
Abstract
Bone mineral, adipose tissue and energy metabolism are interconnected by a complex and multilevel series of networks. Calcium and phosphorus are utilized for insulin secretion and synthesis of high energy compounds. Adipose tissue store lipids and cholecalciferol, which, in turn, can influence calcium balance and energy expenditure. Hormones long-thought to solely modulate energy and mineral homeostasis may influence adipocytic function. Osteoblasts are a target of insulin action in bone. Moreover, endocrine mediators, such as osteocalcin, are synthesized in the skeleton but regulate carbohydrate disposal and insulin secretion. Finally, osteoblasts and adipocytes originate from the same mesenchymal progenitor. The mutual crosstalk between osteoblasts and adipocytes within the bone marrow microenvironment plays a crucial role in bone remodeling. In the present review we provide an overview of the reciprocal control between bone and energy metabolism and its clinical implications.
Similar content being viewed by others

Introduction
Significance of the energy-skeletal remodeling relationship
Ionized calcium (Ca2+), the principal cation in the skeleton, is a key element for the maintenance of cell membrane stability and permeability. Circulatory levels of ionized calcium are rigidly maintained independently of fed or fast state, as well as rest or exercise. The skeleton, the rigid structure necessary for posture and movement, is the storage depot and ultimate source of calcium for other homeostatic functions. Bone modeling and remodeling require significant energy for the skeleton to grow and to maintain strength. The activity of bone renovation elicited by osteocytes, osteoclasts and osteoblasts allows the hard tissue to combine flexibility and the capacity to support continuous mechanical stress, with the release of calcium into the circulation. This process requires significant substrate to generate ATP for osteoclasts to resorb bone, and for osteoblasts to synthesize collagen.
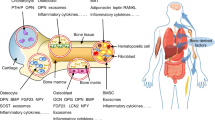
Until recently, energy metabolism was analyzed in specialized and restricted functional tissues (1–3). In that scheme, the liver was the central site for glucose production, muscles were related to adaptation by alternating their use of substrates (i.e., free fatty acids or glucose, depending on the fed state), the adipose tissue was an inert site for fat deposition and the central nervous system (CNS) was considered the preferential site for glucose disposal. Moreover, regulation was thought to be restricted to classical endocrine tissues (pancreas, adrenal and pituitary), CNS and the autonomous nervous system. That fractional view neglected the contribution of other active sites, such as the gastrointestinal tract (4) and bone (5,6), to the disposal of energy, and reflected an incomplete understanding of the complex network connecting those tissues to the endocrine and peripheral nervous systems. Advances in the investigation of molecular and cellular biology, associated with the use of genetically modified mice (Figure 1), revealed the omnipresence of endocrine tissue in the control of the energy metabolism. The interplay between bone cells and adipocytes, and the interaction of the skeleton with the central and autonomic nervous system are so intense and so diverse that it has only been recently that the networks involved in mineral balance have been delineated.

Evidence of the interrelationship between bone and adipose tissue obtained through the use of genetically modified mice. vdr null= vitamin D receptor null mouse, FGF23 null= Fibroblast growth factor-23 null mouse, leptin null= leptin null mouse, esp null= osteotesticular protein tyrosine phosphatase null mouse, ocn null= osteocalcina null mouse and ob ΔIR= insulin receptor deletion specifically in osteoblast.
Friedman and colleagues, using contemporary (gene silencing) and classical (parabiosis) methodologies, showed that leptin, an adipokine, controlled energy expenditure, reproductive capacity and appetite through both central and peripheral pathways (7,8). Subsequently, the Karsenty group proposed that leptin modulated bone remodeling through a central relay in the brain stem and hypothalamus (9,10). These observations revolutionized the field of bone biology and raised new and important questions that started with fat as an endocrine organ.
The amount, location and type of adipose tissue influences both intermediary metabolism and skeletal remodeling. While rare lipoatrophic disorders are associated with insulin resistance (11,12), dyslipidemia and decreased glucose tolerance, the same combination of symptoms occurs with excessive accumulation of adipose tissue (13,14). Moreover, triglyceride storage in adipocytes triggers a complex series of events, including adipokine and cytokine secretion that can attract inflammatory cells to the interstitium (15,16). Previous studies in mice have shown that the expression of macrophage chemo-attractant proteins rises significantly following one week of a high fat diet (17). It is not surprising, therefore, to consider skeletal changes that may occur when adipose tissue becomes an endocrine organ. In the present review, we review the intense relationship between mineral and energy metabolism, bone and adipose tissue, and the sharing of endocrine/paracrine modulators by both calcified and soft tissue.
Calcium, phosphorus, calciotropic hormones and fibroblast growth factor 23 (FGF-23): relationship to energy metabolism
Calcium and phosphorus
The major elements of mineral metabolism, calcium and phosphorus, are tightly linked to intermediary metabolism (18,19). In several cells, intracellular calcium works as a second messenger, mediating the effect of membrane signals on the secretion from neurons, exocrine tissue and endocrine glands (e.g. insulin and epinephrine). Circulating calcium levels are tightly controlled through the mineral hormones, PTH, 1,25-dihydroxyvitamin D and calcitonin. It is recognized that calcium and the cAMP messenger system are related, and both are integrated in such a way that the net cellular response to a given stimulus is determined by a complex interplay between these systems. For example, in the pancreatic β-cell, cytoplasmic calcium levels rise as a direct consequence of glucose metabolism, via closure of KATP channels, triggering Ca2+ entry and subsequent Ca2+-mediated exocytosis of insulin granules (20). A recent study tested an experimental model of the metabolic syndrome, submitting rats to a high ingestion of carbohydrate by drinking water supplemented with sucrose in 20% solution (21). After 24 weeks, the rats exhibited central obesity, hyperinsulinemia, insulin resistance and increased blood pressure. KATP channels, in isolated patches of β cells from those rats, had an increased sensitivity to ATP in comparison to controls. However, the authors observed increased variability in calcium flow compared with cells from control animals. The authors hypothesized that the decrease in calcium flow may represent a feature of β cell subpopulations as they became exhausted by the long-term high sucrose diet (21). Other studies observed increased intracellular ionized calcium in platelets, red blood cells and adipocytes of type 2 diabetic patients. Additionally, an association between increased intracellular ionized calcium and decreased insulin-stimulated uptake of glucose was observed in rat adipocytes. Taken together, disturbances in calcium may be associated with disorders in insulin secretion and in insulin resistance (22).
Phosphorus is a major component of important organic molecules including nucleic acids, cyclic nucleotides, phospholipids and a number of intermediaries in energy metabolism. In contrast to calcium, the circulatory levels of phosphorus vary widely and are influenced by age, sex, diet, pH and a set of hormones (23). Almost all the phosphorous in humans is found in bone, and only a 0.1% exists as inorganic phosphate anions in extracellular fluids. However, the regulation of serum phosphate is particularly important, since alterations in phosphorous homeostasis are involved in metabolic disorders. Indeed, maintaining phosphate balance is of crucial biological importance; acute hypophosphatemia can cause myopathy, cardiac dysfunction, and hematological abnormalities, while chronic hypophosphatemia affects bone mineralization, leading to rickets and osteomalacia. On the other hand, hyperphosphatemia is associated with secondary hyperparathyroidism, a common feature of patients with chronic renal disease.
Recent observational data have linked phosphate levels and phosphate-regulatory factors with morbidity and mortality, suggesting an intriguing link between the phosphate axis and the ageing processes (24). In fact, it has been verified that changes in phosphate concentration affect diverse processes such as glucose metabolism, insulin sensitivity, and oxidative stress. Diet restriction has been known to suppress aging and extend life span in experimental organisms (e.g., fruit flies, rodents, and nonhuman primates) (25). Although phosphorus is a mineral which exists abundantly in nature, its tightly controlled regulatory circuit may influence longevity. In support of that hypothesis is the existence of two calciotropic hormones (PTH and FGF-23), with a powerful effect on urinary loss of phosphorus. In mammals, a low phosphate diet causes changes in metabolism similar to those induced by diet restriction. Animals under diet restriction reduce blood insulin levels to adapt to reduced carbohydrate availability and alter the expression of insulin-responsive genes, which leads to changes in glucose metabolism, including increased gluconeogenesis and decreased glycolysis (26). In comparison, while a low phosphate diet does not reduce blood insulin levels, it also alters the expression of insulin-responsive genes in an identical manner to that induced by diet restriction, leading to increased gluconeogenesis and decreased glycolysis (27). Interestingly, hypophosphatemia can induce moderate insulin resistance through mechanisms that have not been delineated (28). However, increased insulin resistance does not necessarily translate into Type 2 diabetes or a shorter life span.
In addition to its involvement in glucose metabolism and insulin sensitivity, inorganic phosphate has been shown to increase oxidative stress both in vivo and in vitro. Human vascular endothelial cells cultured in high phosphate medium have higher levels of cellular reactive oxygen species (ROS) than those cultured in normal phosphate medium (29). Hyperphosphatemia has been causally associated to poor outcomes in chronic kidney disease (CKD). Indeed, recent studies have identified hyperphosphatemia as a major risk of death in CKD patients, even when the blood levels of phosphate are within the normal range (30). A comprehensive discussion of the metabolic effects of hyperphosphatemia is beyond the scope of the present review; our intention is merely to call attention to the connection between energy metabolism and mineral metabolism.
Calciotropic hormones and FGF-23
Obesity, a complex metabolic disorder acquired as a consequence of genetic and environmental factors, is phenotypically characterized by an increased proportion of fat mass and an array of metabolic, hormonal, and inflammatory disorders. This ominous combination is often a prelude to diabetes mellitus, cardiovascular disease and surprisingly bone fragility. Obese patients also have significant alterations in their mineral metabolism. First, adipose tissue sequestrates vitamin D, decreasing substrate availability for the synthesis of 25-hydroxyvitamin D (25-OHD) (31,32). PTH and calcitriol (1,25-(OH)2D) are usually elevated in obese patients (33). Not surprisingly, since serum 25-OHD is the most appropriate parameter for the diagnosis of hypovitaminosis D, there is a negative correlation between 25-OHD circulatory levels and insulin resistance (34). Moreover, several observational studies have demonstrated an inverse relationship between serum 25-OHD and Type 2 diabetes mellitus. However, observational studies and meta-analyses are insufficient to elucidate the role of vitamin D deficiency, as cause or consequence of obesity and insulin resistance.
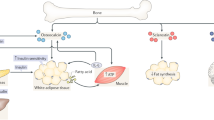
Different experimental approaches have been used to show that the most biologically active metabolite of vitamin D, 1,25-(OH)2D ultimately has adipogenic activity. Severe vitamin D resistance or deficiency in mice (vitamin D receptor and cyp27B1 knockout mice) leads to a lean phenotype and a better metabolic response to a diabetogenic diet than wild type mice (35–37). Moreover, one allele deletion of the VDR gene is sufficient to determine a lean phenotype in male mice, though it has no impact on bone mass (38), suggesting that moderate insensitivity to calcitriol has greater impact on energy metabolism than on bone mass in male mice. Wong et al constructed a transgenic mouse equipped with the expression of human VDR specifically in adipocytes (aP2-h VDRTg). In contrast to VDR null mice, which have a lean phenotype, the transgenic aP2-h VDRTg shows excessive weight gain, without an increment in food intake. The increase in fat mass was mainly due to a markedly reduced energy expenditure, which was correlated with decreased locomotive activity and reduced fatty acid β-oxidation and lipolysis in adipose tissue of the transgenic mice (39). The expression of carnitine palmitoil transferase 1 and 2 (involved in the transport of long chain fatty acid to the mitochondria for β-oxidation), hexokinase (the first step of glucose phosphorilation), UCP2 and UCP3 (involved in adaptive thermogenesis) genes were significantly reduced in white adipose tissue of those transgenic mice (39). In accordance with these experiments, clinical investigations have shown that high calcium favors weight loss maintenance when compared to a isocaloric normocalcemic diet (40,41). Zemel hypothesized that the inhibition of 1,25-(OH)2D synthesis by increased calcium intake is directly involved in the limitation of weight gain (41). In a previous study, Zemel et al showed that 1,25-(OH)2D, by increasing Ca2+ in the cytosol, stimulates lipogenesis and inhibits lipolysis. The increase of (Ca2+) within murine and human adipocytes also stimulates the expression and activity of fatty acid synthase (FAS), a step-limiting enzyme in lipogenesis (42–44) (Figure 2).

Leptin the adipocyte-derived peptide for the modulation of energy expenditure also stimulates the endocrine production of FGF-23 by osteocytes. FGF-23 acts directly on the kidney provoking urinary loss of phosphorus and decreases the synthesis of 1,25-(OH)2D. The calciotropic hormone 1,25-(OH)2D for the stimulation of calcium and phosphorus absorption, stimulates lipogenesis in the adipose tissue.
In sum, vitamin D is a key molecule for mineral homeostasis due to its essential action on the intestine promoting calcium and phosphorus absorption, stimulating bone remodeling and modulating PTH and FGF-23 synthesis. Additionally, vitamin D is a pleiotropic hormone; its nuclear receptor is ubiquitously distributed throughout the organism. Further study is necessary to delimit the physiological role of vitamin D on energy metabolism and the ultimate effect of vitamin D metabolites on the emergence of obesity and the metabolic syndrome.
PTH serum levels are commonly elevated in obese patients and there are data showing a positive correlation between body mass index and PTH serum levels (45–47). Importantly, the association of PTH, with body weight seems to occur independently of vitamin D status. Grethen et al recently evaluated patients scheduled for bariatric surgery and observed increased serum levels of PTH and FGF-23 and decreased circulatory levels of 1,25-(OH)2D. Additionally, they verified the absence of a significant relationship between PTH and 25-OHD (47). On the other hand, there was a significant correlation between PTH and leptin, and multiple regression analysis demonstrated that serum leptin was the highest predictive serum variable for PTH with Ca providing only a small negative predictive value. There are no experiments that fully support or refute the causal effect of leptin on PTH secretion. There are studies showing that obese patients with primary hyperparathyroidism have greater circulatory levels of PTH and tumor size than non-obese patients, indicating that leptin might stimulate parathyroid cell mass (48,49).
In addition to the adipose tissue-PTH-leptin connection, there is another relevant link between PTH and energy metabolism; there may be an association between diabetes mellitus and primary hyperparathyroidism (19,28,50). Epidemiological studies suggest there is a higher prevalence of diabetes mellitus in primary hyperparathyroidism; patients harboring primary hyperparathyroidism have a three-fold higher rate of Type 2 diabetes than the expected prevalence in a general population (51). Both hypercalcemia and hypophosphatemia may lead to insulin resistance in primary hyperparathyroidism. Interestingly, there is no improvement in glucose tolerance after successful parathyroidectomy in PHPT (52). Therefore, PTH, the primordial hormone for the maintenance of calcium homeostasis, concomitantly influences energy metabolism.
FGF-23 is an endocrine hormone secreted by osteocytes and osteoblasts. The classical effects of FGF-23 on the kidney and the parathyroid glands are mediated by its binding to FGF receptors (FGFRs) complexed to the co-receptor klotho. The primary physiological actions of FGF-23 are to stimulate phosphaturia by down-regulating luminal expression of sodium—phosphate co-transporters in the proximal tubule and thereby reduce systemic levels of 1,25-dihydroxyvitamin D by directly inhibiting the renal 1-a hydroxylase (Figure 2) and stimulate the catabolic 24-hydroxylase, and inhibit parathyroid hormone (PTH) secretion (53,54,55). Therefore, FGF-23 is a component of an endocrine feedback loop between bone and kidney, lowering circulating phosphate through a direct renal phosphaturic effect and an indirect mechanism (down-regulating intestinal calcium and phosphorus absorption via suppression of 1,25-(OH)2D3 production) (56). Silencing of FGF-23 function in gene-targeted (FGF-23−/−) mice results in a complex phenotype characterized by severely impaired bone mineralization, hypercalcemia, hyperphosphatemia, highly elevated serum 1,25-(OH)2D3 levels due to increased renal 1α-hydroxylase expression, shortened life span, growth retardation, hypogonadism, emphysema, vascular calcifications, extensive soft tissue calcifications, atrophy of the intestinal villi, skin, thymus, and spleen, together with hypoglycemia and profoundly increased peripheral insulin sensitivity (57). Similarly to FGF-23−, klotho-deficient mice have extremely high serum levels of 1,25-(OH)2D3 and share a similar metabolic phenotype (58). Reducing vitamin D activity in klotho mutant mice by feeding them a vitamin D-deficient diet has resulted in the disappearance of ectopic calcifications, gain of fertility and, most importantly, prolonged survival, suggesting that the premature aging-like features in klotho mutant mice are downstream events resulting from increased activity of vitamin D. Furthermore, when vitamin D activity was genetically ablated from Fgf-23 null mice by deleting the 1α-hydroxylase (CYP27B1) gene, most of the premature aging-like features in Fgf-23 null mice were rescued (59).
Experimental models do not encompass all the alterations observed in humans showing disorders in FGF-23 secretion. For example, circulating FGF23 is positively associated with disease progression, heart hypertrophy and mortality in CKD. In addition, a recent study in two large cohorts of elderly subjects in Sweden reported a strong association between circulating FGF23 and obesity, as well as adverse lipid metabolism (60). Although, the authors had no compelling evidence for the causal link of FGF-23 with obesity, they call attention to several aspects of the metabolic effect of FGF-23. First, phosphate-lowering therapy with sevelamer in CKD patients leads to an improved blood lipid profile, including higher HDL and lower LDL, which speculatively could be attributed to a reduction in net serum phosphate balance in addition to intestinal binding of bile salts (61). Second, the authors observed associations between FGF-23 and fat mass and FGF-23 with serum leptin (58). Leptin inhibits the overall production of 25-OHD 1α-hydroxylase (CYP27B1) and 24-hydroxylase (CYP24) in the kidneys of ob/ob mice, and also corrects hypercalcemia and hyperphosphatemia in these animals (62). In addition, it was found that mouse renal tubule cells did not show suppression of 1α-hydroxylase mNA expression after exposure to leptin, suggesting that leptin regulates this mRNA via some other mechanism (62). Leptin can directly stimulate FGF-23 synthesis by bone cells in ob/ob mice, suggesting that inhibition of renal 1,25-(OH)2D3 synthesis in these mice is at least partially due to elevated bone production of FGF-23. This hypothesis was reinforced by Tsuji et al (2010), verifying that exposure to leptin (200 ng·mL−1) for 24 hours stimulated FGF-23 expression in primary cultured rat osteoblasts (63). Additionally, they also observed that administration of leptin (4 mg·kg−1 i.p. at 12-hour intervals for 2 days) in ob/ob mice markedly increased the serum FGF-23 concentration, while significantly reducing the serum levels of calcium, phosphate, and 1,25-(OH)2D (63).
Collectively, the above mentioned evidence substantiates the interdependence of the endocrine control of mineral and energy metabolism, as well as bone and adipose tissue. While calciotropic hormones and FGF-23 are finely tuned to the control of mineral metabolism, they also impact and are influenced by hormonal factors directly involved in the modulation of energy metabolism.
Bone and energy metabolism
Bone and body composition
Epidemiologic studies indicate that there is an interaction between body fat and bone mass, leading to an initial perception of a positive role of adiposity on bone strength (64). However, despite a considerable body of areal BMD data, there remains controversy as to whether fat has a positive or detrimental effect on bone in both the pediatric and adult populations. There are a number of cross-sectional studies suggesting that fat mass may have a negative effect on bone during childhood and adolescence (65,66). However, there remains a need for longitudinal studies of the relationship between fat and bone from childhood to adulthood to determine whether there are key stages during which excessive fat limits bone mass accrual. The detrimental effect of excessive mechanical loading on bone from excess fat mass in children has been based on the increased risk of slipped capital femoral epiphysis (67) and tibia vary in obese children (68). But, major concerns that excess fat mass may have a detrimental effect on bone mass acquisition in children originated from observations that obese children were overrepresented in fracture groups in studies that sought to evaluate the prevalence of fractures in children (69–71). Cross-sectional studies in different age groups have also alluded to a potential change in the relationship between fat and bone from early childhood into adolescence. The Avon Longitudinal Study of Parents and Children (ALSPAC) project from Bristol in the UK is a long-term health research project that enrolled more than 14 000 mothers during pregnancy in 1991 and 1992, and subsequently studied the health and development of their children (72). Cross-sectional analysis of the relationship between fat mass and bone in this cohort of children at 9.9 years demonstrated a strong positive relationship between total body fat mass and total body (minus the skull) bone mass and area, before and after adjustment for height and lean mass. However, as girls progressed through puberty, the positive relationship between fat and bone mass was attenuated and subsequently reversed; the same relationship was not observed in boys as there were an insufficient number of boys who had progressed far enough into puberty to qualify (72). There are other studies suggesting that an increase in fat mass, particularly intra-abdominal adiposity, may be detrimental to the growing skeleton, particularly during the pubertal period (73–75).
In adults, low body weight has detrimental effects on bone mass and is associated with enhanced fracture risk (76). Additionally, rapid weight loss leads to bone loss in sites that support weight, such as lumbar spine and proximal femur, but also in regions free of mechanical stress (i.e., 1/3 radius) (77,78). However, closer examination of the changes in body composition and in the hormonal profiles of the adipose tissue in obesity leads to some degree of ambiguity. For example, several studies have shown that the rate of fracture in obese postmenopausal women is far greater than would be predicted by normal or high bone mineral density often seen in obese patients (79).
In an audit of postmenopausal women presenting with low trauma fracture to a Fracture Liaison Service, the prevalence of obesity was 28% (80), whilst in the Global Study of Osteoporosis in Women (GLOW) the incidence of low trauma fractures was similar in obese and non-obese postmenopausal women (79). The distribution of fracture sites differ between obese and non-obese women; fractures of the leg, ankle, and humerus being reported more commonly in obese women whereas fractures of the hip, wrist and pelvis are less prevalent in this group (81,82). The Study of Osteoporotic Fractures BMD of 1377 examined obese postmenopausal women encompassing patients from the cohort with and without nonvertebral fractures. The results showed that obese postmenopausal women who sustained nonvertebral fractures had significantly lower BMD on average than obese women without fracture, and were more likely to have a past history of fracture (83).
Visceral adipose tissue (VAT), which has been tied to insulin resistance, has also been evaluated in relation to bone mass. Although, previous study suggested a positive correlation between visceral and BMD (84), recently published data indicate a detrimental effect of VAT on bone mass. In attempt to circumvent imprecise measurement of visceral fat, Russel et al used MRI to accurately assess visceral fat in 30 adolescent girls (85). VAT was found to be a negative predictor of bone mass. Another study, with adolescent girls, supported the negative effect of visceral fat on BMD (86). In Korean healthy men and women, it was found that visceral fat, measured by computed tomography, was independently and negatively associated with bone mass (87). Aligned with these studies, Bredella et al evaluated in 68 obese (BMI=36.7±4.2 kg·m−2) premenopausal women the differential effects of abdominal fat depots and muscle on trabecular BMD of the lumbar spine (88). Quantitative computed tomography (QCT) was used to assess body composition and lumbar trabecular BMD. There was an inverse association between BMD and VAT, independent of age and BMI (88). Finally, in unpublished work, Shane and colleagues have shown that trunk fat, as measured by DXA, was inversely related to trabecular bone volume fraction and bone formation rate by histomorphometry (Cohen, 2012 submitted).
While the stereotype of androgenic and gynecoid patterns of obesity drew attention to visceral and subcutaneous fat as unhealthy and healthy adipose tissue, it is clear that white fat accumulation in other sites may also have adverse effects (89). For instance, intra muscular fat impairs muscle performance, and is directly involved in myocyte insulin resistance (90–92). However, two other types of adipose tissue impact energy metabolism and bone homeostasis. The brown adipose tissue (BAT), enriched with iron-mitochondria for non-shivering thermogenesis, is directly connected to temperature control in neonates. Recent evidence indicates its persistence in some adults (93,94) and a positive relationship between BAT volume and bone mass (95). The other adipose depot, bone marrow, is involved in bone remodeling by its effects, direct or indirect, on marrow mesenchymal stem cell differentiation.
Postmenopausal, senescence and glucocorticoidinduced osteoporosis are all associated with low bone mass and marrow adipose tissue (MAT) accumulation (96). Not surprisingly, aging, menopause and hypercortisolism are associated with gain of adipose tissue, especially in the visceral depot. Nonetheless, there are other circumstances that influence bone marrow micro-environmental changes, including hematopoietic, skeletal and adipogenic tissues. The transition of the hematopoietic marrow “red marrow” to one enriched in adipocytes, “yellow marrow”, takes place early in life, during bone accrual. Consequently, bone mass gain is not irreconcilable with adipogenesis in the bone marrow (97). Indeed, in appropriate physiological conditions, bone marrow adipocytes might be part of a favorable condition for bone mass formation by supporting ‘local’ energy utilization and secreting paracrine factors to stimulate osteoblast activity (98). Noteworthy, growth spurt occurs in a coordinated process that allows overall acquisition of bone, lean and fat mass.
During caloric restriction in young mice, and anorexia nervosa in humans, marrow adipose tissue increased in contraposition to the catabolic state in other compartments. Eckuland et al described unequivocal augmentation of marrow fat in a group of adolescents with anorexia nervosa in comparison to a control group (99). In another study, the investigation of the hormonal/metabolic profile of patients with anorexia nervosa showed normal insulin sensitivity and increased levels of serum adiponectin in relation to normal weight control women (100). The relevance of marrow fat expansion in conditions of negative caloric balance still is speculative; a comprehensive discussion has been recently published by Devlin (2010) (101).
Cellular interactions
The bone marrow is a complex microenvironment, comprising of different cell types (e.g., macrophages, adipocytes, fibroblasts), which together secrete a specialized extracellular matrix. Critical for hematopoietic stem cell (HSC) maintenance, osteoblasts line the inner surface of the trabecular bone (102). When osteoblast-HSC contact is lost, the HSCs progress to form myeloid and lymphoid progenitors (103). The lymphoid lineage produces B, T and NK cells, whereas the myeloid lineage produces granulocytes, erythrocytes and platelets (101). By age 20, the appendicular skeleton is nearly all converted from red to fatty yellow marrow. In the axial skeleton, hematopoiesis continues into adulthood, but there is fatty infiltration of the vertebral bodies with aging (101). Changes in mammalian HSCs may affect the progression of MSCs into osteoblasts or adipocytes (104,105,106). For example, low bone mass is a feature of individuals with hemolytic anemia, a clinical condition associated with enhanced marrow adiposity (107–109).
Osteoblasts and adipocytes share a common progenitor cell, the mesenchymal stromal cell, (MSC), which also serves as a source of progenitors for marrow fibroblasts, chondrocytes, and supporting stroma for hematopoietic cells (110,111). Lineage allocation of marrow MSCs towards either adipocytes or osteoblasts is a finely tuned event in which lineage-specific transcription factors (such as Runx2 and Osterix for osteoblasts and PPARg2 for adipocytes) play critical roles. Importantly, in some but not all situations, lineage allocation of MSCs towards either of these cell types is considered to be mutually exclusive; i.e. activation of PPARg2 leads to enhanced adipogenesis at the expense of osteoblastogenesis and is associated with reduced expression and function of osteogenic transcription factors such as Dlx5, Msx2, Runx2 and Osterix (112,113). In line with this, suppression of PPARγ is reported to stimulate osteoblastogenesis and represses adipogenesis (114). These observations are also consistent with the findings from aged mice models, where marrow adiposity is increased, bone mass is reduced, and there is enhanced PPARg2 expression (115). Following the expansion of preadipocytes, MSCs differentiate into mature adipocytes under the tight control of multiple transcription factors, including C/EBPβ/δ and PPARγ. Among these, PPARγ, a nuclear receptor and transcription factor, plays a central role in adipogenesis, as evidenced by the finding that the loss of PPARγ in mouse embryonic fibroblasts leads to a complete absence of adipogenic capacity (116).
Surrounding trabecular bone, bone marrow is laden with multipotent stem cells. Indeed, most progenitor and differentiated cells originate in bone marrow, and directly or indirectly, contribute to overall energy metabolism. For example, enucleated erythroid cells are specialized to transport oxygen, a crucial component for aerobic oxidation of energetic substrates in the mitochondria. Osteoclasts can secrete inflammatory factors, which could induce insulin resistance. Surprisingly, osteoclasts contain more mitochondria than any other cell type, supporting the premise that energy utilization is an essential element in bone remodeling. Although less attention has focused on energy utilization in osteoblasts, it is clear that substrate utilization is essential for a step up in mitochondrial biogenesis during collagen synthesis. Moreover, differentiation of MSCs into osteoblasts is related to increased oxidative phosphorylation, as ATP is essential for collagen matrix synthesis.
Leptin and osteocalcin: fat-bone cross-talk
Exclusively produced by adipocytes, leptin secretion is positively correlated with adipose tissue mass. Leptin crosses the blood-encephalic barrier to stimulate its receptor in the hypothalamus, thereby triggering complex actions related to energy modulation, pubertal onset and control of bone remodeling. Mice with congenital absence of leptin (ob/ob mice) or the leptin receptor (db/db mice) exhibit a complex phenotype, including obesity and high bone mass in the vertebrae (117,118). This occurs despite hypogonadism and hypercortisolism, conditions that would tend to lower bone mass in mice.
The combination of increased bone mass with leptin deficiency or resistance, respectively, in ob/ob and db/db mice serves as a basis to conclude that leptin acts centrally to inhibit the accumulation of bone mass through the sympathetic nervous system. In accordance with this observation there are studies showing: 1) mice harboring a mutation that leads to a partial gain of function in leptin signaling exhibit normal appetite, but an osteoporotic phenotype (119,120); 2) neuron-specific deletion of Lepr induces the bone phenotype of ob/ob mice, whereas an osteoblast-specific deletion has no such effect (117,119), 3) the skeletal phenotype of ob/ob mice can be corrected by leptin administration into the third ventricle (121), and 4) chemical lesion of neurons in the ventromedial hypothalamus recapitulates the bone changes observed in leptin-deficient mice (10). On the other hand, there are other studies showing that leptin has a peripheral effect of direct stimulation of bone formation. In vitro, it was demonstrated that leptin stimulates osteoblastic bone formation (122) and decreases osteoclastogenesis (123). In support of that data, it was demonstrated in vivo by the experimental administration of leptin in rodents, that leptin increases bone mass (124). In the clinical field, the peripheral beneficial effect of leptin may be defended by the increased levels of leptin in obese patients, which usually show high bone mass, and decreased leptin in young women with anorexia nervosa. Although these are important results, obesity and anorexia nervosa are complex disorders characterized by several hormonal disturbances. Thus, it is hard to attribute the bone profile in obesity and anorexia nervosa to alterations only in serum levels of leptin.
While leptin is the adipocyte messenger for the connection between adipose tissue and bone, osteocalcin is the molecule that communicates signals from bone to adipose tissue. Osteocalcin is a small noncollagenous peptide produced predominantly by osteoblasts. Osteocalcin is carboxylated post-translationally on three glutamic acid residues in a vitamin K-dependent manner by the enzyme gamma c-carboxylase. The product, the amino acid c-carboxyglutamic acid, has the capacity to bind to calcium. Decarboxylation decreases the hydroxyapatite-binding affinity of osteocalcin. Undercarboxylated osteocalcin that enters the circulation regulates energy metabolism, through increases in β-cell proliferation, insulin secretion and insulin sensitivity.
Insulin response in target tissues is strongly dependent on its capacity to trigger the tyrosine kinase activity of its own receptor. Moreover, insulin signaling may be negatively modulated by the existence of intracellular tyrosine phosphatase (125). Esp encodes a tyrosine phosphatase, osteotesticular protein tyrosine phosphatase (OST-PTP), and is expressed in a limited number of cells (e.g., osteoblasts, sertoli cells and embryonic stem cells). The silencing of Esp provokes hypoglycemia due to hyperinsulinemia, with increased serum levels of C-peptide associated with normal glucagon concentrations (126). In addition to increased glucose tolerance and insulin sensitivity, these mice exhibited an increase in mitochondrial area and mitochondrial proteins, such as Mcad, acyl-coA, UCP-2, PGC1α and NFR1, in the gastrocnemius muscle, compatible with augmented mitochondrial activity in Esp−/− mouse (127). Esp−/− mice show a lean phenotype, which most likely occurs as consequence of increased levels of adiponectin.
In addition to enhanced circulatory levels of adiponectin, Esp−/− mice also have increased serum levels of undercarboxylated osteocalcin. In view of the finding that the expression of the osteocalcin gene and the serum levels of osteocalcin were normal in these mice, it seems that OST-PTP acts on the decarboxylation of osteocalcin and its entry into the bloodstream. In contrast to the Esp−/− mice, the deletion of the osteocalcin gene confers a decrease in insulin secretion and an increase in circulatory glucose levels. Ocn−/− mice have an impairment in insulin sensitivity and a decreased number of pancreatic β-cells. Moreover, the metabolic phenotype of Esp−/− is completely reversed by crossing those mice with mice lacking one allele of Ocn (125). As expected, administration of recombinant uncarboxylated osteocalcin to Ocn−/− mice corrected the glucose intolerance and insulin secretion.
Finally, the connection between bone and energy metabolism can further be exemplified by studies showing bone loss in experimental and clinical states of insulinopenia, such as diabetes mellitus type 1 (DM1). In both cases, low serum levels of osteocalcin or decreased osteocalcin expression in bone reflect impaired bone formation and a fundamental defect in osteoblasts function during states of insulin deficiency. As noted, additional evidence of bone regulation by insulin has been acquired through specific genetic deletion of insulin receptors in osteoblasts (128). Insulin receptor silencing resulted in low bone mass and was associated with increased expression of Twist2 and decreased expression of osteocalcin and Runx2 (129). The transcriptional factor Twist2 acts as a cellular inhibitor of Runx2; the latter is a key determinant of osteoblast differentiation.
Conclusion
The skeleton utilizes energy for remodeling and growth, whereas adipose tissue stores major nutrients necessary for ultimate fuel consumption. Similarly, although osteoblasts and adipocytes arise from the same multipotent progenitor, the former utilizes energy and has enhanced oxidative phosphorylation, while the latter stores energy and has reduced substrate requirements. Not surprisingly, the main hormones directly involved in the control of mineral and energy metabolism actually are molecules that also interconnect hard and soft tissues. A complex signaling network between osteoblasts and adipocytes participates closely in the modulation of energy metabolism and bone remodeling, respectively. Understanding bone-fat interactions requires a comprehensive analysis of the physiology of skeleton, its regulatory role in intermediary metabolism, and its utilization of energy. This aspect of bone biology promises that more provocative data will emerge showing the integrative role of the skeleton in metabolic homeostasis.
References
Jackson RA, Roshania RD, Hawa MI, Sim BM, DiSilvio L . Impact of glucose ingestion on hepatic and peripheral glucose metabolism in man: an analysis based on simultaneous use of the forearm and double isotope techniques. J Clin Endocrinol Metab. 1986;63:541–549.
Paula FJ, Pimenta WP, Saad MJ, Paccola GM, Piccinato CE, Foss MC . Sex-related differences in peripheral glucose metabolism in normal subjects. Diabete Metab. 1990;16:234–239.
Ferrannini E . Physiology of glucose homeostasis and insulin therapy in type 1 and type 2 diabetes. Endocrinol Metab Clin North Am. 2012;41:25–39.
Muñoz R, Carmody JS, Stylopoulos N, Davis P, Kaplan LM . Isolated duodenal exclusion increases energy expenditure and improves glucose homeostasis in diet-induced obese rats. Am J Physiol Regul Integr Comp Physiol. 2012;303:R985–R993.
Digirolamo DJ, Clemens TL, Kousteni S . The skeleton as an endocrine organ. Nat Rev Rheumatol. 2012;8:674–683.
Guntur AR, Rosen CJ . Bone as an endocrine organ. Endocr Pract. 2012;18:758–762
Friedman JM . Leptin at 14 y of age: an ongoing story. Am J Clin Nutr. 2009;89:973S–979S.
Zhang Y, Proenca P, Maffei M, Barone M, Leopold L, Friedman JM . Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432.
Elefteriou F, Takeda S, Ebihara K, Magre J, Patano N, Kim CA, Ogawa Y, Liu X, Ware SM, Craigen WJ, Robert JJ, Vinson C, Nakao K, Capeau J, Karsenty G . Serum leptin level is a regulator of bone mass. Proc Natl Acad Sci U S A. 2004;101:3258–3263.
Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, Armstrong D, Ducy P, Karsenty G . Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;11:305–317.
Chan JL, Oral EA . Clinical classification and treatment of congenital and acquired lipodystrophy. Endocr Pract. 2010;16:310–323.
Garg A, Agarwal AK . Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta. 2009;1791:507–513.
Rowe MW, Bergman RN, Wagenknecht LE, Kolberg JA . Performance of a multi-marker diabetes risk score in the Insulin Resistance Atherosclerosis Study (IRAS), a multi-ethnic US cohort. Diabetes Metab Res Rev. 2012;28:519–526.
Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, Rinfret S, Schiffrin EL, Eisenberg MJ . The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56:1113–1132.
Zhang HM, Chen LL, Wang L, Xu S, Wang X, Yi LL, Chen D, Wu ZH, Zhang JY, Liao YF, Shang J . Macrophage infiltrates with high levels of Toll-like receptor 4 expression in white adipose tissues of male Chinese. Nutr Metab Cardiovasc Dis. 2009;19:736–743.
Sun K, Kusminski CM, Scherer PE . Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101.
Chen Z, Torrens JI, Anand A, Spiegelman BM Friedman JM . Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. 2005;1:93–106.
Haap M, Heller E, Thamer C, Tschritter O, Stefan N, Fritsche A . Association of serum phosphate levels with glucose tolerance, insulin sensitivity and insulin secretion in non-diabetic subjects. Eur J Clin Nutr. 2006;60:734–739.
Kautzky-Willer A, Pacini G, Niederle B, Schernthaner G, Prager R . Insulin secretion, insulin sensitivity and hepatic insulin extraction in primary hyperparathyroidism before and after surgery. Clin Endocrinol (Oxf). 1992;37:147–155.
Rorsman P, Renstrom E . Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045.
Velasco M, Larque C, Gutiérrez-Reyes G, Arredondo R, Sanchez-Soto C, Hiriart M . Metabolic syndrome induces changes in KATP-channels and calcium currents in pancreatic β-cells. Islets. 2012;4:302–311.
Marcinkowski W, Zhang G, Smogorzewski M, Massry SG . Elevation of (Ca2+)i of renal proximal tubule cells and downregulation of MRNA of PTH-PTHrP, V1a and AT1 receptors in kidneys of diabetic rats. Kidney Int. 1997;51:1950–1955.
Donate-Correa J, Muros-de-Fuentes M, Mora-Fernández C, Navarro-González JF . FGF23/Klotho axis: phosphorus, mineral metabolism and beyond. Cytokine Growth Factor Rev. 2012;23:37–46.
Kuro-o MA . Potential link between phosphate and aging—lessons from Klotho-deficient mice. Mech Ageing Dev. 2010;131:270–275.
Masoro, EJ . Dietary restriction-induced life extension: a broadly based biological phenomenon. Biogerontology. 2006;7:153–155.
Kayo T, Allison DB, Weindruch R, Prolla TA . Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc Natl Acad Sci U S A. 2001;98:5093–5098.
Xie, W, Tran, TL, Finegood, DT, van de Werve, G . Dietary P(i) deprivation in rats affects liver cAMP, glycogen, key steps of gluconeogenesis and glucose production. Biochem J. 2000;352:227–232.
Paula FJ, Plens AE, Foss MC . Effects of hypophosphatemia on glucose tolerance and insulin secretion. Horm Metab Res. 1998;30:281–284.
Di Marco GS, Hausberg M, Hillebrand U, Rustemeyer P, Wittkowski W, Lang D, Pavenstädt H . Increased inorganic phosphate induces human endothelial cell apoptosis in vitro . Am J Physiol Renal Physiol. 2008;294:F1381–1387.
Rucker D, Tonelli M . Cardiovascular risk and management in chronic kidney disease. Nat Rev Nephrol. 2009;5:287–296.
Arunabh S, Pollack S, Yeh J, Aloia JF . Body fat content and 25-hydroxyvitamin D levels in healthy women. J. Clin. Endocrinol. Metab. 2003;88:157–161.
Wortsman J, Matsuoka LY, Chen TC, Lu Z, Holick MF . Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr. 2000;72:690–693.
Reinehr T, De Sousa G, Alexy U, Kersting M, Andler W . Vitamin D status and parathyroid hormone in obese children before and after weight loss. Eur J Endocrinol. 2007;157:225–232.
Bellia A, Marinoni G, D'Adamo M, Guglielmi V, Lombardo M, Donadel G, Gentileschi P, Lauro D, Federici M, Lauro R, Sbraccia P . Parathyroid Hormone and Insulin Resistance in Distinct Phenotypes of Severe Obesity: A Cross-Sectional Analysis in Middle-Aged Men and Premenopausal Women. J Clin Endocrinol Metab. 2012;97:724–732.
Narvaez CJ, Matthews D, Broun E, Chan M, Welsh J . Lean phenotype and resistance to diet-induced obesity in vitamin D receptor knockout mice correlates with induction of uncoupling protein-1 in white adipose tissue. Endocrinology. 2009;150:651–666.
Wong KE, Szeto FL, Zhang W, Ye H, Kong J, Zhang Z, Sun XJ, Li YC . Involvement of the vitamin D receptor in energy metabolism regulation of uncoupling proteins. Am J Physiol Endocrinol Metab. 2009;296:E820–E828.
Streicher C, Zeitz U, Andrukhova O, Rupprecht A, Pohl E, Larsson TE, Windisch W, Lanske B, Erben RG . Long-term Fgf23 deficiency does not influence aging, glucose homeostasis, or fat metabolism in mice with a nonfunctioning vitamin D receptor. Endocrinology. 2012;153:1795–1805.
De Paula FJ, Dick-de-Paula I, Bornstein S, Rostama B, Le P, Lotinun S, Baron R, Rosen CJ . VDR haploinsufficiency impacts body composition and skeletal acquisition in a gender-specific manner. Calcif Tissue Int. 2011;89:179–191.
Wong KE, Kong J, Zhang W, Szeto FL, Ye H, Deb DK, Brady MJ, Li YC . Targeted expression of human vitamin D receptor in adipocytes decreases energy expenditure and induces obesity in mice. J Biol Chem. 2011;286:33804–33810.
M.B. Zemel, Role of calcium and dairy products in energy partitioning and weight management, Am. J. Clin. Nutr. 2004;79:907S–912S.
Kelishadi R, Zemel MB, Hashemipour M, Hosseini M, Mohammadifard N, Poursafa P . Can a dairy-rich diet be effective in long-term weight control of young children? J Am Coll Nutr. 2009;28:601–610.
Zemel MB, Kim JH, Woychik RP, Michaud EJ, Kadwell SH, Patel IR, Wilkison WO . Agouti regulation of intracellular calcium: role in the insulin resistance of viable yellow mice. Proc Natl Acad Sci USA. 1995;92:4733–4737.
Shi H, Norman AW, Okamura W H, Sen A, Zemel MB . 1alpha,25-dihydroxyvitamin D3 inhibits uncoupling protein 2 expression in human adipocytes, FASEB J. 2002;16:1808–1810.
Xue B, Greenberg AG, Kraemer FB, Zemel MB . Mechanism of intracellular calcium inhibition of lipolysis in human adipocytes, FASEB J. 2001;15:2527–2529.
Bell NH, Epstein S, Greene A, Shary J, Oexmann MJ, Shaw S . Evidence for alteration of the vitamin D-endocrine system in obese subjects. J Clin Invest. 1985;76:370–373
Grethen E, McClintock R, Gupta CE, Jones R, Cacucci BM, Diaz D, Fulford AD, Perkins SM, Considine RV, Peacock M . Vitamin D and hyperparathyroidism in obesity. J Clin Endocrinol Metab. 2011;96:1320–1326
Grethen E, Hill KM, Jones R, Cacucci BM, Gupta CE, Acton A, Considine RV, Peacock M . Serum leptin, parathyroid hormone, 1,25-dihydroxyvitamin D, fibroblast growth factor 23, bone alkaline phosphatase, and sclerostin relationships in obesity. J Clin Endocrinol Metab. 2012;97:1655–1662.
Adam MA, Untch BR, Danko ME, Stinnett S, Dixit D, Koh J, Marks JR, Olson JA Jr. . Severe obesity is associated with symptomatic presentation, higher parathyroid hormone levels, and increased gland weight in primary hyperparathyroidism. J Clin Endocrinol Metab. 2010;95:4917–4924.
Cheng SP, Doherty GM, Chang YC, Liu CL . Leptin: the link between overweight and primary hyperparathyroidism? Med Hypotheses. 2011;76:94–96.
Paula FJ, Rosen CJ . Obesity, diabetes mellitus and last but not least, osteoporosis. Arq Bras Endocrinol Metabol. 2010;54:150–157.
Taylor WH, Khaleeli AA . Coincident diabetes mellitus and primary hyperparathyroidism. Diabetes Metab Res Rev. 2001;17:175–180.
Khaleeli AA, Johnson JN, Taylor WH . Prevalence of glucose intolerance in primary hyperparathyroidism and the benefit of parathyroidectomy. Diabetes Metab Res Rev. 2007;23:43–48.
Wolf M . Forging forward with 10 burning questions on FGF23 in kidney disease. J Am Soc Nephrol. 2010;21:1427–1435.
Juppner H . Phosphate and FGF-23. Kidney Int. 2011;121:S24–S27.
Quarles LD . Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–3828.
Bergwitz C, Jüppner H . Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104
Wolf M . Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;82:737–747
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T . Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568.
Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y . Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17:2393–2403.
Mirza MA, Alsioö J, Hammarstedt A, Erben RG, Michaëlsson K, Tivesten A, Marsell R, Orwoll E, Karlsson MK, Ljunggren O, Mellström D, Lind L, Ohlsson C, Larsson TE . Circulating fibroblast growth factor-23 is associated with fat mass and dyslipidemia in two independent cohorts of elderly individuals. Arterioscler Thromb Vasc Biol. 2011;31:219–227.
Burke SK, Dillon MA, Hemken DE, Rezabek MS, Balwit JM . Meta-analysis of the effect of sevelamer on phosphorus, calcium, PTH, and serum lipids in dialysis patients. Adv Ren Replace Ther. 2003;10:133–145.
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T . Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001;98:6500–6505.
Tsuji K, Maeda T, Kawane T, Matsunuma A, Horiuchi N . Leptin stimulates fibroblast growth factor 23 expression in bone and suppresses renal 1α,25-dihydroxyvitamin D3 synthesis in leptin-deficient mice. J Bone Miner Res. 2010;25:1711–1723.
Reid IR . Fat and bone. Arch Biochem Biophys. 2010;503:20–27.
Dimitri P, Wales J, Bishop N . Fat and bone in children-differential effects of obesity on bone size and mass according to fracture history. J Bone Miner Res. 2010;25:527–536.
Goulding A, Grant AM, Williams SM . Bone and body composition of children and adolescents with repeated forearm fractures. J Bone Miner Res. 2005;20:2090–2096.
Loder R, Aronson D, Greenfield M . The epidemiology of bilateral slipped capital femoral epiphysis. A study of children in Michigan. J Bone Joint Surg. 1993;75:1141–1147.
Davids JR, Huskamp M, Bagley AM . A dynamic biomechanical analysis of the etiology of adolescent tibia vara. J Pediatr Orthop. 1996;16:461–468.
Goulding A, Jones IE, Taylor RW, Williams SM, Manning PJ . Bone mineral density and body composition in boys with distal forearm fractures: a dual-energy x-ray absorptiometry study. J Pediatr. 2001;139:509–515.
Skaggs DL, Loro ML, Pitukcheewanont P, Tolo V, Gilsanz V . Increased body weight and decreased radial cross-sectional dimensions in girls with forearm fractures. J Bone Miner Res. 2001;16:1337–1342.
Manias K, McCabe D, Bishop N . Fractures and recurrent fractures in children; varying effects of environmental factors as well as bone size and mass. Bone. 2006;39:652–657.
Clark EM, Ness AR, Tobias JH . Avon Longitudinal Study of Parents and Children Study Team. Adipose tissue stimulates bone growth in prepubertal children. J Clin Endocrin Metab. 2006;91:2534–2541.
Dimitri P, Bishop N, Walsh JS, Eastell R . Obesity is a risk factor for fracture in children but is protective against fracture in adults: A paradox. Bone. 2012;50:457–466.
Foley S, Quinn S, Jones G . Tracking of bone mass from childhood to adolescence and factors that predict deviation from tracking. Bone 2009;44:752–757.
Nagasaki K, Kikuchi T, Hiura M, Uchiyama M . Obese Japanese children have low bone mineral density after puberty. J Bone Miner Metab. 2004;22:376–381.
De Laet C, Kanis JA, Odén A, Johanson H, Johnell O, Delmas P, Eisman JA, Kroger H, Fujiwara S, Garnero P, McCloskey EV, Mellstrom D, Melton LJ 3rd, Meunier PJ, Pols HA, Reeve J, Silman A, Tenenhouse A . Body mass index as a predictor of fracture risk: a meta-analysis. Osteoporos Int. 2005;16:1330–1338.
Pereira FA, De Castro JA, dos Santos JE, Foss MC, Paula FJ . Impact of marked weight loss induced by bariatric surgery on bone mineral density and remodeling. Braz J Med Biol Res. 2007;40:509–517.
Borges NC, Vasconcellos RS, Carciofi AC, Gonçalves KN, Paula FJ, Filho DE, Canola JC . DXA, bioelectrical impedance, ultrasonography and biometry for the estimation of fat and lean mass in cats during weight loss. BMC Vet Res. 2012;8:111.
Compston JE, Watts NB, Chapurlat R, Cooper C, Boonen S, Greenspan S, Pfeilschifter J, Silverman S, Díez-Pérez A, Lindsay R, Saag KG, Netelenbos JC, Gehlbach S, Hooven FH, Flahive J, Adachi JD, Rossini M, Lacroix AZ, Roux C, Sambrook PN, Siris ES ; Glow Investigators. Obesity is not protective against fracture in postmenopausal women: GLOW. Am J Med. 2011;124:1043–1050.
Holmberg, AH, Johnell O, Nilsson PM, Nilsson J, Berglund G, Akesson K . Risk factors for fragility fracture in middle age. A prospective population-based study of 33 000 men and women. Osteoporos Int. 2006;17:1065–1077.
Spaine LA, Bollen SR . ‘The bigger they come…’: the relationship between body mass index and severity of ankle fractures. Injury. 1996;27:687–689.
Premaor MO, Pilbrow L, Tonkin C, Parker RA, Compston J . Obesity and fractures in postmenopausal women. J Bone Miner Res. 2010;25:292–297.
Premaor MO, Ensrud K, Lui L, Parker RA, Cauley J, Hillier TA, Cummings S, Compston JE ; Study of Osteoporotic Fractures. Risk factors for nonvertebral fracture in obese older women. J Clin Endocrinol Metab. 2011;96:2414–2421.
Yamaguchi T, Kanazawa I, Yamamoto M, Kurioka S, Yamauchi M, Yano S, Sugimoto T . Associations between components of the metabolic syndrome versus bone mineral density and vertebral fractures in patients with type 2 diabetes. Bone. 2009;45:174–179.
Russell M, Mendes N, Miller KK, Rosen CJ, Lee H, Klibanski A, Misra M . Visceral fat is a negative predictor of bone density measures in obese adolescent girls. J Clin Endocrinol Metab. 2010;95:1247–1255.
Campos RM, Lazaretti-Castro M, Mello MT, Tock L, Silva PL, Corgosinho FC, Carnier J, Piano Ad, Sanches PL, Masquio DC, Tufik S, Dâmaso AR . Influence of visceral and subcutaneous fat in bone mineral density of obese adolescents. Arq Bras Endocrinol Metabol. 2012;56:12–18.
Choi HS, Kim KJ, Kim KM, Hur NW, Rhee Y, Han DS, Lee EJ, Lim SK . Relationship between visceral adiposity and bone mineral density in Korean adults. Calcif Tissue Int. 2010;87:218–225.
Bredella MA, Torriani M, Ghomi RH, Thomas BJ, Brick DJ, Gerweck AV, Harrington LM, Breggia A, Rosen CJ, Miller KK . Determinants of bone mineral density in obese premenopausal women. Bone. 2011;48:748–754.
Motyl KJ, Dick-de-Paula I, Maloney AE, Lotinun S, Bornstein S, De Paula FJ, Baron R, Houseknecht KL, Rosen CJ . Trabecular bone loss after administration of the second-generation antipsychotic risperidone is independent of weight gain. Bone 2012;50:490–498.
Bosma M, Hesselink MK, Sparks LM, Timmers S, Ferraz MJ, Mattijssen F, van Beurden D, Schaart G, de Baets MH, Verheyen FK, Kersten S, Schrauwen P . Perilipin 2 improves insulin sensitivity in skeletal muscle despite elevated intramuscular lipid levels. Diabetes. 2012;61:2679–2690.
Brumbaugh DE, Crume TL, Nadeau K, Scherzinger A, Dabelea D . Intramyocellular lipid is associated with visceral adiposity, markers of insulin resistance, and cardiovascular risk in prepubertal children: the EPOCH study. J Clin Endocrinol Metab. 2012;97:E1099–E1105.
Ryan AS, Buscemi A, Forrester L, Hafer-Macko CE, Ivey FM . Atrophy and intramuscular fat in specific muscles of the thigh: associated weakness and hyperinsulinemia in stroke survivors. Neurorehabil Neural Repair. 2011;25:865–872.
Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR . Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517.
van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ . Cold activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508.
Bredella MA, Fazeli PK, Freedman LM, Calder G, Lee H, Rosen CJ, Klibanski A . Young women with cold-activated brown adipose tissue have higher bone mineral density and lower Pref-1 than women without brown adipose tissue: a study in women with anorexia nervosa, women recovered from anorexia nervosa, and normal-weight women. J Clin Endocrinol Metab. 2012;97:E584–E590.
Rosen CJ, Bouxsein ML . Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol. 2006;2:35–43.
Bredella MA . Perspective: the bone-fat connection. Skeletal Radiol. 2010;39:729–773.
Kawai M, De Paula FJ, Rosen CJ . New insights into osteoporosis: the bone-fat connection. J Intern Med. 2012;272:317–329.
Ecklund K, Vajapeyam S, Feldman HA, Buzney CD, Mulkern RV, Kleinman PK, Rosen CJ, Gordon CM . Bone marrow changes in adolescent girls with anorexia nervosa. J Bone Miner Res. 2010;25:298–304.
Karczewska-Kupczewska M, Straczkowski M, Adamska A, Nikołajuk A, Otziomek E, Górska M, Kowalska I . Insulin sensitivity, metabolic flexibility, and seruma diponectin concentration in women with anorexia nervosa. Metabolism 2010;59:473–477.
Devlin MJ . Why does starvation make bones fat? Am J Hum Biol. 2011;23:577–585.
Askmyr M, Sims NA, Martin TJ, Purton LE . What is the true nature of the osteoblastic hematopoietic stem cell niche? Trends Endocrinol Metab. 2009;20:303–309.
Fuchs E, Tumbar T, Guasch G . Socializing with the neighbors: stem cells and their niche. Cell. 2004;116:769–778.
Shen Y, Nilsson SK . Bone, microenvironment and hematopoiesis. Curr Opin Hematol. 2012;19:250–255.
Isern J, Méndez-Ferrer S . Stem cell interactions in a bone marrow niche. Curr Osteoporos Rep. 2011;9:210–218.
Takubo K, Suda T . Roles of the hypoxia response system in hematopoietic and leukemic stem cells. Int J Hematol. 2012;95:478–483.
Franco CB, Paz-Filho G, Gomes PE, Nascimento VB, Kulak CA, Boguszewski CL, Borba VZ . Chronic obstructive pulmonary disease is associated with osteoporosis and low levels of vitamin D. Osteoporos Int. 2009;20:1881–1887.
Forni GL, Perrotta S, Giusti A, Quarta G, Pitrolo L, Cappellini MD, D'Ascola DG, Borgna Pignatti C, Rigano P, Filosa A, Iolascon G, Nobili B, Baldini M, Rosa A, Pinto V, Palummeri E . Neridronate improves bone mineral density and reduces back pain in β-thalassaemia patients with osteoporosis: results from a phase 2, randomized, parallel-arm, open-label study. Br J Haematol. 2012;158:274–282.
Baldanzi G, Traina F, Marques Neto JF, Santos AO, Ramos CD, Saad ST . Low bone mass density is associated with hemolysis in Brazilian patients with sickle cell disease. Clinics (Sao Paulo). 2011;66:801–805.
Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM . Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–49.
Minguell JJ, Erices A, Conget P . Mesenchymal stem cells. Exp Biol Med (Maywood). 2001;226:507–520.
Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B . PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. 2009;106:232–246.
Ichida F, Nishimura R, Hata K, Matsubara T, Ikeda F, Hisada K, Yatani H, Cao X, Komori T, Yamaguchi A, Yoneda T . Reciprocal roles of MSX2 in regulation of osteoblast and adipocyte differentiation. J Biol Chem. 2004;279:34015–34022.
Kang S, Bennett CN, Gerin I, Rapp LA, Hankenson KD, Macdougald OA . Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282:14515–14524.
Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B . Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. 2007;148:2669–2680.
Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, Eto K, Tsubamoto Y, Okuno A, Murakami K, Sekihara H, Hasegawa G, Naito M, Toyoshima Y, Tanaka S, Shiota K, Kitamura T, Fujita T, Ezaki O, Aizawa S, Nagai R, Tobe K, Kimura S, Kadowaki T . PPARgamma mediates high fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 1999;4:597–609.
Karsenty G, Ferron M . The contribution of bone to whole-organism physiology. Nature. 2012;481:314–320.
Bouillon R, Decallonne B . The White adipose tissue connection with calcium and bone homeostasis. J Bone Miner Res. 2010;25:1707–1710.
Björnholm M, Münzberg H, Leshan RL, Villanueva EC, Bates SH, Louis GW, Jones JC, Ishida-Takahashi R, Bjørbaek C, Myers MG Jr. . Mice lacking inhibitory leptin receptor signals are lean with normal endocrine function. J Clin Invest. 2007;117:1354–1360.
Shi Y, Yadav VK, Suda N, Liu XS, Guo XE, Myers MG Jr., Karsenty G . Dissociation of the neuronal regulation of bone mass and energy metabolism by leptin in vivo . Proc Natl Acad Sci U S A. 2008;105:20529–20533.
Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G . Leptin inhibits boneformation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207.
Goldeladze JO, Drevon CA, Syversen U, Reseland JE . Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis and mineralization: Impact on Differentiation markers, apoptosis and osteoclastic signaling. J Cell Biochem. 2002;85:825–836.
Holloway WR, Collier FM, Aitken CJ, Myers DE, Hodge JM, Malakellis M, Gough TJ, Collier GR, Nicholson GC . Leptin inhibits osteoclast generation. J Bone Miner Res. 2002;17:200–209.
Burguera B, Hofbauer LC, Thomas T, Gori F, Evans GL, Khosla S, Riggs BL, Turner RT . Leptin reduces ovariectomy-induced bone loss in rats. Endocrinology. 2001;142:3546–3553.
Clemens TL, Karsenty G . The osteoblast: an insulin target cell controlling glucose homeostasis. J Bone Miner Res. 2011;26:677–680.
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G . Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–469.
Hood DA, Mechanisms of exercises-induced mitochondrial biogenesis in skeletal muscle. Applied Physiol Nutr Metab. 2009;34:465–472.
Fulzele K, Clemens TL . Novel functions for insulin in bone. Bone. 2012;50:452–456.
Fulzele K, Riddle RC, DiGirolamo DJ, Cao X, Wan C, Chen D, Faugere MC, Aja S, Hussain MA, Brüning JC, Clemens TL . Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Paula, F., Rosen, C. Bone Remodeling and Energy Metabolism: New Perspectives. Bone Res 1, 72–84 (2013). https://doi.org/10.4248/BR201301005
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.4248/BR201301005
This article is cited by
-
Reduced bone formation and increased bone resorption drive bone loss in Eimeria infected broilers
Scientific Reports (2023)
-
Official position of the Brazilian Association of Bone Assessment and Metabolism (ABRASSO) on the evaluation of body composition by densitometry: part I (technical aspects)—general concepts, indications, acquisition, and analysis
Advances in Rheumatology (2022)
-
Metabolic activities affect femur and lumbar vertebrae remodeling, and anti-resorptive risedronate disturbs femoral cortical bone remodeling
Experimental & Molecular Medicine (2021)
-
Upper Airway Obstruction Elicited Energy Imbalance Leads to Growth Retardation that Persists after the Obstruction Removal
Scientific Reports (2020)
-
The Benefits of Strength Training on Musculoskeletal System Health: Practical Applications for Interdisciplinary Care
Sports Medicine (2020)
