
Abstract
Mitochondrial localization of p53 was observed in stressed and unstressed cells. p53 is involved in DNA repair and apoptosis. It exerts physical and functional interactions with mitochondrial DNA and DNA polymerase γ (pol γ). The functional cooperation of p53 and pol γ during DNA synthesis was examined in the mitochondrial fraction of p53-null H1299 cells, as the source of pol γ. The results show that p53 may affect the accuracy of DNA synthesis in mitochondria: (1) the excision of a misincorporated nucleotide increases in the presence of (a) recombinant wild-type p53 (wtp53); (b) cytoplasmic fraction of LCC2 cells expressing endogenous wtp53 (but not specifically pre-depleted fraction); (c) cytoplasmic extract of H1299 cells overexpressing wtp53, but not exonuclease-deficient mutant p53-R175H. (2) Mitochondrial extracts of HCT116(p53+/+) cells display higher exonuclease activity compared with that of HCT116(p53−/−) cells. Addition of exogenous p53 complements the HCT116(p53−/−) mitochondrial extract mispair excision. Furthermore, the misincorporation was lower in the mitochondrial fraction of HCT116(p53+/+) cells as compared with that of HCT116(p53−/−) cells. (3) Irradiation-induced mitochondrial translocation of endogenous p53 in HCT116(p53+/+) cells correlates with the enhancement of error-correction activities. Taken together, the data suggest that p53 in mitochondria may be a component of an error–repair pathway and serve as guardian of the mitochondrial genome. The function of p53 in DNA repair and apoptosis is discussed.
Similar content being viewed by others


Main
The tumor suppressor protein p53 represents a central factor for the maintenance of genome stability and for the suppression of cancer.1 p53 is present at low levels in unstressed cells, but after exposure to various stress signals, the protein is stabilized and activated by a series of post-translational modifications. p53 is involved in the induction of cell-cycle arrest and apoptosis through transcriptional activation of target genes.2 p53 displays multicompartmental functions in the cell. Interestingly, in response to multiple death stimuli, mitochondrial p53 targeting occurs in a wide spectrum of cell types, where it elicits a series of responses that can promote an apoptosis through a transcription-independent mechanism.3, 4 Several lines of evidence support a connection between p53 and mitochondrion. Mitochondrial p53 levels are proportional to total p53 levels, and a small fraction of protein is associated with mitochondrial DNA (mtDNA) in the absence of exogenous stress, thus implying a non-apoptotic function for mitochondrial p53.5 Furthermore, translocation of p53 to mitochondria was observed in various cells (e.g., MCF-7 and HCT116) independent of apoptosis.6 p53 may be a component of a stress response pathway that involves the upregulation of mitochondrial repair in mouse liver and cancer cells, suggesting the potential role of p53 in maintaining mtDNA stability.7, 8
Mitochondrial DNA, a 16.5-kb circular double-stranded molecule, is replicated by an assembly of enzymes and proteins consisting of DNA polymerase γ (pol γ), ssDNA-binding protein, DNA helicase and various accessory proteins and transcription factors.9 Mitochondrial DNA is prone to mutations, as it is localized near the inner mitochondrial membrane in which reactive oxygen species are generated. In addition, mtDNA lacks histone protection and highly efficient DNA repair mechanisms.10 The mutations in mtDNA, commonly found in many tumors, lead to genomic instability (mutator phenotype), increased cell survival or cancer in humans.11 The mutation rate of mtDNA is estimated to be about 20- to 100-fold higher than that of nuclear DNA.12 Pol γ is responsible for the replication of mtDNA in eukaryotic cells and is implicated in all repair processes.9 In humans, pol γ consists of two subunits, a 140 kDa catalytic subunit (with DNA polymerase and exonuclease activities) and a smaller 55 kDa subunit. The polymerase activity is essential for mtDNA maintenance and its exonuclease activity is essential for mtDNA integrity.13, 14, 15 The mutagenic mechanisms were shown to be replication errors caused by misinsertion (as a result of a dNTP excess) or decreased proofreading efficiency (as a result of next nucleotide effect or mutations in the catalytic subunit of pol γ). Balanced as well as unbalanced dNTP accumulations are mutagenic; mutation rates and mutational spectra could be influenced by dNTP levels in mitochondria.16, 17 Mice expressing an error-prone form of pol γ lacking the 3′ → 5′ exonuclease activity exert a three- to five-fold increase in the level of point mutations and undergo accelerated aging.18 Mutations in mtDNA occur in some rapidly growing tumors, including tumors harboring p53 mutations.19 Therefore, it seems plausible that mutation of p53 may be directly or indirectly linked with loss of mtDNA genomic stability.
p53 exhibits multiple activities in DNA repair.20 Several groups showed 3′ → 5′ exonuclease activity intrinsic to p53, which may act as an external proofreader for errors introduced by exonuclease-deficient DNA polymerases.21, 22, 23, 24, 25, 26 This activity provides a molecular basis for p53 involvement in DNA replication machinery complexes where proofreading is necessary. p53 is localized in mitochondria to the inside face of the inner membrane, the compartment in which mtDNA is located.3, 27 It exerts physical and functional interaction with mtDNA and pol γ in response to mtDNA damage induced by exogenous and endogenous insults.28, 29 Hence, it was of interest to elucidate the functional cooperation between p53 and pol γ during DNA synthesis in mitochondria. Our results show that the presence of p53 in mitochondria, provided by recombinant p53 or endogenous p53, increases the excision of the wrong nucleotides incorporated into DNA. These data suggest that p53 in mitochondria may affect the accuracy of DNA synthesis by acting as an external proofreader, thus reducing the production of polymerization errors.
Results
We investigated the functional interaction between p53 and pol γ during the DNA synthesis catalyzed by the replication apparatus in the mitochondrial fractions of human p53-null H1299 cells (H1299mit), as a model system. The lack of the mitochondrial locations of endogenous p53 was verified by western blot (WB) analysis with specific antibodies against p53 (Figure 1a). As the DNA polymerization activity used mitochondrial extracts as the source of pol γ, it was important to rule out the possibility that the observed polymerization activity might be because of a contamination of the extracts with nuclear DNA polymerases. The composition and purity of the mitochondrial fraction was confirmed by the absence and presence of nuclear and mitochondrial marker proteins, for example c-jun and cytochrome c, respectively (Figure 1a).

Incorporation and excision of wrong nucleotide dGTP in the mitochondrial fractions of H1299 cells in the presence of p53. (a) Analysis of p53 levels in various fractions of H1299 and LCC2 cells by western blotting. Protein samples (20 μg) from mitochondrial (mit), nuclear (nuc) and cytoplasmic (cyt) fractions of these cells were subjected to SDS-PAGE. p53 protein expression was detected by the Do-1 anti-human p53 mAb. The distribution of the nuclear marker c-jun or the mitochondrial marker cyt-c was analyzed to ascertain the purity of each fraction. (b) The correctly paired dsDNA substrate (lane 1) was incubated with 0.5 mM dGTP and H1299mit (5 μg) without p53 (lanes 2 and 3) or in the presence of p53-GST fusion protein (250 ng) (lanes 4 and 5) or GST protein (400 ng) (lanes 6 and 7). After 10-min incubation at 37° C, 5 μl aliquots were withdrawn and analyzed on 16% polyacrylamide gel, as described in Materials and Methods. (c) Experimental scheme for the analysis of excision of incorporated wrong nucleotide in mitochondrial extracts. The correctly paired 5′-end-labeled template/primer was incubated with wrong dGTP and H1299mit for 20 min. Aliquots were taken for analysis of misincorporation by PAGE. After an initial 20-min incubation, reaction mixture was further incubated in the absence or presence of p53. Aliquots were taken at various times and analyzed by PAGE, to follow after excision of the incorporated mispair in mitochondrial extracts in the absence or presence of p53. (d) The incorporation of dGTP opposite the template T was examined with H1299mit. After incubation of DNA template/primer (lane 1) for 10 min (lane 2) and 20 min (lane 3), the reaction mixture was further incubated in the absence (lanes 4 and 5) or presence (lanes 6 and 7) of p53-GST fusion protein (250 ng) or preheated p53-GST (lane 8), or p53-GST and Do-1 anti-human p53 mAb (lane 9) or p53-GST and anti-horse IgG (H+L) (lane 10). Aliquots were taken after 20 min (lanes 4 and 6) and 40 min of incubation (lanes 5, 7–10). The positions of the 16- and 17mer primers are indicated by arrows
The functional interaction between p53 and pol γ was assessed in the presence of low dNTP concentration, to avoid the mispair extension by the enzyme after misincorporation at a high dNTP concentration. Notably, proofreading 3′ → 5′ exonuclease activity is enhanced in less-stable DNA regions leading to a reduction in base substitution error frequencies in AT- versus GC-rich sequences.30 Therefore, DNA sequences were chosen to provide an AT-rich environment from a template target site to maximize proofreading. Combining these approaches enabled us to elucidate the function of p53 in DNA synthesis in mitochondria under optimal reaction conditions.
Misincorporation in mitochondria in the presence of p53
Normal intramitochondrial dNTP pools are highly asymmetric and may reduce by three-fold the fidelity of DNA synthesis by pol γ due to increased formation of dGTP:T mispairs and inefficient correction by proofreading.31 In H1299mit, the incorporation of dGTP opposite the template T was observed with standing-start dsDNA substrate (wherein the target template residue immediately follows the 3′-terminal end of the primer) (Figure 1b, lanes 2 and 3). The 17- and 18mer products were accumulated following the production of the T:G mispair. However, the addition of recombinant p53-GST fusion protein to the mitochondrial extracts resulted in a decrease in the number of mispaired nucleotides (lanes 4 and 5). In the control experiment, there was no reduction of 17- and 18mer products in the presence of GST protein alone (lanes 6 and 7), indicating that the GST moiety does not contribute to the observed decrease in misincorporation.
A problem in determining the functional cooperation of the p53 exonuclease and pol γ during DNA synthesis in mitochondria is that pol γ is an exonuclease-proficient enzyme. Hence, the impact of p53 on the lesser incorporation of the wrong nucleotide by pol γ was tested in a sequential reaction experiment (Figure 1c). The strategy was to follow after the excision of the incorporated mispair in the absence or presence of p53. After 20 min incubation, pol γ in H1299mit exerts dGTP misinsertion, thus creating a substrate for the excision step (Figure 1d, lanes 2 and 3). The reaction mixture was further incubated in the absence or presence of recombinant p53-GST fusion protein. The accumulation of 17mer product in the absence of p53 indicates that T:G mispair is inefficiently corrected by 3′ → 5′ exonuclease-proficient mitochondrial pol γ (lanes 4 and 5). However, the efficient removal of the polymerization error following a misincorporation event was observed in the presence of p53-GST; the amount of 17mer products significantly decreased and products lower than 16mer were formed. The intensity of the 17mer band decreased with incubation time (lanes 6 and 7). These results were reproduced four times with separate preparations of mitochondrial extracts. Notably, the excision of the 17mer product was unaffected by the addition of the boiled, non-active p53-GST protein (lane 8). Furthermore, exonuclease reactions with p53-GST performed in the presence of the p53-specific antibodies (Do-1) (lane 9), but not with the nonspecific anti-horse IgG (H+L) antibodies (lane 10), demonstrated the decrease in excision of the T:G terminal mispair.
Proofreading in mitochondria in the presence of p53 protein
Excision of the wrong nucleotide may occur by either an intrinsic intramolecular mechanism (without dissociation of the DNA from the enzyme) or an external intermolecular mechanism (following the release of the DNA from the enzyme). We performed two experiments to elucidate the involvement of p53 in error correction in mitochondria.
First, to determine whether the removal of the incorporated mispair (Figure 2a, lane 1) involves the dissociation of the DNA from the enzyme, the reaction mixture was further incubated with p53-GST in the absence or presence of unlabeled ssDNA, dsDNA (sequence-specific or non-sequence-specific substrates for p53) or ssRNA trap. The low efficiency of T:G mispair excision by pol γ alone (Figure 2a, lane 2) was unaffected in the presence of DNA or RNA trap (data not shown). However, although incubation with p53-GST results in an efficient excision of the incorporated dGTP and decrease of 17mer product (lane 3), the substantial reduction in exonucleolytic excision of the mispair was observed in the presence of either DNA (lanes 4, 5 and 7) or RNA trap (lane 6). Evidently, following misincorporation, the dissociation of the DNA–polymerase complex occurs. Consequently, the mispaired 17mer product may be accessible for excision by external p53 exonuclease.

Error correction in H1299mit in the presence of p53. (a) The incorporation of dGTP opposite the template T was examined with H1299mit in the presence of p53-GST and unlabeled competitor DNA or RNA. After initial incubation of DNA template/primer for 20 min (lane 1), the reaction mixture was further incubated in the absence (lane 2) or presence of p53-GST (250 ng) (lanes 3–7). Lane 3 – incubation with p53-GST. Lane 4 – incubation with p53-GST and ssDNA. Sequence of the ssDNA trap is 5′-ATTTCACATCTGACTA-3′. Lane 5 – incubation with p53-GST and dsDNA containing 3′-terminal A:A mispair (see legend for 2b). Lane 6 – incubation with p53-GST and ssRNA. Sequence of the ssRNA trap is 5′-AUUUAUUUAUUAUUUUAUUAUUUAA-3′. Lane 7 – incubation with p53-GST and dsDNA (sequence-specific substrate, sense: 5′-AGTCCTTGTACAGGGTTGTACAACTCGA-3′; antisense: 5′-TCGAGTTGTACAACCCTGTACAAGGACT-3′). Aliquots were taken after 40 min of incubation. The position of the 16mer primer is indicated by an arrow. (b) Three versions of primers were used that were identical except for the 3′-terminal nucleotide (N), which is A, C or T. The reaction mixture contained template-primer with 3′-terminal A:A (lanes 1–4) or A:C mispair (lanes 5–8) or correct A:T base pair (lanes 9–11) and H1299mit. Lanes 1 and 5 – incubation in the presence of 0.5 mM dATP. Lanes 2 and 6 – incubation in the presence of 0.5 mM dATP and p53-GST. Lanes 3 and 7 – incubation in the presence of 0.5 mM dATP and dTTP. Lanes 4 and 8 – incubation with p53-GST in the presence of 0.5 mM dATP and dTTP. Correctly paired (A:T) substrate (lane 9) was incubated with H1299mit in the absence (lane 10) or presence of p53-GST (lane 11). The position of the 16mer primer is indicated by an arrow
Second, we examined the proofreading in mitochondria in the presence of p53. Mismatch-containing DNA requires the efficient excision of the terminal mispair, correct replacement of the mismatched nucleotide and the transfer of the primer to the polymerase active site for normal polymerization–primer extension. The polymerization activity of pol γ in H1299mit was tested with DNA template-primers containing 3′-terminal A:A or A:C mispair, by incubation with either dATP or dATP+dTTP, in the absence or presence of p53-GST. No elongation products were detected with H1299mit and mismatch-containing template-primers (A:A or A:C) in the presence of dATP (Figure 2b, lanes 1 and 5, respectively) or dATP+dTTP (lanes 3 and 7, respectively). Apparently, pol γ was unable to extend mispairs without an efficient correction of the mismatch by exonuclease activity of the enzyme. No detectable extension of the 3′-terminal mispairs occurred with p53-GST/H1299mit complex in the presence of dATP only as well (lanes 2 and 6). However, both mispaired primers were elongated by the p53-GST/H1299mit complex in the presence of dATP+dTTP, giving rise to a 17mer product (lanes 4 and 8, respectively). Remarkably, p53 itself displays no DNA polymerization activity.22 Furthermore, there is no increase in the polymerization activity of pol γ, as the extension of A:T correctly paired template-primer by pol γ in the presence of dATP was unaffected by p53-GST (lanes 10 and 11). The proofreading results imply a functional association between polymerization (by pol γ) and excision (by external p53 exonuclease) activities, which act in a coordinated manner during DNA synthesis. Presumably, the elongation (lanes 4 and 8) occurs after initial hydrolysis of the terminally mispaired nucleotide by p53-GST, correction of the mismatch by dTTP and subsequent extension from the new, correctly paired 3′ terminus (A:T) by pol γ.
Misincorporation in mitochondria in the presence of endogenous p53
Sequential incorporation and excision of T:G mispair by pol γ in H1299mit was further evaluated in the presence of endogenous p53 provided by cytoplasmic extract of LCC2 cells (LCC2cyt) expressing wild-type p53 (wtp53) (Figure 3a, lane 1) with intrinsic exonuclease activity.25 After misincorporation of dGTP (Figure 3b, lanes 2 and 3), the reaction mixture was further incubated in the absence (lanes 4 and 5) or presence of LCC2cyt (lanes 6 and 7). The results show the efficient excision of the incorporated T:G mispair in the presence of LCC2cyt (expressing high level of exonuclease activity). The amount of the degraded products significantly increased with incubation time, and products lower than 16mer were formed (lanes 6 and 7). In control experiments, we verified no excision of the mispair with H1299cyt (lanes 8 and 9). The specific depletion of p53 from the LCC2cyt following immunoprecipitation by Do-1 anti-p53 antibody (Figure 3a, lane 2) is concomitant with a marked reduction in the removal of the T:G mispair (Figure 3b, lane 10). Nonspecific immunoprecipitation by the anti-horse IgG (H+L) (Figure 3a, lane 3) did not affect the extent of excision observed with LCC2cyt (Figure 3b, lane 11). The efficient removal of the T:G mispair after addition of LCC2cyt was apparently due to the presence of functional p53 protein.

Incorporation of wrong nucleotide dGTP in the mitochondrial fraction of H1299 cells in the presence of cytoplasmic fraction of LCC2 cells. (a) Cytoplasmic fractions of LCC2 cells (5 μg) (lane 1) expressing high levels of p53 or immunodepleted by either Do-1 anti-human p53 mAb (lane 2) or anti-horse IgG (H+L) (lane 3) were assayed for p53 expression by WB. (b) The incorporation of dGTP opposite the template T was examined with H1299mit (5 μg). After incubation of DNA template/primer (lane 1) for 10 min (lane 2) and 20 min (lane 3), the reaction mixture was further incubated in the absence (lanes 4 and 5) or presence of LCC2cyt (5 μg) (lanes 6 and 7) or LCC2cyt immunodepleted by Do-1 anti-human p53 mAb (lanes 8 and 9) or by anti-horse IgG (H+L) (lanes 10 and 11). Aliquots were taken after 20 min (lanes 4, 6, 8 and 10) and 40 min (lanes 5, 7, 9 and 11) of incubation. The position of the 16mer primer is indicated by an arrow
In vivo correlation between mitochondrial p53 and enhanced exonuclease activity
Mitochondrial localization of p53 has been observed in several systems.3, 4, 5, 6 p53 was detected within the inner mitochondrial compartment, where mtDNA and mtBER components localize.7, 8, 28 It was of interest to evaluate the incorporation and excision of the wrong nucleotide by mitochondrial extracts derived from isogenic HCT116 cells. p53 was detected in the mitochondrial fraction of HCT116 (p53+/+) cells (p53+/+)mit), but not of HCT116 (p53−/−) cells (p53−/−)mit) with Do-1 anti-human p53 mAb (Figure 4a). To define the localization of p53 within mitochondria, we tested the sensitivity of mitochondria-associated p53 to proteinase K. (p53+/+)mit was incubated with proteinase K to degrade external protein, with or without Triton X-100, and mitochondrial proteins were examined by WB for p53 and Bcl-2. As shown in Figure 4b, Bcl-2, which is associated with the external mitochondrial membrane, was completely degraded, regardless of whether Triton X-100 was added or not. However, p53 protein was highly protected from proteinase K in the absence of detergent, indicating that it is located in the inner mitochondrial compartment.

Exonuclease activity in the mitochondrial fraction of HCT116 cells. (a) Mitochondrial fractions (10 μg) of HCT116 (p53−/−) and HCT116 (p53+/+) cells were tested for p53 expression by Do-1 anti-human p53 mAb. Cytochrome c was blotted as a protein loading control. (b) p53 detection in submitochondrial fraction. HCT116 (p53+/+)mit (control – C) was treated with 50 ng/ml proteinase K (PK) with or without 0.5% Triton (TX) at room temperature for 30 min. Reaction was examined by WB with antibodies anti-Bcl-2 and -p53. (c) Dose response. The exonuclease activity in (p53−/−)mit (lanes 2–5) and (p53+/+)mit (lanes 6–9) was analyzed with 3′-terminal A:A mispair containing DNA/DNA template/primer (lane 1), under standard exonuclease assay conditions by increasing the amount of mitochondrial protein extracts. Lanes 2 and 6 – incubation with 2 μg of extracts. Lanes 3 and 7 – incubation with 5 μg of extracts. Lanes 4 and 8 – incubation with 10 μg of extracts. Lanes 5 and 9 – incubation with 20 μg of extracts. The reaction mixtures were incubated for 10 min. (d) Time course of 3′-terminal nucleotide excision with (p53−/−)mit (lanes 1–5) or (p53+/+)mit (lanes 6–10) was tested by incubation with 3′-terminal A:A mispair containing DNA/DNA template/primer. The reaction was started by the addition of mitochondrial fractions (5 μg). After 2 min (lanes 1 and 6), 5 min (lanes 2 and 7), 10 min (lanes 3 and 8), 30 min (lanes 4 and 9) and 60 min (lanes 5 and 10) of incubation at 37°C, 5 μl aliquots were withdrawn and analyzed on a 16% polyacrylamide gel. (e) The mitochondrial extracts of (p53−/−) (lanes 1–4) and (p53+/+) (lanes 5–8) cells were tested for 3′ → 5′ exonuclease activity with dsDNA substrates containing 3′-teminal mispair A:A (lanes 1 and 5), A:C (lanes 2 and 6), A:G (lanes 3 and 7) or correct A:T pair (lanes 4 and 8). (f) Incorporation of wrong nucleotide dGTP (lanes 1 and 2) or correct nucleotide dATP (lanes 3 and 4) was analyzed with (p53−/−)mit (lanes 1 and 3) or (p53+/+)mit (lanes 2 and 4). Sequences of template-primers – see legend to Figure 2b. The position of the 16mer primer is indicated by an arrow
Mitochondrial extracts of HCT116 (p53+/+) and HCT116 (p53−/−) cells, tested for exonuclease activity, showed the removal of 3′-terminal A:A mispair in a dose-dependent manner (Figure 4c). However, the substrate was degraded more efficiently by (p53+/+)mit (lanes 6–9) than by (p53−/−) mit (lanes 2–5). Time course analysis demonstrated that the excision of A:A mispair occurs faster and more efficiently with (p53+/+)mit than with (p53−/−)mit (Figure 4d). Furthermore, under non-polymerization conditions, (p53+/+)mit displays higher 3′-terminal nucleotide excision activity with 3′ incorrect (A:A, A:C or A:G) or correct (A:T) template-primers (Figure 4e, lanes 5–8) than (p53−/−)mit (lanes 1–4), with characteristic preferential excision of purine–purine mispairs (e.g., A:A and A:G) over purine–pyrimidine mispairs (e.g., A:C).22, 25 Interestingly, incubation of the correctly paired DNA substrate with (p53+/+)mit results in less incorporation of wrong dGTP opposite the template T (Figure 4f, lane 2), as compared with that with the same amount of (p53−/−)mit (lane 1). Remarkably, both fractions had comparable DNA synthesis activities during the incorporation of correct dATP nucleotide (lanes 3 and 4). Thus, the low level of misincorporation with (p53+/+)mit is not due to the low level of polymerization activity of pol γ, but rather due to the presence of p53.
Immunoprecipitation of (p53+/+)mit with the anti-p53 antibody showed a correspondence between the level of p53 present in the lysate and exonuclease activity (lane 3 in Figure 5a and b, respectively). Conversely, nonspecific depletion did not affect either p53 expression or exonuclease activity (lane 2 in Figure 5a and b, respectively). The contribution of endogenous p53 to the high level of exonuclease activity in p53(+/+)mit was confirmed by the supplementation of exogenous p53 in (p53−/−)mit. Although supplementation with H1299mit (increasing the amount of pol γ) elicited no increase in excision of wrong nucleotide (Figure 5c, lane 3), addition of recombinant p53 (lane 6), or cellular fractions harboring endogenous wtp53, for example LCC2cyt (lane 4) or (p53+/+)mit (lane 5), stimulated efficient excision of incorporated mispair.

3′ → 5′ Exonuclease activity in p53-depleted HCT116 (p53+/+)mit and excision of the incorporated wrong nucleotide in HCT116 (p53−/−)mit. (a) Mitochondrial fractions of HCT116 (p53+/+) cells (10 μg) (lane 1) immunodepleted by anti-horse IgG (H+L) (lane 2) or Do-1 anti-human p53 mAb (lane 3) were assayed for p53 expression by western blotting. Cytochrome c was blotted as a protein loading control. (b) Excision of the 3′-terminal A:A mismatch containing DNA/DNA template/primer was examined with 5 μg of HCT116 (p53+/+)mit (lane 1) or HCT116 (p53+/+)mit immunodepleted by anti-horse IgG (H+L) (lane 2) or HCT116 (p53+/+)mit immunodepleted by Do-1 anti-human p53 mAb (lane 3). (c) Excision of the incorporated wrong dGTP nucleotide in HCT116 (p53−/−)mit (lane 1) was examined in the absence (lane 2) or presence of H1299mit (lane 3), LCC2cyt (lane 4), HCT116 (p53+/+)mit (lane 5) or p53-GST (250 ng) (lane 6). The position of the 16mer primer is indicated by an arrow
Our studies illustrated that cytoplasmic fractions of H1299 or HCT116 cells overexpressing wtp53, but not exonuclease-deficient mutant p53-R175H, had significantly higher levels of exonuclease activity, than those transfected with the control vector, supporting the likelihood that p53 in cytoplasm is linked to observed exonuclease activity.32 These cytoplasmic fractions were used to further define the contribution of endogenous p53 exonuclease to the error correction in H1299mit. The results illustrate the efficient excision of the incorporated T:G mispair in the presence of H1299cyt overexpressing wtp53 (Figure 6, lanes 3 and 4). No efficient excision was observed in the presence of H1299cyt transfected with the control vector (lanes 1 and 2) or exonuclease-deficient mutant p53-R175H (lanes 5 and 6). However, the excision of the incorporated T:G mispair (lane 7) was also detected in the presence of H1299cyt overexpressing mutant p53-R174K (lanes 8 and 9) expressing high levels of exonuclease activity (data not shown). Notably, arginine 174 is not located in or near the DNA-binding site.33

Excision of the incorporated wrong nucleotide in HCT116 (p53−/−) mitochondria in the presence of cytoplasmic fractions of p53-transfected cells. Excision of the incorporated wrong dGTP nucleotide in HCT116 (p53−/−)mit was examined in the presence of cytoplasmic fractions of H1299 cells (1 μg) transfected with control vector (lanes 1, 2 and 7) or wtp53 (lanes 3 and 4), mutant p53-R175H expression vector (lanes 5 and 6) or mutant p53-R174K expression vector (lanes 8 and 9). Aliquots were taken after 20 min (lanes 1, 3, 5 and 8) and 40 min (lanes 2, 4, 6, 7 and 9) of incubation. The position of the 16mer primer is indicated by an arrow
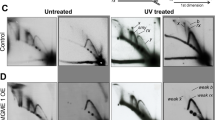
Irradiation (IR) induces the mitochondrial translocation of p53 in various cell lines (e.g., MCF-7 and HCT116) that are highly resistant to apoptosis.6 Mitochondrial p53 comprised small amount (∼2%) of the total induced p53. The absence of activated apoptotic pathway in HCT116(p53+/+) cells under p53-inducible conditions, for example IR, permits the use of mitochondrial fractions as a suitable experimental system for the analysis of p53 exonuclease activity in the context of a cellular stress. Consistent with the published results and using appropriate combination of antibodies, immunofluorescent staining of p53 in HCT116(p53+/+) cells showed increased p53 levels in IR-treated cells (predominantly in nucleus) compared with control untreated cells (Figure 7a). Interestingly, increased mitochondrial localization of p53 was observed as a yellow signal in the overlay panel. IR-induced p53 translocation to the mitochondria was further documented by WB analysis (Figure 7b). The evaluation of the mitochondrial fractions for the exonuclease activity revealed the increase in constitutive A:A mispair excision capacity following the IR treatment and p53 translocation to mitochondria (Figure 7c, lanes 1 and 2). Furthermore, lower incorporation of wrong nucleotide dGTP (lanes 3 and 4), but not correct dATP (lanes 5 and 6), (opposite the template T), was observed with IR-treated (p53+/+)mit as compared with that with the same amount of untreated (p53+/+)mit. Apparently, pol γ is not responsible for the observed increased error-correction activities in (p53+/+)mit following the IR event, as there was no difference in mispair excision, misincorporation or correct polymerization between the mitochondrial fractions of untreated and IR-treated HCT116(p53−/−) cells (Figure 7d, lanes 7–12, respectively).

Irradiation induces translocation of p53 to mitochondria of HCT116 (p53+/+) cells and increases the error-correction activities. (a) Semiconfocal microscopic analysis of intracellular localization of p53 in untreated and IR-treated cells (24 h post-IR). Cells were labeled for p53 (green). Nuclear and mitochondrial expression of p53 was tested by staining the cells with DAPI (blue) and the antimitochondrial protein, OxPhos complex V inhibitor protein (F1F0-ATPase IP) (red), respectively. Localization of p53 in mitochondria was evidenced by the yellow signal in the overlay panel. (b) WB analysis of the mitochondrial fractions (10 μg) of untreated and IR-treated cells for the status of p53. The mitochondrial protein cyt-c was used as a loading control. (c) Mitochondrial extracts of untreated and IR-treated (24 h post-IR) HCT116 (p53+/+) and HCT116 (p53−/−) cells were examined for either 3′-terminal A:A mispair excision activity (lanes 1, 2 and 7, 8, respectively), incorporation of wrong dGTP nucleotide opposite the template T (lanes 3, 4 and 9, 10, respectively) or correct dATP (lanes 5, 6 and 11, 12, respectively). Sequences of template-primers – see legend to Figure 2b. The position of the 16mer primer is indicated by an arrow
Discussion
The current studies aimed to evaluate the potential involvement of p53 in mtDNA repair. Several lines of experimental evidence show that p53 has an impact on the accuracy of mtDNA synthesis, which are listed below. (1) The excision of incorporated wrong nucleotide in H1299mit increases in the presence of recombinant p53. The contribution of p53 depended on the integrity of the protein, as there was no increase in exonuclease activity in the presence of heat-inactivated p53. (2) Efficient mispair excision was detected in the presence of LCC2cyt expressing endogenous p53 protein. The enhanced exonuclease activity was greatly reduced in pre-depleted LCC2cyt with specific anti-p53 antibodies. (3) The mispair excision is lower in (p53−/−)mit as compared with that in (p53+/+)mit. The lower activity is probably related to the absence of p53. The complementation of exonuclease activity in HCT116(p53−/−)mit after the addition of recombinant p53 or fractions harboring endogenous p53 suggests the direct contribution of external p53 exonuclease activity. (4) The incorporation of wrong nucleotide is lower in (p53+/+)mit compared with that in (p53−/−)mit, although p53 status did not influence the incorporation of correct nucleotide. (5) Increased removal of mispair was observed with H1299cyt overexpressing wtp53, but not exonuclease-deficient mutant p53-R175H. (6) The mitochondrial translocation of endogenous p53 following the IR stress stimuli correlates with the increase in DNA repair activities, for example increased mispair excision and low misincorporation. These findings, taken together with the fact that p53 is located in the inner mitochondrial compartment (Figure 4b), support the view that p53 in mitochondria may contribute a proofreading function by excising potential errors at the 3′ termini.
Proofreading during DNA synthesis in mitochondria can be carried out by intramolecular or intermolecular processes. When the substrate has a mispaired primer terminus, DNA dissociation seems generally more rapid than exonucleolytic digestion. The dissociation rate is increased by 10-fold in the presence of a mispaired versus correctly paired 3′ terminus, providing an opportunity for exonucleolytic hydrolysis upon rebinding of different DNA polymerase or external exonuclease.14, 15 The observed decrease in mispair excision in mitochondria with p53 in the presence of a DNA or RNA trap confirms that the enzyme dissociates before excision of DNA and that the misinserted nucleotides can be corrected by an error–repair pathway involving external exonuclease (Figure 2a). The observed functional interaction between pol γ and p53 in mitochondria with preformed 3′-terminally mispaired substrates, under conditions that mimic a situation of intermolecular editing during DNA synthesis, points to a strict mechanism of mismatch correction before polymerization (Figure 2b). The logical interpretation of the functional coordination is that nucleotide misinsertion results in polymerase dissociation from the template-primer, leaving the 3′-terminal mispair accessible to p53. Upon excision of the mispair, the p53 probably dissociates, thus allowing the pol γ to reassociate with the correct 3′ terminus and resume DNA synthesis (Figure 8). Our findings are consistent with recent reports demonstrating the increase in the polymerization product in the presence of p5328 and provide a mechanism by which p53 could influence the accuracy of DNA synthesis.

A model for error correction in mitochondria in the presence of p53. The incorporation of wrong nucleotide into mitochondrial DNA ( ) results in DNA polymerase γ dissociation from the template-primer, leaving the 3′-terminal mispair accessible to p53. Upon excision of the mispair, the p53 exonuclease dissociates, thus allowing DNA polymerase γ to reassociate with the correct 3′ terminus and resume DNA synthesis
) results in DNA polymerase γ dissociation from the template-primer, leaving the 3′-terminal mispair accessible to p53. Upon excision of the mispair, the p53 exonuclease dissociates, thus allowing DNA polymerase γ to reassociate with the correct 3′ terminus and resume DNA synthesis
We have shown the differences in p53 exonuclease activity in nuclear and cytoplasmic compartments.25 Evidently, p53 in nucleus, cytoplasm and mitochondria harbors distinct post-translational modifications, which may be involved in the regulation of the subcellular localization and functions of p53.5 Notably, methylated and acetylated p53s are restricted to the nucleus, whereas mitochondrial p53 is not acetylated. Hence, it may be interesting to further investigate the involvement of post-translational modifications in the regulation of the ability of p53 to serve as an exonuclease in nucleus, cytoplasm and mitochondria.
The possibility that p53 can enhance the accuracy of pol γ would present two challenges for p53 in mitochondria: (i) p53 protein must have an access to DNA polymerization complex and (ii) the level of p53 in mitochondria and the ratio of p53 exonuclease/pol γ must be high.
(i) The p53 protein physically interacts with mtDNA and pol γ.28 Identification of a putative p53-binding sequence within the human mitochondrial 16S rDNA region implies that p53 may be involved in DNA replication through binding mtDNA.34 Recent studies have reported that mtDNA and various proteins involved in its replication and maintenance (e.g., pol γ, mtTFA) may all associate with an inner membrane fraction.27 p53 can enhance the binding of mtTFA to cisplatin-damaged DNA by 10- to 20-fold, thus pointing to the function of p53 in mtDNA damage repair.29 Furthermore, p53 enhances BER through direct interaction with the repair complex in an inner mitochondrial membrane subfraction.8 The fact that p53 localizes to the mitochondria and interacts with mtDNA and pol γ, taken together with our observations that the presence of p53 decreases the amount of incorporation of wrong nucleotide in H1299mit, suggests that p53 may be the fidelity-enhancing component of DNA replication machinery in mitochondria.
(ii) The polymerase/exonuclease ratio serves as an important enzymatic ‘marker’ of polymerase fidelity.35 The proofreading efficiency increases on decreasing the ratio between polymerase and exonuclease. In mitochondria, the change in ratio could be achieved through p53 gene induction (increase in p53 concentration), p53 targeting (increase in local mitochondrial concentration) or reduction in polymerization efficiency of pol γ due to mutations. The accuracy of DNA synthesis in mitochondria might respond to alterations in composition of replication complex. The mtDNA synthesis may be a dynamic process with p53 component binding and dissociating the polymerization complex during DNA synthesis, thus affecting the polymerase/exonuclease (p53) ratio. Cellular responses to DNA damage include repair processes that act coordinately, before, during and after DNA replication, to maintain genomic stability. p53 is able to excise 3′-terminal nucleotides during the ongoing DNA synthesis that is coupled with DNA polymerization and following direct binding to template-primer that is independent of DNA polymerase, thus increasing the potency of involvement of the protein during DNA replication by acting as an external proofreader.23 Consequently, the presence of p53 in mitochondria, by carrying these properties, may be relevant to the accuracy of DNA synthesis by pol γ.
Mitochondrial localization of p53 was observed in unstressed cells, at the onset of p53-dependent apoptosis and in p53-independent apoptosis systems.3, 5 The exact function of p53 in mitochondria remains incomplete, as p53 may serve a triggering function for mitochondrial-driven apoptosis and mtDNA repair.36 Although there is no correlation between mitochondrial p53 accumulation and apoptosis susceptibility, our results demonstrate increased DNA repair responses following the IR-stress stimuli and enhancement of endogenous p53 in mitochondria, for example increased mispair excision and low misincorporation. In DNA repair pathways, key proteins may serve dual functions in DNA repair and apoptosis, independently or simultaneously. A causal relationship exists between inefficient mtDNA repair and the induction of apoptosis.37 It is of interest to elucidate p53 exonuclease activity in mitochondria under conditions that provoke p53-dependent apoptosis following DNA damage. The switch from repair to apoptosis may occur when DNA damage (hypermutation) overwhelms repair capacity or following the interaction of p53 with other proapoptotic proteins. These possibilities need to be further investigated.
DNA pol γ in mitochondria is highly sensitive to inhibition by nucleoside analogs (NAs) used in the treatment of HIV infection. Incorporation of NAs leads to mitochondrial toxicity in antiviral therapy. Polymerase γ exonuclease is inefficient in removing these drugs.38 Earlier studies have demonstrated the efficient removal of NA from the 3′ terminus in the presence of p53.39, 40 p53 in mitochondria can play an important function as an external proofreader in the removal of 3′-terminal NAs, thus decreasing their potential for chain termination (manuscript in preparation).
In view of these observations, it seems reasonable to hypothesize that p53 in mitochondria might provide a proofreading function for pol γ during mtDNA replication. The functional coupling between the p53 exonuclease and pol γ should decrease nucleotide misinsertion, thus reducing the production of polymerization errors.
Materials and Methods
Materials
The recombinant purified glutathione-S-transferase (GST) and p53-GST fusion proteins expressed in Escherichia coli were obtained from LabVision Cooperation.
Cell lines and culture medium
H1299 cells were grown in the presence of RPMI-1640+10% fetal calf serum (FCS). The LCC2 cells (subclone derived from MCF-7) were grown in DMEM lacking phenol red+10% charcoal-stripped FCS. The colorectal cancer cells isogenic for p53, HCT116 (p53+/+) and HCT116 (p53−/−) were grown in McCoy's medium supplemented with 10% FBS. All lines were maintained in a humidified 95% air, 5% CO2 atmosphere.
Preparation of mitochondrial fraction
Mitochondria were prepared as described.3 Briefly, cells washed in TD buffer were centrifuged (2500 r.p.m. at 4°C) and resuspended in MgRSB buffer (1.5 mM MgCl2, 10 mM NaCl, 10 mM Tris (pH 7.5)) and incubated for 10 min. Swollen cells were disrupted in a glass Dounce homogenizer to yield approximately 95% of free nuclei. Mitochondria were pelleted from the washed homogenate, resuspended in a manitol–sucrose buffer, layered over a 1–2 M discontinuous sucrose gradient and spun at 4°C for 30 min at 22 000 r.p.m. The mitochondria were collected from the 1.0 to 1.5 sucrose interphase.
Western blotting
Equal amounts of total protein of mitochondrial or cytoplasmic fractions were subjected to WB analysis as described.25 The following antibodies were used: Do-1 monoclonal antibody for p53 (Oncogene), antibodies for cytochrome c (Oncogene) and polyclonal antibody for c-jun (Santa Cruz Biotechnology). Immunoprecipitation for p53 was performed as previously described.25
Template-primers
The sequences of the template-primers used for the experiments are depicted in figures. The primers were end labeled at the 5′-end with T4 polynucleotide kinase (Fermentas) and [γ-32P] ATP. Unincorporated radioactivity was removed by using G-25 microspin columns (Pharmacia Biotech.), according to the manufacturer's instructions. The end-labeled primers were annealed to the template DNA as described.22
Exonuclease/polymerase-coupled assays
DNA primer extension assay used allowed simultaneous detection of both degradation (exonucleolysis) and extension (polymerization). Pol γ hydrolyzes 3′-terminal mismatched nucleotides from DNA under conditions of either absence or presence of DNA synthesis13, 14 although the rate of exonucleolytic degradation is lower under polymerization conditions than in the absence of DNA synthesis. Time course analysis was performed at various dNTP and mitochondrial protein concentrations to obtain equal activities of exonuclease and polymerase (data not shown). For site-specific nucleotide misinsertion, a correctly paired template-primer substrate was used to analyze the dGTP misincorporation opposite the T residues at position 10 of the DNA template. The incubation mixture (10 μl) contained 50 mM Tris HCl (pH 7.5), 5 mM MgCl2, 1 mM DTT, 0.1 mg/ml BSA, 5′-end-labeled substrates, nucleotide and mitochondrial protein extracts. The 3′ → 5′ exonuclease activity was detected by excision of the 3′-terminal nucleotide, resulting in the conversion of the 16mer 5′-end-labeled oligonucleotide to 15mer, 14mer and smaller oligonucleotides, as determined by increased mobility during electrophoresis. The reaction products (polymerization or excision) were analyzed by electrophoresis through 16% PAGE as described.22 The dNTPs used were of the highest purity available (Pharmacia Biotech.) with no detectable traces of contamination by other dNTPs. The variables, including reaction time and amounts of extracts, are given in the legends to the figures. Degradation or extension of the 5′-end-labeled primers was detected by autoradiography.
Immunofluorescence microscopy
HCT116(p53+/+) cells were exposed to ionizing radiation (20 gray), using a Gammacell 1000 Elite. Cells were fixed with 4% (w/v) paraformaldehyde in PBS for 20 min and permeabilized with 0.1% Triton X-100 for 5 min at room temperature. Immunostaining was performed using mouse anti-p53 monoclonal antibody (DO-1; Santa Cruz, USA) at a dilution of 1 : 100, followed by incubation with secondary goat anti-mouse antibody (DyLight 488) (Pierce Biotechnology, USA) diluted 1 : 100. Mitochondrial staining was performed using mouse monoclonal anti-OxPhos complex V inhibitor protein (F1F0-ATPase IP) (Invitrogen, USA) at a dilution of 1 : 100. DAPI staining was added in the mounting gel. Fluorescent images were obtained using an Olympus IX81 inverted microscope equipped with X-cite 120 Halogen light source.
Proteinase K treatment
Mitochondrial extracts were treated with 50 ng/ml proteinase K with or without 0.5% Triton X-100 at room temperature for 30 min. The reaction was stopped by the addition of 0.1 mM protease inhibitor mixture and then analyzed by WB with antibodies to p53 and Bcl-2.
Abbreviations
- H1299mit:
-
mitochondrial fraction of H1299 cells
- HCT116 (p53−/−)mit:
-
mitochondrial fraction of HCT116 (p53−/−) cells
- HCT116 (p53+/+)mit:
-
mitochondrial fraction of HCT116 (p53+/+) cells
- LCC2cyt:
-
cytoplasmic fraction of LCC2 cells
- mtDNA:
-
mitochondrial DNA
- pol γ:
-
DNA polymerase γ
- wtp53:
-
wild-type p53
References
Lane DP . Cancer: p53, guardian of the genome. Nature 1992; 358: 15–16.
Prives C, Hall PA . The p53 pathway. J Pathol 1999; 187: 112–126.
Marchenko ND, Zaika A, Moll UM . Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 2000; 275: 16202–16212.
Sansome C, Zaika A, Marchenko ND, Moll UM . Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett 2001; 488: 110–115.
Mahyar-Roemer M, Fritzsche C, Wagner S, Laue M, Roemer K . Mitochondrial p53 levels parallel total p53 levels independent of stress response in human colorectal carcinoma and glioblastoma cells. Oncogene 2004; 23: 6226–6236.
Essmann F, Pohlmann S, Gillissen P, Daniel PT, Schulze-Osthoff K, Janicke RU . Irradiation-Induced translocation of p53 to mitochondria in the absence of apoptosis. J Biol Chem 2005; 280: 37169–37177.
de Souza-Pinto NC, Harris CC, Bohr VA . p53 functions in the incorporation step inDNA base excision repair in mouse liver mitochondria. Oncogene 2004; 23: 6559–6568.
Chen D, Yu Z, Zhu Z, Lopez CD . The p53 pathway promotes efficient mitochondrial DNA base excision repair in colorectal cancer cells. Cancer Res 2006; 66: 3485–3494.
Kaguni LS . DNA polymerase γ, the mitochondrial replicase. Annu Rev Biochem 2004; 73: 293–320.
Singh KK . Mitochondria damage checkpoint in apoptosis and genome stability. FEMS Yeast Res 2004; 5: 127–132.
Copeland WC, Ponamarev MV, Nguyen D, Kunkel TA, Longley MJ . Mutations in DNA polymerase gamma cause error-prone DNA synthesis in human disorders. Acta Biochim Pol 2003; 50: 155–167.
Pesole G, Gissi C, De Chirico A, Saccone C . Nucleotide substitution rate of mammalian mitochondrial genomes. J Mol Evol 1999; 48: 427–434.
Kunkel T, Sonu A . Exonucleolytic proofreading enhances the fidelity of DNA synthesis by chick embryo DNA polymerase gamma. J Biol Chem 1988; 263: 4450–4459.
Johnson A, Johnson K . Fidelity of nucleotide incorporation by human mitochondrial DNA polymerase. J Biol Chem 2001a; 276: 38090–38106.
Johnson A, Johnson K . Exonuclease proofreading by human mitochondrial DNA polymerase. J Biol Chem 2001b; 276: 38097–38107.
Nishigaki Y, Marti R, Copeland WC, Hirano M . Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. J Clin Invest 2003; 111: 1913–1921.
Mathews CK . DNA precursor metabolism and genomic stability. FASEB J 2006; 20: 1300–1314.
Trifunovic A, Wredenberg A, Falkrnberg M, Spelbrink JN, Rovio AT, Bruder CE et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Nature 2004; 429: 417–423.
Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM et al. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000; 287: 2017–2019.
Albrechtsen N, Dornreiter I, Grosse F, Kim E, Wiesmuller L, Deppert W . Maintenance of genomic integrity by p53: complementary roles for activated p53. Oncogene 1999; 18: 7708–7717.
Huang P . Excision of mismatched nucleotides from DNA: a potential mechanism for enhancing DNA replication fidelity by the wild-type p53 protein. Oncogene 1998; 17: 261–270.
Bakhanashvili M . Exonucleolytic proofreading by p53 protein. Eur J Biochem 2001a; 268: 2047–2054.
Bakhanashvili M . p53 enhances the fidelity of DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. Oncogene 2001b; 20: 7635–7644.
Melle C, Nasheuer H . Physical and functional interactions of the tumor suppressor protein p53 and DNA polymerase α-primase. Nucleic Acids Res 2002; 30: 1493–1499.
Lilling G, Novitsky E, Sidi Y, Bakhanashvili M . p53-associated 3′ → 5′ exonuclease activity in nuclear and cytoplasmic compartments of cells. Oncogene 2003; 22: 233–245.
Bakhanashvili M, Novitsky E, Lilling G, Rahav G . p53 in cytoplasm may enhance the accuracy of DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. Oncogene 2004; 23: 6890–6899.
Stuart JA, Mayard S, Hashiguchi K, Souza-Pinto NC, Bohr VA . Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulated fraction. Nucleic Acids Res 2005; 33: 3722–3732.
Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H et al. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA pol γ. EMBO J 2005; 24: 3482–3492.
Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Yoshida T, Itoh H et al. P53 physically interacts with mitochondrial transcription factor A and differentially regulates binding to damaged DNA. Cancer Res 2003; 63: 3729–3734.
Wang Z, Lazarov E, O'Donnell M, Goodman MF . Resolving a fidelity paradox: why Escherichia coli DNA polymerase II makes more base substitution errors in AT- compared with GC-rich DNA. J Biol Chem 2002; 277: 4446–4454.
Song S, Pursell ZF, Copeland WC, Longley MJ, Kunkel TA, Mathews CK . DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc Natl Acad Sci USA 2005; 102: 4990–4995.
Bakhanashvili M, Gedelovich R, Grinberg S, Rahav G . Exonucleolytic degradation of RNA by the tumor suppression protein p53 in cytoplasm. J Mol Med 2008; 86: 75–88.
Ashur-Fabian O, Avivi A, Trakhtenbroat L, Adamsky K, Cohen M, Kajakaro G et al. Evolution of p53 in hypoxia-stressed spalax mimics human tumor mutation. Proc Natl Acad Sci USA 2004; 101: 12236–12241.
Heyne K, Mannebach S, Wuertz E, Knaup KZ, Mahyar-Roemer M, Roemer K . Identification of a putative p53 binding sequence within the human mitochondrial genome. FEBS Lett 2004; 578: 198–202.
Goodman MF, Fygenson DK . DNA polymerase fidelity: from genetics toward a biochemical understanding. Genetics 1998; 148: 1475–1482.
Nithipongvanitch R, Ittarat W, Velez JM, Zhao R, Clair DKS, Oberlay TD . Evidence for p53 as guardian of the cardiomyocyte mitochondrial genome following acute adriamycin treatment. J Histochem Cytochem 2007; 55: 629–639.
LeDoux SP, Druzhyna NM, Hollensworth SB, Harrison KF, Wilson GL . Mitochondrial DNA repair: a critical player in the response of cells of the CNS to genotoxic insults. Neuroscience 2007; 145: 1249–1259.
Feng JY, Johnson AA, Johnson KA, Anderson KS . Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J Biol Chem 2001; 276: 23832–23837.
Feng L, Achanta G, Pelicano H, Zhang W, Plunkett W, Huang P . Role of p53 in cellular response to anticancer nucleoside analog-induced DNA damage. Int J Mol Med 2000; 5: 597–604.
Bakhanashvili M, Novitsky E, Rubinstein E, Levy I, Rahav G . Excision of nucleoside analogs from DNA by p53 protein, a potential cellular mechanism of resistance to inhibitors of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother 2005; 49: 1576–1579.
Acknowledgements
This research was supported by grant from Israel Cancer Association. We thank Novitsky Elena for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by L Scorrano
Rights and permissions
About this article
Cite this article
Bakhanashvili, M., Grinberg, S., Bonda, E. et al. p53 in mitochondria enhances the accuracy of DNA synthesis. Cell Death Differ 15, 1865–1874 (2008). https://doi.org/10.1038/cdd.2008.122
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.122
