
Abstract
Parathyroid hormone (PTH) secretion is characterized by an ultradian rhythm with tonic and pulsatile components. In healthy subjects, the majority of PTH is secreted in tonic fashion, whereas approximately 30% is secreted in low-amplitude and high-frequency bursts occurring every 10–20 min, superimposed on tonic secretion. Changes in the ultradian PTH secretion were shown to occur in patients with primary and secondary osteoporosis, with skeletal effects depending on the reciprocal modifications of pulsatile and tonic components. Indeed, pathophysiology of spontaneous PTH secretion remains an area potentially suitable to be explored, particularly in those conditions such as secondary forms of osteoporosis, in which conventional biochemical and densitometric parameters may not always give reliable diagnostic and therapeutic indications. This review will highlight the literature data supporting the hypothesis that changes of ultradian PTH secretion may be correlated with skeletal fragility in primary and secondary osteoporosis.
Similar content being viewed by others

Introduction
Pulsatility is a fundamental property of the majority of hormone secretion1 and denotes the recurrence of individual punctuated events (busts, peaks or pulses) interrupting a more or less constant baseline process.1 Many hormones have a pulsatile component to their secretory profile and pulsatility is believed to modulate target organ responsiveness.2,3 Interestingly, there is intra-individual variability in number and/or amplitude and/or frequency of hormone pulses with potential modifications in the target tissue responsiveness.1 In fact, growth hormone (GH) pulse size and frequency may be influenced by several hormones such as cortisol and testosterone, physiological such as fasting and aging, and pathological conditions such as obesity and diabetes.3–9 Luteinizing hormone pulsatility is the prototype of dual amplitude and frequency regulation, whereas thyrotropin and adrenocorticotropin secretory patterns are examples of triple control of pulsatility with size, frequency and number bursts being influenced to several signals.1 Although pituitary gland is the paradigm of pulsatile hormone secretion under control of several hypothalamic neuroendocrine signals, also the peripheral glands can secrete hormones in a pulsatile manner.
In this paper, we focus on parathyroid hormone (PTH) secretion, dealing with physiological, pathophysiological and clinical aspects related to the pulsatile secretion of the hormone.
Physiology of PTH secretion
Circadian and seasonal secretion
Parathyroid glands secretory activity shows seasonal and circadian fluctuations, which are associated with changes in serum calcium, phosphate and bone turnover.10 In fact, PTH levels usually decline about 20% below the annual mean during the summer, reflecting circannual rythms of vitamin D and bone turnover.11 Moreover, it has been described a circadian rhythm for PTH with a more pronounced peak in the early morning, a nadir in the late morning and a second, lower peak in the afternoon, likely related to calcium, phosphate and calcitriol changes which can be modified by nutritional intake of these components.12
Pulsatile ultradian secretion
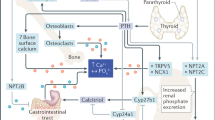
Spontaneous PTH secretion is also characterized by an ultradian rhythm with tonic and pulsatile components. In healthy young subjects, the majority of PTH (about 70%) is secreted in tonic fashion, whereas approximately 30% is secreted in low-amplitude and high-frequency bursts occurring every 10–20 min, superimposed on tonic secretion.13–17 PTH pulsatile secretion is highly sensitive to changes in ionized calcium and calcitriol serum levels.10 Calcium exerts its effects on the parathyroid gland through binding to a cell surface receptor, the calcium-sensing receptor.18 Acute hypocalcemia induces a selective, several-fold increase in bursts frequency and amplitude, whereas hypercalcemia suppresses the PTH pulsatile secretion component, as does prolonged calcitriol therapy. Indeed, calcitriol in synergism with ionized calcium is the major physiological inhibitor of PTH secretion.19 Conversely, vitamin D deficiency or resistance might contribute, indirectly (throughout the reduction in calcium absorption) and even directly to PTH hypersecretion.20
PTH effects on target tissues
The pulsatile secretion of PTH may have a role in physiological regulation of bone metabolism and structure. Although there are no animal models investigating specifically the skeletal effects of pulsatile PTH secretion, there is experimental evidence that intermittent administration of pharmacological doses of PTH has a bone anabolic effect, while continuous PTH administration is detrimental for the skeleton due to stimulation of bone resorption.21 Consistently with these experimental findings, daily injections of PTH produce significant increase in bone mass and decrease in fracture risk in patients affected by osteoporosis.22,23 The initial effect of PTH intermittent administration is the stimulation of bone modeling, which prompts to an increase in bone formation markers, whereas the subsequent stimulation of bone remodeling is characterized clinically also by an increase in bone resorption markers: this sequence gives rise to the so called ‘anabolic window’ during which the osteoanabolic actions of PTH take place.24 Comparable anabolic effects may be produced by treatment with short-acting antagonists of calcium sensing receptor, the so-called calcilytics, which induce an acute stimulation of PTH secretion by the parathyroid gland mimicking a pulsatile secretory pattern which may lead to improvement of bone mineral density (BMD) and decrease in fracture risk.25,26 Over the recent years, there has been convincing evidence that PTH exerts extra-skeletal effects, mainly on cardiovascular system and glucose metabolism.27–29 However, it is still unknown whether and how the pulsatile PTH secretion may influence these extra-skeletal targets.
Assessment of PTH pulsatility
Accurate characterization of hormonal pulses requires specific and sensitive assays, intensive schedule of blood sampling, and validated and objective methods of pulse analysis.13,17,30,31 Due to the intrinsic characteristics of PTH physiological secretion with very frequent and small pulses, PTH pulsatility studies are complex to perform and this explains in part the lack of published data on this topic. PTH is measured with immunoradiometric assays (sensitivity about 1 pg·mL−1; intra- and inter-assay coefficients of variation between 4% and 6%). PTH half-life is approximately 2–5 min and PTH pulses are estimated to occur with a frequency of 10–20 min. Therefore, in all the experimental studies on PTH pulsatility so far carried out, two milliliters of blood are withdrawn (samples are analyzed in duplicate) every 2–3 minutes for 6 h, usually from 9:00 a.m. to 3:00 p.m., in fasting subjects. Plasma PTH levels need to be analyzed by a deconvolution analysis in order to obtain an accurate estimate of secretion and clearance. The most representative parameters of this analysis are: basal PTH secretion rate (pg·mL−1·min−1), secretory bursts half-duration (minutes), maximal amplitude of secretory bursts (pg·mL−1·min−1) and the total number of secretory bursts. Plasma PTH levels can also be subjected to an approximate entropy analysis, which provides a measure of the orderliness of the release process.
PTH pulsatility in skeletal diseases
Primary hyperparathyroidism
Primary hyperparathyroidism (PHPT) is a common endocrine disorder characterized primarily by hypercalcemia and elevated levels of PTH. The common clinical presentation of PHPT has changed over the last two–three decades from an overt disease characterized by hypercalcemic symptoms and kidney stones to a mild or even asymptomatic disease.32 Although PHPT occurs at all ages, the majority of patients are postmenopausal women, in whom a variable degree of bone loss is commonly found, with a greater involvement of cortical sites. Increased fracture risk has been reported at both vertebral and non-vertebral sites in PHPT.33
PHPT is accompanied by qualitative and quantitative abnormalities of PTH secretion (Table 1). Harms et al.34 measured PTH in nine female patients with PHPT, compared with 10 postmenopausal control women. The analysis of pulsatile secretory pattern revealed significant increase in total PTH secretion per hour, basal PTH secretion per hour and average PTH secretion per pulse in PHPT patients as compared to the control subjects, without significant differences in the number of bursts between the two groups in which the pulsatile secretion accounted for about 50% of the total secretion.34 The cross-correlation of PTH and calcium indicated an impaired feedback regulation in PHPT. This study did not give insights on the possible clinical impact of PTH pulsatility on skeletal and extra-skeletal targets in PHPT patients.
Post-menopausal osteoporosis
Osteoporosis is a chronic, progressive bone disease in which bone resorption exceeds bone formation, leading to a reduction in BMD, disruption of bone microarchitecture and increase in fracture risk.35 The incidence of osteoporosis increases with age and it occurs most frequently in postmenopausal women due to the cessation in ovarian activity with consequent increase in bone resorption.36 Very few cross-sectional studies have investigated spontaneous PTH secretion in patients with post-menopausal osteoporosis, reporting discordant results30,37 (Table 1). Harms et al.37 observed a decrease in basal secretion and pulse amplitude of PTH during estrogen replacement therapy in early post-menopausal women, whereas Samuels et al.30 did not find relevant differences in structure and modality of PTH secretion in postmenopausal women versus young adults. Indeed, in all of these studies the number of subjects was too low to draw meaningful conclusions. In fact, it has to be taken into account the possible variability induced by several factors, such as nutritional and vitamin D status, daily calcium intake, duration and age of menopause.
Glucocorticoid-induced osteoporosis (GIO)
GIO is the most common cause of secondary osteoporosis.38 Fractures are often asymptomatic and occur in about 30%–50% of patients receiving chronic glucocorticoid therapy.39 Vertebral fractures occur early after exposure to glucocorticoids, and they tend to occur at higher BMD levels than in women with postmenopausal osteoporosis.40 Glucocorticoids impair the replication, differentiation and function of osteoblasts and induce the apoptosis of mature osteoblasts and osteocytes.38 These effects lead to a suppression of bone formation. Glucocorticoids also increase bone resorption by increasing osteoclastogenesis.38 In addition to the direct effects of glucocorticoids on bone cells, other skeletal effects are mediated by neuroendocrine changes, such as functional GH deficiency, hypogonadism and possible qualitative abnormalities of PTH secretion.40 Most explanatory diagrams of GIO include a secondary, compensatory, rise in PTH which is believed to be due to the effects of glucocorticoids in limiting calcium absorption in the gastrointestinal tract (by decreasing the expression of specific calcium channels in the duodenum) and in facilitating renal calcium excretion.40 Some studies have not found an association between GIO and increased PTH levels; conversely, other studies have shown that patients with GIO did exhibit frankly elevated PTH serum levels.41 These data can be reconciled thinking to the relatively low clinical relevance of baseline PTH levels, since the hormone is secreted in a pulsatile fashion. However, very few data are so far available on the effect of chronic glucocorticoid excess on spontaneous fluctuations of PTH in humans. Bonadonna et al.17evaluated spontaneous PTH pulsatile secretion in patients chronically treated with pharmacological amounts of glucocorticoids (Table 1). They enrolled six adult male patients (aged 31–64 years) receiving chronic (>6 months) therapy with glucocorticoids (daily dosage >7.5 mg of prednisone or dose equivalent of other corticosteroid) and compared their spontaneous PTH secretion with that of a control group of 10 age- and sex-matched normal subjects. In the glucocorticoid-treated group, the PTH tonic secretory rate was reduced; however, there was an increase in the fractional pulsatile PTH secretion in glucocorticoid-treated vs normal subjects. Mean overall PTH concentration, as well as mean integrated area, was similar among normal and glucocorticoid-treated subjects. Therefore, it can be concluded that chronic glucocorticoid treatment induces a redistribution of spontaneous PTH secretory dynamics by reducing the amount released in tonic fashion and increasing the amount released as pulses. The clinical importance of this finding is unclear: a compensatory mechanism cannot be excluded although unable to restore the physiological bone metabolism (the alteration of which could only be overcome by much larger PTH peaks such as those obtained by exogenous administration). Other possible explanations include direct effect of glucocorticoids on parathyroid glands or subtle changes in ionized calcium levels. The decrease of circulating osteocalcin induced by glucocorticoids could also be hypothesized to feedback at the parathyroid gland level.
Acromegaly
GH and IGF-1 are anabolic hormones for bone with important regulating effects of bone homeostasis throughout life.42 Both GH deficiency and acromegaly are traditionally considered as causes of skeletal fragility. Patients with GH deficiency have low bone turnover osteoporosis,42,43 whereas patients with acromegaly have increased bone turnover, as determined by changes in biochemical markers, calcium kinetics, and bone histomorphometry.44 Chronic GH/IGF-I excess in acromegaly is associated with high bone turnover, which in turn can cause bone loss and skeletal fractures, despite modestly decreased or normal BMD, as measured by DXA.45 Several cross-sectional and prospective studies demonstrated that acromegaly is associated with an increased prevalence and incidence of radiological and morphometric vertebral fractures.45–48 Biochemical activity of acromegaly is significantly correlated with the risk of vertebral fractures, but incident fractures are shown to occur also in patients with controlled disease, in relationship with the coexistence of untreated hypogonadism and prevalent vertebral fractures at baseline.48 Also in acromegaly, as expected based on previous considerations, data on PTH are not consistent, since they have been shown to be normal or slightly increased at the diagnosis of disease and they may also be influenced by medical treatment with somatostatin analogs.49,50 Mazziotti et al.31 evaluated whether GH excess in acromegaly could modify spontaneous pulsatile PTH secretion (Table 1). Five male patients with newly diagnosed active acromegaly and eight healthy subjects were evaluated by 3-min blood sampling for 6 h. Plasma PTH concentrations were evaluated by multiparameter deconvolution analysis and plasma PTH release profiles were also subjected to an approximate entropy estimate. PTH pulse half-duration was significantly longer and PTH pulse mass tended to be greater in acromegaly patients versus healthy subjects. So it can be hypothesized that GH excess may affect PTH secretory dynamics in patients with acromegaly, but potentially negative bone effects of the modifications of PTH secretory pattern in acromegaly still remain to be investigated.
Conclusions
Assessment of spontaneous PTH secretion is made complex by its physiological pattern which requires very frequent and prolonged sampling with consequently elevated number of PTH samples to assay. Therefore, studies in this area are experimental and difficult to undertake and may cause discomfort to patients. Due to these limitations and despite the currently very limited amount of relevant informations which can be given by baseline PTH values, this approach has found no or very limited space in the assessment of patients with bone metabolic diseases. This procedure has been so far confined to small research studies that can only be defined proof of concept studies often unfortunately giving conflicting results. Nevertheless, pathophysiology of spontaneous PTH secretion remains an interesting area to be explored, particularly in those conditions such as secondary forms of osteoporosis, in which conventional biochemical and densitometric parameters may not always give reliable diagnostic and therapeutic indications.
References
Veldhuis JD, Keenan DM, Pincus SM . Motivations and methods for analyzing pulsatile hormone secretion. Endocr Rev 2008; 29: 823–864.
Samuels MH, Veldhuis JD, Henry P, Ridgway EC . Pathophysiology of pulsatile and copulsatile release of thyroid-stimulating hormone, luteinizing hormone, follicle-stimulating hormone, and alpha-subunit. J Clin Endocrinol Metab 1990; 71: 425–432.
Wehrenberg WB, Giustina A . Basic counterpoint: mechanisms and pathways of gonadal steroid modulation of growth hormone secretion. Endocr Rev. 1992; 13: 299–308.
Brogan RS, Fife SK, Conley LK, Giustina A, Wehrenberg WB . Effects of food deprivation on the GH axis: immunocytochemical and molecular analysis. Neuroendocrinology 1997; 65: 129–135.
Giustina A, Bresciani E, Bussi AR et al. Characterization of the paradoxical growth hormone inhibitory effect of galanin in acromegaly. J Clin Endocrinol Metab 1995; 80: 1333–1340.
Giustina A, Veldhuis JD . Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev 1998; 19: 717–797.
Mazziotti G, Giustina A . Glucocorticoids and the regulation of growth hormone secretion. Nat Rev Endocrinol 2013; 9: 265–276.
Giustina A, Mazziotti G . Impaired growth hormone secretion associated with low glucocorticoid levels: an experimental model for the Giustina effect. Endocrine 2014; 47: 354–356.
Giustina A, Berardelli R, Gazzaruso C, Mazziotti G . Insulin and GH–IGF-I axis: endocrine pacer or endocrine disruptor? Acta Diabetol 2014; doi: 10.1007/s00592-014-0635-6.
Schmitt CP, Hömme M, Schaefer F . Structural organization and biological relevance of oscillatory parathyroid hormone secretion. Pediatr Nephrol 2005; 20: 346–351.
Woitge HW, Knothe A, Witte K et al. Circaannual rhythms and interactions of vitamin D metabolites, parathyroid hormone, and biochemical markers of skeletal homeostasis: a prospective study. J Bone Miner Res 2000; 15: 2443–2450.
Markowitz ME, Arnaud S, Rosen JF, Thorpy M, Laximinarayan S . Temporal interrelationships between the circadian rhythms of serum parathyroid hormone and calcium concentrations. J Clin Endocrinol Metab 1988; 67: 1068–1073.
Samuels MH, Veldhuis J, Cawley C et al. Pulsatile secretion of parathyroid hormone in normal young subjects: assessment by deconvolution analysis. J Clin Endocrinol Metab 1993; 77: 399–403.
Harms HM, Kaptaina U, Külpmann WR, Brabant G, Hesch RD . Pulse amplitude and frequency modulation of parathyroid hormone in plasma. J Clin Endocrinol Metab 1989; 69: 843–851.
Kitamura N, Shigeno C, Shiomi K et al. Episodic fluctuation in serum intact parathyroid hormone concentration in men. J Clin Endocrinol Metab 1990; 70: 252–263.
Schmitt CP, Schaefer F, Bruch A et al. Control of pulsatile and tonic parathyroid hormone secretion by ionized calcium. J Clin Endocrinol Metab. 1996; 81: 4236–4243.
Bonadonna S, Burattin A, Nuzzo M et al. Chronic glucocorticoid treatment alters spontaneous pulsatile parathyroid hormone secretory dynamics in human subjects. Eur J Endocrinol 2005; 152: 199–205.
Brown EM, Gamba G, Riccardi D et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 1993; 366: 575–800.
Schmitt CP, Schaefer F, Huber D et al. 1,25(OH)2-vitamin D3 reduces spontaneous and hypocalcemia-stimulated pulsatile component of parathyroid hormone secretion. J Am Soc Nephrol 1998; 9: 54–62.
Gram J, Junker P, Nielsen HK, Bollerslev J . Effects of short-term treatment with prednisolone and calcitriol on bone and mineral metabolism in normal men. Bone 1998; 23: 297–302.
Hock JM, Gera I . Effects of continuous and intermittent administration and inhibition of resorption on the anabolic response of bone to parathyroid hormone. J Bone Miner Res 1992; 7: 65–72.
Slovik DM, Rosenthal DI, Doppelt SH et al. Restoration of spinal bone in osteoporotic men by treatment with human parathyroid hormone (1–34) and 1,25-dihydroxyvitamin D. J Bone Miner Res 1986; 1: 377–381.
Cipriani C, Irani D, Bilezikian JP . Safety of osteoanabolic therapy: a decade of experience. J Bone Miner Res 2012; 27: 2419–2428.
Bilezikian JP . Combination anabolic and antiresorptive therapy for osteoporosis: opening the anabolic window. Curr Osteoporos Rep 2008; 6: 24–38.
Shinagawa Y, Inoue T, Katsushima T et al. Discovery of a potent and short-acting oral calcilytic with a pulsatile secretion of parathyroid hormone. ACS Med Chem Lett 2010; 2: 238–242.
Cabal A, Mehta K, Ross DS et al. A semimechanistic model of the time-course of release of PTH into plasma following administration of the calcilytic JTT-305/MK-5442 in humans. J Bone Miner Res 2013; 28: 1830–1836.
Procopio M, Barale M, Bertaina S et al. Cardiovascular risk and metabolic syndrome in primary hyperparathyroidism and their correlation to different clinical forms. Endocrine 2014; 47: 581–589.
Walker MD, Fleischer JB, Di Tullio MR et al. Cardiac structure and diastolic function in mild primary hyperparathyroidism. J Clin Endocrinol Metab 2010; 95: 2172–2179.
Mazziotti G, Maffezzoni F, Doga M, Hofbauer LC, Adler RA, Giustina A . Outcome of glucose homeostasis in patients with glucocorticoid-induced osteoporosis undergoing treatment with bone active-drugs. Bone 2014; 67: 175–180.
Samuels MH, Veldhuis JD, Kramer P et al. Episodic secretion of parathyroid hormone in postmenopausal women: assessment by deconvolution analysis and approximate entropy. J Bone Miner Res 1997; 12: 616–623.
Mazziotti G, Cimino V, de Menis E et al. Active acromegaly enhances spontaneous parathyroid hormone pulsatility. Metabolism 2006; 55: 736–740.
Bilezikian JP, Brandi ML, Rubin M, Silverberg SJ . Primay hyperparathyroidism: new concepts in clinical, densitometric and biochemical features. J Intern Med 2005; 257: 6–17.
Vignali E, Viccica G, Diacinti D et al. Morphometric vertebral fractures in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab 2009; 94: 2306–2312.
Harms HM, Schlinke E, Neubauer O et al. Pulse amplitude and frequency modulation of parathyroid hormone in primary hyperparathyroidism. J Clin Endocrinol Metab 1994; 78: 53–57.
Mazziotti G, Bilezikian JP, Canalis E, Cocchi D, Giustina A . New understanding and treatments for osteoporosis. Endocrine 2012; 41: 58–69.
North American Menopause Society. The role of calcium in peri- and postmenopausal women: 2006 position statement of the North American Menopause Society. Menopause 2006; 13: 862–877; quiz 878-880.
Harms HM, Neubauer O, Kayser C et al. Pulse amplitude and frequency modulation of parathyroid hormone in early postmenopausal women before and on hormone replacement therapy. J Clin Endocrinol Metab 1994; 78: 48–52.
Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A . Glucocorticoid-induced osteoporosis: an update. Trends Endocrinol Metab 2006; 17: 144–149.
Angeli A, Guglielmi G, Dovio A et al. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: a cross-sectional outpatient study. Bone 2006; 39: 253–259.
Canalis E, Mazziotti G, Giustina A, Bilezikian JP . Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int 2007; 18: 1319–1328.
Rubin MR, Bilezikian JP . The role of parathyroid hormone in the pathogenesis of glucocorticoid-induced osteoporosis: a re-examination of the evidence. J Clin Endocrinol Metab 2002; 87: 4033–4041.
Giustina A, Mazziotti G, Canalis E . Growth hormone, insulin-like growth factors, and the skeleton. Endocr Rev 2008; 29: 535–559.
Mazziotti G, Bianchi A, Bonadonna S et al. Increased prevalence of radiological spinal deformities in adult patients with GH deficiency: influence of GH replacement therapy. J Bone Miner Res 2006; 21: 520–528.
Giustina A, Casanueva FF, Cavagnini F et al. Pituitary Society and the European Neuroendocrine Association. Diagnosis and treatment of acromegaly complications. J Endocrinol Invest 2003; 26: 1242–1247.
Mazziotti G, Biagioli E, Maffezzoni F et al. Bone turnover, bone mineral density and fracture risk in acromegaly: a meta-analysis. J Clin Endocrinol Metab 2014; doi: 10.1210/jc.2014–2937.
Bonadonna S, Mazziotti G, Nuzzo M et al. Increased prevalence of radiological spinal deformities in active acromegaly: a cross-sectional study in postmenopausal women. J Bone Miner Res 2005; 20: 1837–1844.
Mazziotti G, Bianchi A, Bonadonna S et al. Prevalence of vertebral fractures in men with acromegaly. J Clin Endocrinol Metab 2008; 93: 4649–4655.
Mazziotti G, Bianchi A, Porcelli T et al. Vertebral fractures in patients with acromegaly: a 3-year prospective study. J Clin Endocrinol Metab 2013; 98: 3402–3410.
Legovini P, de Menis E, Breda F et al. Long-term effects of octreotide on markers of bone metabolism in acromegaly: evidence of increased serum parathormone concentrations. J Endocrinol Invest 1997; 20: 434–438.
Kamenický P, Mazziotti G, Lombès M, Giustina A, Chanson P . Growth hormone, insulin-like growth factor-1, and the kidney: pathophysiological and clinical implications. Endocr Rev 2014; 35: 234–281.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Chiavistelli, S., Giustina, A. & Mazziotti, G. Parathyroid hormone pulsatility: physiological and clinical aspects. Bone Res 3, 14049 (2015). https://doi.org/10.1038/boneres.2014.49
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2014.49
