded DNA nicking. Moreover, cyt c release is independent of Bid or Bax/Bak. We further showed that GzmF impairs mitochondrial electron transport to abolish ATP generation. Intracellular ATP decline may contribute to a failure of apoptosome formation, leading to caspase inactivation.
Results
GzmF induces cell death
Murine GzmF cDNA was cloned from a mouse thymus and subcloned into pET26b. Recombinant GzmF was expressed in E. coli and refolded from inclusion bodies as previously described.19 The inactive counterpart, Ser195AlaGzmF (S-AGzmF), was generated by a point mutation of the active site, Ser 195, to Ala. The Gzms were purified with nickel chelation columns, followed by cation exchange chromatography. Proteins were verified by mass spectrometry (data not shown) and purity (>95%) was monitored by Coomassie brilliant blue staining (Figure 1a left panel).

GzmF induces target cell death. (a) Production of recombinant GzmF and enzymatic assay. Recombinant GzmF and point-mutated S-AGzmF were expressed and refolded in inclusion bodies and activated by enterokinase treatment. Samples were separated by 12% SDS-PAGE and visualized by Coomassie brilliant blue staining (left panel). proGzmF, GzmF with enterokinase cleavage sequence. Recombinant GzmF (50 nM) showed enzymatic activity toward the synthetic substrate (left panel). Suc-Phe-Leu-Phe-pNA (FLF-pNA). GzmF had no activity toward PTSY-pNA (substrate of GzmH), AAF-pNA (substrate of tripeptidyl peptidase I and II), AAPF-pNA (substrate of cathepsin G) and AAPL-pNA (substrate of GzmM). These data are representative of three independent experiments. (b) Yac-1 cells were preincubated with Na251CrO4 at 37°C for 1 h. 51Cr-labeled cells were treated with 0.5 μM GzmF, S-AGzmF, GzmC and GzmB plus Ad at 37°C for 6 h. Released 51Cr was counted by Microbeta counter (PerkinElmer) and presented as means±S.D. Cells were mock-treated or treated with Ad and 0.5 μM GzmF alone as negative controls. These data represent three separate experiments. (c) GzmF causes phosphatidylserine externalization. Yac-1 cells (2 × 105) were incubated with Ad, 1 μM GzmF alone, 1 μM S-AGzmF plus Ad, 1 μM GzmB or GzmC plus Ad and with increased concentrations of GzmF plus Ad at 37°C for 6 h. Cells were stained with FITC-conjugated Annexin V (AV) and PI for flow cytometry assay. These data are representative of four separate experiments. Staurosporine (STP) was used as a positive control. (d) Percentages of AV single-positive versus AV/PI double-positive cells. The black bar and the gray bar represent AV single-positive and AV/PI double-positive cells, respectively. These data were calculated from three experiments shown as means±S.D.
A previous report showed that native GzmF had activity toward Suc-Phe-Leu-Phe-Sbzl.20 Recombinant GzmF was able to cleave the synthetic substrate, Suc-Phe-Leu-Phe–pNA, and showed strong enzymatic activity (Figure 1a right panel), which was in agreement with the previous report. The enzymatic inactive mutant GzmF (S-AGzmF) had no activity toward Suc-Phe-Leu-Phe-pNA. We also utilized a set of synthetic substrates to define the catalytic specificity of recombinant GzmF. GzmF had no activity against other synthetic substrates, including AAF-pNA (substrate for tripeptidylpeptidase I and II), AAPF-pNA (substrate for cathepsin G), PTSY-pNA (substrate for GzmH), AAPL-pNA (substrate for GzmM), IETD-AFC (substrate for GzmB), Phe-Arg-AMC (substrate for cathepsin L) and Z-Arg-Arg-AMC (substrate for cathepsin B), as well as against a series of caspase substrates, VDVAD-AFC, DEVD-AFC and LEHD-AFC (Figure 1a right panel, data not shown).
To determine the function of GzmF, Gzm delivery agent replication-deficient adenovirus type V (Ad) was used to deliver GzmF into target tumor cells. The adenovirus-mediated delivery of Gzms was developed by Froelich laboratory and this system proved useful for loading Gzms into target tumor cells.9, 21, 22 GzmF was loaded into 51Cr-labeled Yac-1 cells with an optimal dose of Ad for a 6 h-specific cytotoxicity assay. We found that GzmF caused dramatic cytolysis (56.9±9.9%) (Figure 1b). GzmB/Ad- or GzmC/Ad-treated cells were used as positive controls (GzmB, 58.3±3.7%; GzmC, 42.6±14.3%). In contrast, cells treated with Ad and GzmF alone, or with S-AGzmF/Ad, displayed only background levels of 51Cr release. It indicates that GzmF was capable of inducing target cell death and its enzymatic activity is required. These data are representative of at least three separate experiments.
To further characterize the death features of GzmF-induced death, PS externalization was detected by staining with Fluos-coupled Annexin V (AV) and propidium iodide (PI). PS externalization is an early hallmark of apoptotic cells. At a low concentration of 0.25 μM, GzmF was able to cause PS externalization of target Yac-1 cells (46.5%) (Figure 1c). PS externalization reached a peak level at a concentration of 0.7 μM (0.5 μM, 54.3%; 0.7 μM, 66.1%; 0.85 μM, 64.6%; 1.0 μM, 66.8%). Late-stage dead cells (AV+/PI+) kinetically augmented with increasing concentrations of GzmF (0.25 μM, 23.8%; 0.5 μM, 34.1%; 0.7 μM, 46.1%; 0.85 μM, 50.1%; 1.0 μM, 58.4%). Strikingly, at a concentration of 1.0 μM, the number of single AV+ cells was much less than that of AV+/PI+ (8.4 versus 58.4%). These observations are reminiscent of GzmC (18.1 versus 60%). Staurosporine (STP)-treated cells were used as typical apoptosis control. In contrast, GzmB induced much higher single AV+ cells than did AV+/PI+ (58.2 versus 27.7%), which is one of the classical apoptotic features (Figure 1c, d). Mock-treated, GzmF and Ad alone, or S-AGzmF/Ad-treated cells showed only background death (AV+/PI+ <8%). These data represent at least three independent experiments. Similar results were obtained using human Jurkat cells (data not shown). These data suggest that GzmF induces a novel cell death pathway that is distinct from typical apoptosis.
Knockdown of GzmF expression by RNAi reduces LAK cell-mediated caspase-independent cytotoxicity
To determine the patterns of Gzms expression during LAK cell activation, we assessed mRNA levels of Gzms at different times using quantitative RT-PCR. As shown in Figure 2a, GzmB mRNA contents increased with IL-2 stimulation and reached a peak level after day 5. However, GzmF mRNA contents reached its peak level at day 8. These observations were consistent with those of a previous study.18 To investigate whether GzmF has a role in cytotoxic lymphocyte-mediated cytolysis, GzmF was knocked down in LAK cells using short hairpin RNA (shRNA)-expressing adenovirus (Ad-shGzmF). In LAK cells, the GzmF mRNA level was reduced by 60% after Ad-shGzmF treatment. In contrast, the mRNA level of GzmB was not obviously altered (Figure 2b left panel). GFP-expressing adenovirus (Ad-GFP) was used to infect LAK cells as a negative control.

GzmF-silenced LAK cells reduce their cytotoxicity against caspase-inhibited target tumor cells. (a) GzmF mRNA expression is significantly upregulated in LAK cells. IL-2-stimulated LAK cells were harvested and their mRNA levels were determined by quantitative RT-PCR. Gzm mRNA levels were normalized to that of 18S rRNA detected in the same sample. Data shown are representative of three separate experiments as means±S.D. (b) GzmF-silenced LAK cells reduce their cytotoxicity against caspase-inhibited target tumor cells. The GzmF expression was decreased to one-third through Ad-shGzmF treatment (left panel). Ad-GFP-infected LAK cells were used as a negative control. GzmB expression was not obviously changed after Ad-shGzmF treatment. Cytotoxicity against the Yac-1 target cell was assessed as described in Materials and Methods section. Ad-shGzmF-treated LAK cells showed a reduced cytotoxicity against z-VAD-FMK (100 μM)-pretreated Yac-1 cells compared with Ad-GFP control LAK cells (right panel). These data are representative of three experiments as shown with means±S.D.
We found that GzmF silencing did not show a significant decline of cytotoxicity compared with Ad-GFP control LAK cells (data not shown). This suggests the fact that other Gzms may provide fail-safe mechanisms to induce cytolysis of target cells. Our above data confirmed the fact that GzmF induces caspase-independent cell death. To exclude caspase-dependent death induced by GzmB and other caspase-dependent Gzms, Yac-1 cells were preincubated with a caspase pan inhibitor z-VAD, which blocked a caspase-dependent death pathway. We found that GzmF-silenced LAK cells decreased their cytotoxicity against z-VAD-treated Yac-1 cells compared with Ad-GFP control LAK cells (Figure 2b right panel). These results suggest that GzmF has a role in caspase-independent cytolysis induced by LAK cells.
GzmF causes nuclear morphological changes
Chromatin condensation is considered to be a criterion for distinguishing apoptosis from necrosis.23 To determine the nature of GzmF-induced cell death, we visualized the nuclear morphological alterations of target cells by transmission electron microscopy. GzmF caused nuclear membrane collapse and chromatin condensation (Figure 3a). Chromatin was incompletely condensed and segmented at the nuclear periphery, which is distinct from that of GzmB or GzmC. In contrast, GzmB initiated a more compact nuclear condensation to form crescent or spherical shapes (Figure 3a). GzmC caused a condensed and shrunk nucleus. Mock-treated cells were uniform and did not change their nuclear morphology. These observations verify the fact that GzmF triggers a unique death program. Moreover, GzmF-treated cells exhibited obvious alterations in the cytosol. Cellular organelles were damaged by GzmF treatment (Figure 3a). The cytoplasm showed extensive vacuolization. However, GzmB-treated cells did not trigger obvious changes in the cytosol. Taken together, GzmF-induced death features are reminiscent of necroptosis.

GzmF causes nuclear morphological changes with single-stranded DNA nicking. (a) Nuclear morphological changes induced by GzmF. Yac-1 cells were treated by 0.5 μM GzmF, GzmB or GzmC in the presence of Ad at 37°C for 6 h. Cells were harvested and visualized by transmission electron microscopy (magnification × 1700). GzmB/Ad and GzmC/Ad were used as contrast. (b) GzmF does not cause DNA laddering. Yac-1 cells were treated with S-AGzmF (1 μM) and with the indicated concentrations of GzmF plus Ad at 37°C for 6 h. Genomic DNA was extracted and separated on 2% agarose gel stained with ethidium bromide. M, DNA marker. (c) GzmF induces single-stranded DNA nicks. GzmF/Ad-treated Yac-1 cells were radiolabeled with 32P-dATP by Klenow fragment. Genomic DNA was then harvested and visualized by denaturing alkaline gel electrophoresis. These data are representative of at least three independent experiments. (d) GzmF does not cleave SET. Yac-1 cells (3 × 105) were treated with different concentrations of GzmF or GzmB plus Ad. Samples were harvested and detected with anti-SET antibody. Caspase-3 (casp-3) was probed as a positive control for GzmB. β-Actin was used as a loading control
GzmF causes single-stranded DNA nicks
For typical apoptosis, genomic DNA is degraded to 180–200 bp oligonucleosomal fragments. To assess whether GzmF induces DNA fragmentation, we extracted genomic DNA from GzmF-treated cells and visualized it using an agrose gel. As shown in Figure 3b, GzmF failed to cause DNA laddering. GzmB was used as a positive control. We further checked whether ICAD is cleaved by GzmF. We found that GzmF did not degrade ICAD (data not shown). We next detected DNA nicking as previously reported.9 Nicked DNA can be labeled with 32P-dATP by a DNA polymerase I Klenow fragment and visualized with a denaturing alkaline agrose gel. GzmF caused single-stranded DNA nicking (Figure 3c). S-AGzmF/Ad or mock-treated target cells failed to make 32P-dATP incorporation. We previously identified NM23H1 as a DNA nickase to nick DNA after GzmA treatment.24 NM23H1 binds its inhibitor SET to form a complex for blocking the DNase activity of NM23H1. We demonstrated that GzmF did not degrade SET (Figure 3d). SET was not cleaved by GzmB, which was used as a negative control. Caspase-3 activation induced by GzmB was used as a positive control. β-Actin was unchanged as a loading control. This suggests that DNase NM23H1 is not activated in GzmF-induced death.
GzmF initiates mitochondrial swelling, depolarization and ROS generation
Mitochondrial damage is an initial step in the induction of apoptosis. We visualized mitochondrial alterations in GzmF-induced cell death by transmission electron microscopy. GzmF plus Ad treatment caused dilating and swelling of the mitochondria, followed by a loss of the cristae structure and a rupture of membrane (Figure 4a). Mock-treated cells showed a normal state of mitochondria with an elliptical shape and narrow cristae. GzmB caused mitochondrial damage as a positive control.

GzmF targets mitochondria of target cells. (a) Mitochondrial damage was detected by transmission electron microscopy (magnification × 6500). Mitochondria were indicated with white arrows. Yac-1 cells were incubated with 0.5 μM GzmF or GzmB in the presence of Ad at 37°C for 6 h. (b) GzmF induces loss of mitochondrial membrane potential (Δψm). Yac-1 cells were treated with the increased doses of GzmF or GzmB in the presence of Ad at 37°C for 6 h. Treated cells were stained with DiOC6(3) and analyzed by flow cytometry. (c) ROS generation in GzmF-treated cells. Yac-1 cells were preincubated with H2DCF-DA at 37°C for 30 min. Different doses of GzmF were then loaded in the presence of Ad for 2 h and analyzed by flow cytometry. GzmB and H2O2 were used as positive controls. These data are representative of three independent experiments
The above observations indicate that normal mitochondrial functions may be destroyed by GzmF. We used a sensitive dye, DiOC6(3), to monitor the loss of membrane potential (Δψm) with GzmF treatment. We found that GzmF initiated a dose-dependent loss of Δψm (Figure 4b). At a concentration of 1 μM, GzmF resulted in a nearly complete loss of Δψm. Mock-treated, Ad or S-AGzmF/Ad-treated cells showed background changes of Δψm. GzmB/Ad-treated cells were used as a positive control. Reactive oxygen species (ROS) have been considered as critical regulators in the induction of apoptosis. Mitochondria are the main source of intracellular ROS. ROS can be produced after a damage to the mitochondrial inner membrane. To assess ROS production in GzmF-treated cells, Yac-1 cells were probed with an oxidation-sensitive dye, H2DCF-DA, which shows a basal level of fluorescence at a reduced state and is converted into strong fluorescence after oxidation by intracellular ROS. GzmF led to ROS accumulation in a dose-dependent manner (Figure 4c). Mock-treated, Ad or S-AGzmF/Ad-treated cells failed to generate ROS. GzmB/Ad-treated cells were able to produce ROS. H2O2-treated cells were utilized as a positive control. These above data are representative of at least four separate experiments.
GzmF triggers cyt c release from mitochondria to cytosol
Cyt c release from mitochondria to cytosol is a key event during apoptosis. To investigate cyt c release in GzmF-induced cell death, we fractionated mitochondrial and cytosolic sections after GzmF/Ad treatment, followed by probing of cyt c by western blotting. GzmF caused cyt c release in a dose-dependent manner (Figure 5a left panel). To further define the time course of the release of cyt c, 0.5 μM GzmF was loaded with Ad into Yac-1 cells at different times. We found that cyt c release occurred within 2 h and reached a roughly maximal release at that time (Figure 5a right panel), although COX IV, a mitochondria matrix protein, remained unchanged in the treated mitochondrial pellets. Mock-treated, Ad and GzmF alone, or S-AGzmF/Ad-treated cells did not cause cyt c release. GzmB/Ad-treated cells were used as a positive control. We utilized an optimal dose of PFP or streptolysin O (SLO) for delivering Gzms into Yac-1 cells, and similar results were obtained (Figure 5b). GzmB was used as a positive control. These observations were confirmed by confocal microscopy (Figure 5c).

GzmF initiates cytochrome c release. (a) GzmF causes cyt c release in a dose- and time-dependent manner. Yac-1 cells (3 × 105) were treated with indicated concentrations of GzmF (left panel) and with the increased times of GzmF (0.5 μM) (right panel) plus Ad. The same pellet blot was stripped and reprobed with anti-COX IV antibody as a loading control. These data are representative of at least three independent experiments. supern, supernatant. (b) Yac-1 cells were treated with 0.5 μM GzmF plus sublytic PFP or SLO. Cyt c was probed as above. (c) Cyt c release was confirmed by confocal microscopy. MEFs were treated with GzmF or GzmB plus Ad at 37°C for 2 h. Cells were fixed, permeabilized and stained for cyt c. Cyt c staining green fluorescence is shown on the left, Mitotracker with red fluorescence is shown in the middle and the merged image at the right
Cyt c release is independent of Bid, Bax and Bak
To examine whether Bid participates in GzmF-induced cyt c release, mitochondrial and cytosolic fractions were probed with an anti-Bid antibody. Bid remained unchangeable in cytosolic fractions after different doses of GzmF treatment with Ad (Figure 6a). Bid was undetectable in cytosol and tBid appeared in the pellet after GzmB treatment (Figure 6a). These data indicate that Bid was completely cleaved to tBid and the processed tBid translocated into mitochondria in GzmB-loaded cells. Our observations are consistent with previous reports.25, 26 We further showed that Bid was not processed to tBid in GzmF-loaded cells, whereas Bid was activated to tBid by GzmB treatment (Figure 6b). Caspase-3 was probed as a positive control for GzmB. β-Actin was used as a loading control.

Cytochrome c release is independent of Bid or Bax/Bak. (a) GzmF-induced cyt c release is independent of Bid. The same immunoblots from the left panel of Figure 5a were stripped and reprobed with anti-Bid antibody. Full-length Bid detected in supernatants remained unchanged after GzmF treatment. GzmB processed Bid to tBid and tBid translocated to mitochondria. The loading control of COX IV blotting was shown in Figure 5a left panel. (b) GzmF does not process Bid to tBid. Yac-1 cells (3 × 105) were treated with different doses of GzmF or GzmB plus Ad at 37°C for 2 h. Bid was detected by western blotting. Caspase-3 was probed as a positive control for GzmB. β-Actin was used as a loading control. (c) GzmF causes cyt c release in Bid-deficient MEFs. WT and Bid−/− MEFs (3 × 105) were loaded with 0.5 μM S-AGzmF or with GzmF plus Ad at 37°C for 4 h. Cytoplasmic or pellet fractions were separated and probed for cyt c. GzmB and COX IV were used as positive or loading controls, respectively. (d) Cyt c release is independent of Bax/Bak. Bax/Bak WT or DKO MEFs (3 × 105) were treated as above for cyt c detection. GzmB was used as a positive control. COX IV was used as a loading control. (e) Bax cannot be activated in GzmF-treated cells. MEFs were mock treated (left), or treated with 0.5 μM GzmF (middle) or GzmB (right) plus Ad at 37°C for 4 h. Activated Bax was probed by immunofluorescent staining. These data represent at least three separate experiments. Activated Bax with FITC staining is shown in the upper panel, differential interference contrast (DIC) in the middle panel, and the merged image in the bottom panel. Supern, supernatant; casp-3, caspase-3
To further verify whether Bid is required for GzmF-caused cyt c release, Bid WT and knockout mouse embryonic fibroblast (MEFs) were treated by GzmF/Ad to assess cyt c release. Cyt c was still released in Bid-deficient MEFs, a process similar to that of WT MEFs (Figure 6c). Mock-treated, or S-AGzmF/Ad-treated cells did not release cyt c. COX IV was used as a good loading control. Cyt c release still needs the enzymatic activity of GzmF, although GzmB did not induce cyt c release in Bid-deficient MEFs. Bax and/or Bak induce a selective process of outer membrane permeabilization of mitochondria through the formation of channels or pores that allow the selective protein release soluble in the inner membrane space such as cyt c. We then detected whether cyt c is released from GzmF-treated Bax/Bak double-knockout (DKO) MEFs. We found that both Bax/Bak WT and DKO MEFs induced cyt c release from mitochondria (Figure 6d). However, GzmB did not induce cyt c release in Bax/Bak-deficient MEFs. COX IV was used as a good loading control. We further assessed active Bax with a specific Bax antibody in GzmF-loaded cells through confocal microscopy. Activated Bax was validated in GzmB/Ad-loaded cells and not in GzmF/Ad-loaded cells (Figure 6e). Therefore, our findings indicate that GzmF-induced cyt c release is independent of Bid or Bax/Bak.
GzmF-induced cell death is caspase independent
Once cyt c releases into the cytosol, it can bind cytosolic apoptotic protease-activating factor 1 (Apaf-1) and procaspase-9 to form an apoptosome in the presence of ATP, which causes the caspase activation cascade to execute apoptosis.27 As mentioned above, GzmF can cause cyt c release at an early stage of cell death program, hence we presumed that caspase activation has to occur to execute cell death after GzmF treatment. Surprisingly, the caspase pan-inhibitor Z-VAD-FMK did not significantly inhibit GzmF-induced cell death (52.6±3.6 versus 45.2±4.2%, P>0.05) (Figure 7a left panel). Z-VAD-FMK can block GzmB-caused death (56.2±1.8 versus 16.1±0.2%, P<0.01). These observations were confirmed by loading Yac-1 cells with PFP or SLO (Figure 7a right panel). Moreover, we pretreated cells with different caspase inhibitors, including caspase-2 inhibitor Z-VDVAD-FMK, caspase-3 inhibitor Z-DEVD-FMK, caspase-8 inhibitor Z-IETD-FMK or caspase-9 inhibitor Z-LEHD-FMK before GzmF loading. All inhibitors cannot suppress GzmF-induced death (data not shown).

GzmF cannot trigger caspase activation. (a) GzmF-induced death can-not be inhibited by caspase-pan inhibitor Z-VAD-FMK in the presence of Ad (left panel) or PFP/SLO (right panel). Yac-1 cells were pretreated with 100 μM Z-VAD-FMK before GzmF treatment. Z-VAD-FMK can block GzmB-induced apoptosis that was used as a positive control. Total dead cells were calculated by AV+ plus AV+/PI+ cells and are shown as means±S.D. *, P<0.05; **, P<0.01. (b) GzmF does not initiate the caspase activation cascade. Yac-1 cells (4 × 105) were treated with 0.5 μM GzmF and GzmB at the indicated times. Caspase activity was measured in cellular lysates. GzmB/Ad-treated cells were used as a positive control. Data were shown as means±S.D. (c) Caspase inactivation was confirmed by western blotting. Cells (2 × 105) were loaded with GzmF or GzmB as above and caspases 2, 3, 8 and 9 were probed with each specific antibody. Gzms were delivered with Ad (left panel) or sublytic concentrations of PFP or SLO (right panel). Cell death was verified by trypan blue staining
We also measured the activities of caspases in GzmF-loaded cells with specific fluorogenic substrates Ac-DEVD-AFC (caspase-3 and 7), Z-VDVAD-AFC (caspase-2), Z-IETD-AFC (caspase-8) and Ac-LEHD-AFC (caspase-9). GzmF treatment did not activate all the detected caspases (Figure 7b). Cell death was confirmed by trypan blue staining. GzmB activates caspases very early and reaches a peak level within 4 h. We also verified these observations in GzmF-loaded Yac-1 cells in the presence of Ad, PFP or SLO by western blotting (Figure 7c). All these procaspase forms remained unchangeable after GzmF treatment. GzmB could process all procaspase forms to active caspases. β-Actin was probed as a loading control. Taken together, GzmF-induced death did not depend on caspase activation.
GzmF impairs mitochondrial electron transport to disrupt ATP generation
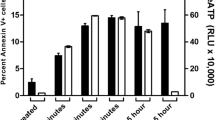
Although cyt c was released from the mitochondria into cytosol, caspase activation did not exist in GzmF-treated target cells. Cytosolic cyt c recruits Apaf-1 and procaspase-9 in an ATP-dependent manner to assemble an apoptosome, leading to caspase activation. We first detected the contents of procaspase-9 after GzmF treatment. Procaspase-9 maintained similar levels in GzmF-treated and mock-treated cells (data not shown). Considering the fact that ATP is essential for the formation of apoptosome, we measured the ATP contents of Gzms-treated Yac-1 cells. After GzmF treatment, cellular ATP concentrations diminished dramatically at an early time point, which dropped to half within 2.5 h (Figure 8a). The ATP decrease was in agreement with dynamic cyt c release after GzmF treatment (Figure 8b). One-fourth of ATP contents were consumed within 1 h of treatment, whereas at this time point, cyt c release was just slightly detectable in the cytosolic fraction. In contrast, GzmB treatment induced a short-term elevation of ATP levels at early stages, which dropped slightly even within 2.5 h (Figure 8a). ATP levels rose continuously to reach a maximal value within 0.5–1 h and then gradually decreased. At this period, GzmB induced a high level of cyt c release (Figure 8b). GzmB caused cyt c release with the elevation of ATP at an early stage. These observations imply that apoptosomes could be formed in GzmB-treated cells. GzmF-induced cell death caused a dramatic decline in ATP contents. ATP decline may favor the formation of inactive Apaf-1 aggregates and may prevent apoptosome assembly.28 β-Actin and COX IV remained unchanged and served as loading controls. These data are representative of at least three independent experiments.

GzmF disrupts mitochondrial electron transport leading to the loss of ATP. (a) Cellular ATP dramatically declines after GzmF treatment. Yac-1 cells (4 × 105) were treated with 0.5 μM GzmF or GzmB plus Ad at 37°C for the increased times. Intracellular ATP levels were assessed as described in the methods. ATP contents were calculated as means±S.D. Data are representative of at least four separate experiments. (b) Dynamic cyt c release with GzmF treatment. The other halves of the treated cells in panel a were probed for cyt c by western blotting. β-Actin and COX IV were used to control protein loading. (c) GzmF abolishes the mitochondrial respiration of target cells. Yac-1 cells (3 × 106) were treated with 0.5 μM GzmF or GzmB plus Ad at 37°C for the indicated times. Oxygen consumption rates were calculated and designated as nmol O2/min (means±S.D.). These data represent at least three separate experiments
ATP is the main energy storage in cells and is mostly generated by mitochondrial electron transport chains. We detected oxygen consumption of target cells after GzmF treatment. We found that cells reduced mitochondrial respiratory rates in a time-dependent manner after GzmF treatment (Figure 8c), whereas GzmB caused little change during the treated time. Therefore, the disruption of mitochondrial respiration might partially contribute to loss of ATP.
Discussion
In this study, we found that murine GzmF can induce a novel cell death program. Our data demonstrated that induction of cell death needs its enzymatic activity and is characterized by PS externalization, nuclear condensation, mitochondrial damage, cyt c release and single-stranded DNA nicking. However, GzmF-induced cell death takes place without caspase activation, although cyt c is released. Moreover, cyt c release is independent of Bid or Bax/Bak. We knocked down the expression of GzmF in LAK cells and observed a reduced cytotoxicity against caspase-inhibited target tumor cells. We further showed that GzmF impairs mitochondrial electron transport to abolish ATP generation. The loss of ATP may contribute to a failure of apoptosome assembly, leading to caspase inactivation.
Granzyme F-induced cell death takes place rapidly. Unlike typical apoptosis, GzmF-induced death has many unique features. AV and PI double-positive cells are predominant in GzmF-induced death. It indicates that membrane integrity is disrupted during the induction of death. Nuclear morphology showed a less compact or an incomplete chromatin condensation that is partially segmented at the nuclear periphery. These features are in agreement with necroptosis, which is a necrotic form of programed cell death.29, 30 For necroptosis, the nuclear morphological changes are not with obvious chromatin condensation, but with chromatin clustering to speckles. We compared nuclear morphological changes and other death features among three death types of apoptosis, necrosis and necroptosis in another of our just published paper.31
The DNA damage is characterized by single-stranded DNA nicking. Indeed, GzmF cannot cleave ICAD, the inhibitor of caspase-activated DNase, leading to DNA fragmentation (data not shown). We previously identified NM23H1 as a DNA nickase.24 It binds its inhibitor SET to form a complex, which is predominantly cytoplasmic. The DNase activity of NM23H1 was released through a degradation of its inhibitor SET after GzmA or GzmK treatment.9 However, GzmF does not degrade SET. It suggests that DNase NM23H1 is not activated in GzmF-induced death. Nevertheless, several possible factors could account for this nuclear damage. Endonuclease G (Endo G) and AIF are considered to be involved in chromatin condensation triggered by several stimuli.32, 33 We found that both Endo G and AIF were released from mitochondria to cytosol after GzmF treatment (data not shown). However, whether Endo G or AIF initiates DNA nicking remains to be further studied.
Mitochondria have a major role in both caspase-dependent and -independent death programs. They contain a number of proapoptotic proteins, such as cyt c, Endo G, AIF and SMAC, which are released into the cytosol in the induction of cell death.34 GzmF initiates mitochondrial damage and results in the release of cyt c. Cyt c release is often triggered by proapoptotic Bcl-2 family members, including Bid, Bax and Bak. These proteins translocate from the cytosol to mitocondria after a death signal. Bid can be processed to tBid, which recruits Bax or Bak to mitochondria through a caspase-dependent machinery, where it is integrated into the outer mitochondrial membrane to trigger the release of cyt c and other proapototic factors. Cyt c release induced by GzmB or GzmK is through the processing of Bid to tBid.22, 25 However, GzmF cannot activate Bid to tBid in the induction of death. Cyt c was still released in Bid−/− and Bax/Bak DKO MEF cells with GzmF treatment but was not released with GzmB treatment. We further found GzmF alone could not mediate cyt c release in isolated mitochondria (unpublished data). These observations suggest that some unknown cellular factors may be required for the release of cyt c during GzmF-induced death.
Cyt c release from the mitochondria to cytoplasm has been considered to be an important event in the activation of downstream caspases during apoptosis. Once cyt c is released into cytosol, it binds Apaf-1, procaspase-9 and ATP/dATP to form an apoptosome, which subsequently activates caspase-3 to execute the caspase activation cascade. Cytosolic ATP level has a critical role in forming an apoptosome or inactive aggregates during this process. In the absence of ATP, cyt c binding to Apaf-1 triggers its inactive aggregation.28 Several reports showed that intracellular ATP levels remain unchanged or even elevated for a short time in STP or in etoposide-induced apoptosis, and then gradually decrease until the very end of the death process.35, 36, 37 We first demonstrated that this phenomenon occurs in GzmB-induced apoptosis (Figure 8a). However, we found that, after GzmF treatment, intracellular ATP diminishes at an early stage, which is much earlier than cyt c release. Therefore, loss of intracellular ATP may fail to assemble apoptosomes, leading to caspase inactivation.
ATP is generated through the oxidative phosphorylation (OXPHOS) system in the mitochondrial inner membrane. The OXPHOS system consists of four respiratory chain complexes (complexes I to IV), which transfer electrons from the reducing equivalent (NADH-FADH2) to molecular oxygen, generating a proton gradient in the mitochondrial intermembrane space, and of the ATP synthase that uses this proton gradient to generate ATP. Complex I is a large multienzyme complex consisting of at least 46 subunits, which is the main source of ROS and the major route of entry of electrons into the respiratory chain. A recent report showed that the p75 subunit of respiratory complex I is degraded as a caspase substrate during apoptosis.38 Cells expressing a noncleavable mutant p75 maintain Δψm and ATP levels during apoptosis. GzmF-induced death takes place without caspase activation. Moreover, we found that the p75 subunit of complex I is not degraded after GzmF treatment, and GzmF cannot directly destroy the respiration complex I activity in isolated mitochondria (data not shown). Nevertheless, GzmF-treated target cells reduce mitochondrial respiratory rates in induction of cell death. These observations demonstrate that GzmF indeed destroys the electron transport of the OXPHOS system. The molecular mechanism of GzmF-mediated ATP decline remains to be further investigated.
Materials and Methods
Recombinant Gzms expression and purification
Murine GzmF was cloned from cDNA templates prepared from a mouse thymus. PCR was performed using the following primers: sense (5′-atgccaccaatcctgattctc-3′) and antisense (5′-cgggtggtttaactcctgtta-3′). Active GzmF cDNA was subcloned into pET26b with an enterokinase cleavage site before N-terminal amino acids IIGG. Inactive counterpart S-AGzmF was generated by point mutation. Both proteins were expressed and refolded from Rosetta (DE3) (Novagen, Darmstadt, Germany) inclusion bodies as described.19 After thoroughly washing and refolding for 3 days, proteins were harvested and activated by enterokinase treatment. The enterokinase was removed and GzmF was purified on a Hitrap ion exchange column (Amersham Biosciences, Pittsburgh, PA, USA). Recombinant human GzmB and murine GzmC were expressed in the same way.
Enzymatic activity of recombinant GzmF
Granzyme F was incubated with synthetic peptide substrates in a buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl) at RT for 30 min. Suc-Phe-Leu-Phe-pNA and Suc-Ala-Ala-Pro-Leu-pNA were purchased from Bachem (Bubendorf, Switzerland). Suc-Ala-Ala-Pro-Phe-pNA, H-Ala-Ala-Phe-pNA, Z-Phe-Arg-AMC and Z-Arg-Arg-AMC were from Sigma-Aldrich (St Louis, MO, USA). Suc-Phe-Thr-Ser-Tyr-pNA was synthesized by Chinese Peptide Company (Hangzhou, China). ODvalues were measured using a Wallac VICTOR reader (PerkinElmer, Waltham, MA USA).
Cell lines, antibodies and reagents
Yac-1 cells were cultured in RPMI 1640 (Gibco, Carlsbad, CA USA), supplemented with 10% FBS, 50 μM β-ME, 100 U/ml penicillin and 100 μg/ml streptomycin. MEFs were grown in DMEM medium supplemented with 10% FBS, 50 μM β-ME and 100 μM asparagine. Commercial antibodies were mouse mAb against β-actin (Sigma, St Louis, MO, USA), cyt c (BD Pharmingen, San Jose, CA, USA), activated Bax (MAb 6A7, BD Pharmingen), caspase 2, caspase 8, caspase 9 (Cell signaling, Danvers, MA, USA), rabbit anti-COX IV (Abcam, Cambridge, UK), anti-caspase 3 (Cell signaling), anti-caspase 3 (Biomol Research, Plymouth Meeting, PA, USA), Alexa488-conjugated donkey anti-mouse IgG (Molecular Probes, Carlsbad, CA, USA) and HRP-conjugate secondary antibodies (Santa Cruz, CA, USA). The ProLong antifade kit and Mitotracker were from Molecular Probes. Caspase inhibitor Z-VAD-FMK and fluorogenic substrates Ac-DEVD-AFC, Z-VDVAD-AFC, Z-IETD-AFC and Ac-LEHD-AFC were from Calbiochem (Darmstadt, Germany). The luminescent ATP cell viability assay kit was from Promega (Madison, WI, USA).
Gzms delivering with adenovirus, PFP or SLO
Replication-deficient adenovirus type V (Vector Gene Technology Company, Beijing, China), human PFP purified from YT cells (PFP, from Alexis Biochemicals, San Diego, CA, USA) and SLO (from Sigma-Aldrich) were utilized for delivering Gzms. A total of 100 PFU/cell of Ad was used to deliver Gzms into target cells as previously reported.21 Sublytic concentrations of PFP or SLO were determined to load Gzms into target cells. PFP was used at a final concentration of 150 ng/ml and SLO was used at 400 ng/ml for Yac-1 cells. For each experiment, SLO was preactivated by 10 mM DTT at RT for 10 min. Yac-1 Cells (2 × 105 in a final volume of 50 μl) were incubated with different concentrations of Gzms for the indicated times.
Cytotoxicity assay
Cytotoxicity of LAK cells was tested against Yac-1 target cells in standard 51Cr release assay. Yac-1 cells (1 × 106) were labeled with 200 μCi 51Cr at 37°C for 1 h. After a complete wash, Yac-1 cells were plated at 1 × 104 cells/well. LAK cells were added at the indicated E/T ratios and after 4 h of incubation, supernatants were harvested and the specific release of 51Cr was measured using MicroBeta counter (PerkinElmer). In some experiments, Yac-1 cells were preincubated with z-VAD-FMK (100 μM) at 37°C for 30 min. Specific cytotoxicity was calculated as [(sample release − spontaneous release)/(maximum release−spontaneous release)] × 100. Maximum release of target cells was measured after treatment with 2% TritonX-100. For in vitro delivery of Gzms, Yac-1 cells were treated with Gzms plus Ad and the specific release of 51Cr was assessed as above.
Laser-scanning confocal microscopy
For cyt c assay, cells were seeded overnight and treated as indicated. The treated cells were washed thrice, then fixed with 4% paraformaldehyde for 1 h at 4°C, permeabilized with 0.05% NP-40 for 20 min and blocked for 30 min with 10% donkey serum in PBS. Anti-cyt c antibody was added for 1 h at RT, and stained with Alexa488-conjugated donkey anti-mouse IgG. Slides were mounted and images were visualized with Olympus FV500 laser-scanning confocal microscopy (Tokyo, Japan). The same procedure was used for Bax activation assay except for permeabilizing with 0.2% Triton X-100.
Transmission electron microscopy
Yac-1 cells (1 × 106) were treated with Gzms and Ad at 37°C for 6 h. The cells were washed twice and fixed with 2% glutaraldehyde on ice for 1 h, then postfixed in 2% osmium tetroxide. After dehydrating with sequential washes in 50, 70, 80, 90 and 100% ethanol, cells were embedded in Spurrs resin. The sections were mounted in copper grids, counterstained with uranyl acetate and lead citrate, then visualized in a JEM-100 CX transmission electron microscopy (Tokyo, Japan). Images were photographed and scanned by Eversmart Jazz+ program (Scitex, Israel).
Quantification of ATP content
Total cellular ATP concentration was assessed using a luminescent ATP cell viability assay kit from Promega. Briefly, Yac-1 cells were treated as indicated, washed carefully and then lyzed with 50 μl lysis buffer. Luminescence was measured for 10 s with a luminometer (Model TD-20/20, Turner BioSystems, Sunnyvale, CA USA). ATP contents are presented as relative luminescence units (RLU).
Cellular caspase activity assay
Caspase activity was measured under the manufacturer's instruction. Briefly, 4x105 Yac-1 cells were treated as indicated, washed twice and lysed with 50 μl lysis buffer (50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% Chaps, 5 mM DTT, 0.1 mM EDTA). Cellular lysates were harvested and incubated with 30 μM caspase substrates at 37°C. Fluorescence was measured using a Fluoroskan Ascent (Type 374, ThermoLabsystems, Waltham, MA, USA). Data were normalized to the level of blank cells and shown as means±S.D.
Cyt c release assay
Cyt c release from mitochondria to cytosol was detected by western blotting. Cells were fractionated with 0.025% digitonin for 5 min on ice. Mitochondrial (pellet) and cytosolic (supernatant) fractions were obtained after 5 min centrifuge at 10000 × g.
Oxygen consumption assessment
Cells were treated as indicated and resuspended in a final concentration of 3 × 106 cells/ml at TD buffer (25 mM Tris-HCl, pH 7.4–7.5 (25°C), 137 mM NaCl, 10 mM KCl, 0.7 mM Na2HPO4). Oxygen consumption was measured using a Respirometer (model RC650, Strathkelvin Instruments, North Lanarkshire, UK). Respiratory rates are designated as nmol O2/min.39
DNA fragmentation and nicking assay
Treated Yac-1 cells were harvested and resuspended in a DNA extraction buffer (100 mM Tris-Cl, pH 8.0, 5 mM EDTA, 0.2 M NaCl, 0.5% SDS and 0.2 μg/μl proteinase K) for 2 h at 52°C. NaCI was added to a final concentration of 1.5 M to precipitate intact genomic DNA. DNA was then precipitated with standard EtOH precipitation procedures and resuspended in 1 × TE plus 10 μg/ml RNase A at 37°C for 2 h and analyzed by 2% agarose gel electrophoresis. For DNA nicking assay, GzmF-treated cells were lysed with 0.5% NP-40 lysis buffer and incubated with 5 U Klenow fragment of DNA polymerase I and 10 μCi 32P-dATP for 1 h at 37°C. Genomic DNA was extracted, washed thoroughly, separated by denaturing alkaline gel electrophoresis and visualized by autoradiograph.
Transmembrane potential and ROS detection
Changes in mitochondrial membrane potential (Δψm) were measured by flow cytometry with DiOC6(3) (Molecular Probes) at a final concentration of 40 nM. Cells were harvested after treatment, washed twice and stained with DiOC6(3) for flow cytometry analysis. Intracellular ROS production was assessed by incubating cells with 20 μM H2DCF-DA at 37 °C for 30 min. Unincorporated dye was removed by completely washing, and cells were then treated as indicated and monitored by flow cytometry.
Generation of LAK cells
For preparation of LAK cells, splenocytes from BALB/c mice were purified by the Ficoll-Hypaque density gradient centrifugation separation. Harvested cells were cultured in six-well plates (1 × 107/well) with complete medium containing 1000 units/ml recombinant human interleukin 2 (rhIL-2). After 5 days, 1 ml fresh medium with rhIL-2 was added for 3 more days of culture.
Quantitative RT-PCR
Total RNA was isolated using Trizol reagent and reverse-transcribed with Superscript II (Invitrogen , Carlsbad, CA, USA). The mRNA content was measured by the Rotor-Gene 6000 analyzer (Corbett Research, Sydney, Australia). Primers specific for each Gzm used in this study were obtained from the literature.18, 40 GzmB sense, 5′-ATCAAGGATCAGCAGCCTGA-3′, and antisense, 5′-TGATGTCATTGGAGAATGTCT-3′; GzmF sense, 5′-TGAGGTTTGTGAAAGATAATG-3′, and antisense, 5′-TCACTGGTGTTGTCCTTATC-3′; 18S rRNA sense, 5′-ACCGCAGCTAGGAATAATGGA-3′, and antisense, 5′-GCCTCAGTTCCGAAAACCA-3′. Relative mRNA levels were calculated by the comparative cycle threshold method and 18S rRNA was used as the internal control for each sample.
RNA silencing of GzmF
Three pairs of RNA sequences against GzmF for RNAi were designed using BLOCK-iT RNAi Designer (Invitrogen) and cloned into pSuper-puro that expresses 19 nt shRNA. One pair of shRNA sequence that efficiently knocked down the expression of GzmF was used in this study. The sense strand of this sequence was GCACTGGAAGCTCAATGAG. Recombinant adenoviruses were constructed according to the manufacturer's instructions (Stratagene, La Jolla, CA, USA). H1 promoter with shRNA was cloned from pSuper-shGzmF by PCR and subcloned into an adenoviral pShuttle vector. The verified pShuttle-H1-shGzmF vector was transformed into BJ5183-AD1 cells, and recombinant pAd-H1-shGzmF adenoviral genome was generated. Recombinant shGzmF-expressing adenoviruses (Ad-shGzmF) were produced in Ad293 cells for RNA-silencing experiments. GFP-expressing adenoviruses (Ad-GFP) were generated in the same way, except that it was under the control of CMV promoter. For infection of LAK cells, splenocytes were plated in six-well dishes at 1 × 107 cells per well plus rhIL-2 (1000 U/ml). At day 5, Ad-shGzmF or Ad-GFP was added to infect LAK cells at 2000 PFU/cell and cultured for an additional 3 or 4 days for GzmF silencing. Infection efficiency was monitored using Ad-GFP through flow cytometry or fluorescence microscopy.
Abbreviations
- Gzm:
-
granzyme
- S-AGzmF:
-
Ser195AlaGzmF
- Ad:
-
replication-deficient adenovirus type V
- PFP:
-
perforin
- SLO:
-
streptolysin O
- MEF:
-
mouse embryonic fibroblast
- STP:
-
staurosporine
- cyt c:
-
cytochrome c
- LAK:
-
lymphokine-activated killer
- shRNA:
-
small hairpin RNA
- AV:
-
annexin V
- PI:
-
propidium iodide
- ROS:
-
reactive oxygen species
- ICAD:
-
inhibitor of caspase-activated DNase
- Apaf-1:
-
apoptotic protease-activating factor 1
References
Russell JH, Ley TJ . Lymphocyte-mediated cytotoxicity. Annu Rev Immunol 2002; 20: 323–370.
Barry M, Bleackley RC . Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol 2002; 2: 401–409.
Grossman WJ, Revell PA, Lu ZH, Johnson H, Bredemeyer AJ, Ley TJ . The orphan granzymes of humans and mice. Curr Opin Immunol 2003; 15: 544–552.
Thomas DA, Du C, Xu M, Wang X, Ley TJ . DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 2000; 12: 621–632.
Waterhouse NJ, Sedelies KA, Browne KA, Wowk ME, Newbold A, Sutton VR et al. A central role for Bid in granzyme B-induced apoptosis. J Biol Chem 2005; 280: 4476–4482.
Lieberman J, Fan Z . Nuclear war: The granzyme A-bomb. Curr Opin Immunol 2003; 15: 553–559.
Fan Z, Beresford PJ, Zhang D, Lieberman J . HMG2 interacts with the nucleosome assembly protein SET and is a target of the cytotoxic T-lymphocyte protease granzyme A. Mol Cell Biol 2002; 22: 2810–2820.
Johnson H, Scorrano L, Korsmeyer SJ, Ley TJ . Cell death induced by granzyme C. Blood 2003; 101: 3093–3101.
Zhao T, Zhang H, Guo Y, Zhang Q, Hua G, Lu H et al. Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ 2007; 14: 489–499.
Hou Q, Zhao T, Zhang H, Lu H, Zhang Q, Sun L et al. Granzyme H induces apoptosis of target tumor cells characterized by DNA fragmentation and Bid-dependent mitochondrial damage. Mol Immunol 2008; 45: 1044–1055.
Lu H, Hou Q, Zhao T, Zhang H, Zhang Q, Wu L et al. Granzyme M directly cleaves inhibitor of caspase-activated DNase (CAD) to unleash CAD leading to DNA fragmentation. J Immunol 2006; 177: 1171–1178.
Kelly JM, Waterhouse NJ, Cretney E, Browne KA, Ellis S, Trapani JA et al. Granzyme M mediates a novel form of perforin-dependent cell death. J Biol Chem 2004; 279: 22236–22242.
Fellows E, Gil-Parrado S, Jenne DE, Kurschus FC . Natural killer cell-derived human granzyme H induces an alternative, caspase-independent cell-death program. Blood 2007; 110: 544–552.
Masson D, Tschopp J . A family of serine esterases in lytic granules of cytolytic T lymphocytes. Cell 1987; 49: 679–685.
Jenne D, Rey C, Haefliger JA, Qiao BY, Groscurth P, Tschopp J . Identification and sequencing of cDNA clones encoding the granule-associated serine proteases granzymes D, E, and F of cytolytic T lymphocytes. Proc Natl Acad Sci USA 1988; 85: 4814–4818.
Sattar R, Ali SA, Abbasi A . Bioinformatics of granzymes: sequence comparison and structural studies on granzyme family by homology modeling. Biochem Biophys Res Commun 2003; 308: 726–735.
Pham CT, MacIvor DM, Hug BA, Heusel JW, Ley TJ . Long-range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci USA 1996; 93: 13090–13095.
Revell PA, Grossman WJ, Thomas DA, Cao X, Behl R, Ratner JA et al. Granzyme B and the downstream granzymes C and/or F are important for cytotoxic lymphocyte functions. J Immunol 2005; 174: 2124–2131.
Hink-Schauer C, Estebanez-Perpina E, Wilharm E, Fuentes-Prior P, Klinkert W, Bode W et al. The 2.2-A crystal structure of human pro-granzyme K reveals a rigid zymogen with unusual features. J Biol Chem 2002; 277: 50923–50933.
Odake S, Kam CM, Narasimhan L, Poe M, Blake JT, Krahenbuhl O et al. Human and murine cytotoxic T lymphocyte serine proteases: subsite mapping with peptide thioester substrates and inhibition of enzyme activity and cytolysis by isocoumarins. Biochemistry 1991; 30: 2217–2227.
Froelich CJ, Orth K, Turbov J, Seth P, Gottlieb R, Babior B et al. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J Biol Chem 1996; 271: 29073–29079.
Zhao T, Zhang H, Guo Y, Fan Z . Granzyme K directly processes bid to release cytochrome c and endonuclease G leading to mitochondria-dependent cell death. J Biol Chem 2007; 282: 12104–12111.
Leist M, Jaattela M . Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2001; 2: 589–598.
Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J . Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 2003; 112: 659–672.
Heibein JA, Goping IS, Barry M, Pinkoski MJ, Shore GC, Green DR et al. Granzyme B-mediated cytochrome c release is regulated by the Bcl-2 family members Bid and Bax. J Exp Med 2000; 192: 1391–1402.
Barry M, Heibein JA, Pinkoski MJ, Lee SF, Moyer RW, Green DR et al. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol Cell Biol 2000; 20: 3781–3794.
Darmon AJ, Ley TJ, Nicholson DW, Bleackley RC . Cleavage of CPP32 by granzyme B represents a critical role for granzyme B in the induction of target cell DNA fragmentation. J Biol Chem 1996; 271: 21709–21712.
Kim HE, Du F, Fang M, Wang X . Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci USA 2005; 102: 17545–17550.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1: 112–119.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3–11.
Zhang H, Zhong C, Shi L, Guo Y, Fan Z . Granulysin induces cathepsin B release from lysosomes of target tumor cells to attack mitochondria through processing of bid leading to necroptosis. J Immunol 2009; 182: 6993–7000.
Wang X, Yang C, Chai J, Shi Y, Xue D . Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 2002; 298: 1587–1592.
Li LY, Luo X, Wang X . Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001; 412: 95–99.
Kroemer G, Galluzzi L, Brenner C . Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163.
Zamaraeva MV, Sabirov RZ, Maeno E, Ando-Akatsuka Y, Bessonova SV, Okada Y . Cells die with increased cytosolic ATP during apoptosis: a bioluminescence study with intracellular luciferase. Cell Death Differ 2005; 12: 1390–1397.
Tsujimoto Y . Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ 1997; 4: 429–434.
Sato T, Machida T, Takahashi S, Murase K, Kawano Y, Hayashi T et al. Apoptosis supercedes necrosis in mitochondrial DNA-depleted Jurkat cells by cleavage of receptor-interacting protein and inhibition of lysosomal cathepsin. J Immunol 2008; 181: 197–207.
Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 2004; 117: 773–786.
Duan S, Hajek P, Lin C, Shin SK, Attardi G, Chomyn A . Mitochondrial outer membrane permeability change and hypersensitivity to digitonin early in staurosporine-induced apoptosis. J Biol Chem 2003; 278: 1346–1353.
Getachew Y, Stout-Delgado H, Miller BC, Thiele DL . Granzyme C supports efficient CTL-mediated killing late in primary alloimmune responses. J Immunol 2008; 181: 7810–7817.
Acknowledgements
We thank Dr. Q Hou and H Zhang, and D Hu and H Tang for their helpful advice; and CC Liu, L Sun and Y Teng for their technical support. Bid KO MEF is a gift from Dr. A Strasser (The Walter and Eliza Hall Institute of Medical Research, Australia); Bax/Bak DKO MEF is generously provided by Dr. Q Chen (Institute of Zoology, CAS). This work was supported from the National Natural Science Foundation of China (30525005, 30830030, 30772496, 30700126), 863 program (2006AA02Z4C9), 973 programs (2006CB504303, 2006CB910901), the Innovative program (KSCX2-YW-R-42) and the Hundred Talents Program of CAS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by; J Tschopp
Rights and permissions
About this article
Cite this article
Shi, L., Wu, L., Wang, S. et al. Granzyme F induces a novel death pathway characterized by Bid-independent cytochrome c release without caspase activation. Cell Death Differ 16, 1694–1706 (2009). https://doi.org/10.1038/cdd.2009.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.101
Keywords
This article is cited by
-
Normality sensing licenses local T cells for innate-like tissue surveillance
Nature Immunology (2022)
-
FADD cleavage by NK cell granzyme M enhances its self-association to facilitate procaspase-8 recruitment for auto-processing leading to caspase cascade
Cell Death & Differentiation (2012)
-
A quarter century of granzymes
Cell Death & Differentiation (2012)
-
Lysosomal chymotrypsin B potentiates apoptosis via cleavage of Bid
Cellular and Molecular Life Sciences (2010)
