
Abstract
Background:
Cancer progression and metastasis is profoundly influenced by protein kinase D1 (PKD1) and metastasis-associated protein 1 (MTA1) in addition to other pathways. However, the nature of regulatory relationship between the PKD1 and MTA1, and its resulting impact on cancer metastasis remains unknown. Here we present evidence to establish that PKD1 is an upstream regulatory kinase of MTA1.
Methods:
Protein and mRNA expression of MTA1 in PKD1-overexpressing cells were determined using western blotting and reverse-transcription quantitative real-time PCR. Immunoprecipitation and proximity ligation assay (PLA) were used to determine the interaction between PKD1 and MTA1. PKD1-mediated nucleo-cytoplasmic export and polyubiquitin-dependent proteosomal degradation was determined using immunostaining. The correlation between PKD1 and MTA1 was determined using intra-tibial, subcutaneous xenograft, PTEN-knockout (PTEN-KO) and transgenic adenocarcinoma of mouse prostate (TRAMP) mouse models, as well as human cancer tissues.
Results:
We found that MTA1 is a PKD1-interacting substrate, and that PKD1 phosphorylates MTA1, supports its nucleus-to-cytoplasmic redistribution and utilises its N-terminal and kinase domains to effectively inhibit the levels of MTA1 via polyubiquitin-dependent proteosomal degradation. PKD1-mediated downregulation of MTA1 was accompanied by a significant suppression of prostate cancer progression and metastasis in physiologically relevant spontaneous tumour models. Accordingly, progression of human prostate tumours to increased invasiveness was also accompanied by decreased and increased levels of PKD1 and MTA1, respectively.
Conclusions:
Overall, this study, for the first time, establishes that PKD1 is an upstream regulatory kinase of MTA1 status and its associated metastatic activity, and that the PKD1-MTA1 axis could be targeted for anti-cancer strategies.
Similar content being viewed by others


Main
Metastasis is a complex physiological process that results in increased plasticity of the cellular phenotypes and underlying molecular pathways, which are crucial for increased invasion. A critical determinant of cancer metastasis is the process of tumour cells dissemination and acquiring invasiveness, also referred to as epithelial-mesenchymal transition (EMT) (Nguyen et al, 2009). The process of EMT is profoundly influenced by the set of regulatory molecules and transcription factors with role in promoting dissemination and invasiveness, leading to metastatic phenotype (Nguyen et al, 2009; Yu et al, 2016). In this context, metastasis-associated protein 1 (MTA1), a major upstream regulator of EMT-related transcription factors, has a critical role in promoting EMT (Pakala et al, 2011). As its identification, MTA1 has evolved into an important metastatic marker for human cancers (Li et al, 2012). Expression of MTA1 is upregulated in a wide variety of human cancers and positively correlates with tumour aggressiveness and metastasis including prostate cancer (Li et al, 2013a). In general, inhibition of MTA1 improves clinical outcome in cancer patients, including of the non-small cell lung and hepatocellular carcinomas (Li et al, 2013b; Deng et al, 2015). Despite these advances, the molecular mechanisms that regulates MTA1 protein in human cancer, including the contribution of protein kinase D1 (PKD1) signalling (see below), remain poorly understood.
The PKD1, a serine-threonine kinase, is downregulated in prostate, breast and colon cancer (Jaggi et al, 2003; Du et al, 2010; Sundram et al, 2012, 2014; Kang et al, 2016). PKD1 belongs to the protein kinase D family of kinases, which are homologous to protein kinase C family of kinases and calcium-calmodulin kinases in the regulatory and kinase domains, respectively (Sundram et al, 2011). PKD1 localises to the cytosol, nucleus, membrane, Golgi and mitochondria (Du et al, 2009; Sundram et al, 2011; Du et al, 2012; Li et al, 2016), and mediates a variety of cellular functions such as signal transduction, membrane trafficking, cell survival, cell migration and proliferation (Rozengurt et al, 2005). PKD1 interacts and directly regulate β-catenin/TCF4 pathway in cancer cells (Sundram et al, 2012, 2014). In addition, PKD1 also interacts and transcriptionally attenuates androgen receptor expression in prostate cancer cells and thus contributes to the progression of androgen-dependent to androgen-independent prostate cancer (Mak et al, 2008). PKD1 and its associated proteins are critical regulators of epithelial phenotypes and tissue development (Ivanova et al, 2008; Bastea et al, 2012; Borges et al, 2013; Coxam et al, 2014). Hence, dysregulation of PKD1 could lead to reversion of epithelial phenotype and increased cancer metastasis through EMT (Zheng et al, 2014; Qin et al, 2015; Durand et al, 2016).
Here we provide experimental evidence to demonstrate that PKD1 is a negative regulator of MTA1, and that PKD1-MTA1 axis promotes EMT and prostate cancer metastasis. For the first time, findings presented here revealed PKD1 as a new upstream modifier of MTA1 oncogene, and represents a potential therapeutic target to inhibit MTA1-driven cancer metastasis.
Materials and methods
Meta-analysis for PKD1 and MTA1 gene expression
We performed meta-analysis for PKD1 (gene name: PRKD1) and MTA1 gene expression data in Oncomine database (Rhodes et al, 2004) (Compendia Bioscience, Ann Arbor, MI, USA). Primary filter of Cancer vs Normal analysis for prostate, breast and colorectal cancer, and data set filter to use mRNA data sets were applied. Studies were selected based on mean P-value of the median gene rank in MTA1 overexpression or PRKD1 under expression patterns across data sets. The log2 median gene intensity of different datasets were extracted and imported into Graphpad Prism v5.0 (Graphpad Software, San Diego, CA, USA) and the matched curve (normal vs cancer) was constructed.
Materials
RPMI-1640 media containing glutamine were supplemented with 10% heat-inactivated FBS (Atlanta Biologics, Atlanta, GA, USA), 1 × 100 mM sodium pyruvate and 100 × Antibiotic and Antimycotic Solution purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). G418 sulphate solution was purchased from MP Biomedicals (Fisher Scientific, Waltham, MA, USA). Cycloheximide, Bryostatin-1 and MG132 were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA). For PKD1 and MTA1 inhibition studies, selective PKD1 siRNA and MTA1 siRNA were purchased from Life Technologies (Thermo Fischer Scientific, Carlsbad, CA, USA).
Cell lines and other materials
LNCaP (ATCC, Manassas, VA, USA) and C4-2 (Urocor, Oklahoma City, OK, USA) were grown in RPMI-1640 (Lonza, Walkersville, MD, USA) media supplemented with 10% FBS (Atlanta Biologicals, Atlanta, GA), Antibiotic and Antimycotic solution. Cells (C4-2 transfected with PKD1 or GFP vector) were grown in G418 selection media. SW480, SW48, MDA-MB231 and MCF7 (ATCC) were grown in DMEM (Lonza) supplemented with 10% FBS, and Antibiotic and Antimycotic solution. Other chemicals were purchased from Sigma (Sigma-Aldrich) unless otherwise mentioned.
Antibodies
Rabbit Polyclonal PKD1 (C-20) (sc-639) and mouse monoclonal MTA1 (A-11) (sc-17773) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit polyclonal MTA1 (A300-280A) was purchased from Bethyl Laboratories Inc. (Montgomery, TX, USA). Phospho-PKD1 (S916) (2051S), PKD1 substrate antibody (4381), K48-linkage-specific polyubiquitin (12805S), α-tubulin (2125) and GAPDH (5174) were purchased from Cell Signaling, Inc. (Danvers, MA, USA). Golgi and trans-Golgi antibodies (611434) (Golgi Sampler Kit; BD Transduction Laboratories, San Jose, CA, USA). Ubiquitin (ab134953) and RANK (ab13918) antibody was purchased from Abcam (Cambridge, MA, USA). β-Actin (A2228) antibody was purchased from Sigma (Sigma-Aldrich). HRP-conjugated secondary antibody was purchased from Promega, Inc. (Madison, WI, USA).
Western blotting
Cancer cells (70–80% confluent) were washed with ice-cold phosphate buffer saline (PBS) and lysed in 2 × SDS lysis buffer. Equivalent amounts of protein samples were electrophoretically resolved on 4–20% SDS–PAGE gels and transferred on a PVDF membrane (Biorad Laboratories, Hercules, CA, USA). Membranes were blocked with 5% BSA or milk in TBST and incubated with primary antibody for overnight at 4 °C. After three subsequent washings, membranes were incubated in secondary antibody and developed with the help of Immobilon Western Chemiluminescent HRP substrate (EMD Millipore, Billerica, MA, USA) after appropriate washing procedure.
Immunofluorescence
Cancer cells were seeded in a four-well chamber (Thermo Scientific, Nunc, Waltham, MA, USA) slides with 1 × 105 cells in each well. The cells were fixed in 2% paraformaldehyde for 15 min, permeabilised for 5 min with Triton-X (0.2%) in PBS solution. After subsequent PBS washes the slides were incubated in PKD1 (1:500), MTA1 (1:1000), GM130 (1:250), p230 (1:250) and Ubiquitin (1:500) antibodies for 1 h at room temperature. After washing with PBS, the slides were incubated with anti-mouse CY3 and anti-rabbit Alexa Fluor 488 (1:200) (Thermo Fisher Scientific, Carlsbad, CA, USA)-labelled secondary antibody, mounted with Vectashield Mounting Medium containing DAPI and then processed for confocal microscopy laser with Zeiss 710 confocal microscope (Zeiss, Oberkochen, Germany). Similarly, cells were fixed, permeabilised and stained lysotracker Red DND-99 (Thermo Fisher Scientific, Carlsbad, CA, USA).
Transfection
Prostate cancer (C4-2) and colon cancer (SW480) cells were serum starved overnight in OPTI-MEM media (Thermo Fisher Scientific, Carlsbad, CA, USA) and then transfected with pEGFP vector or pEGFP vector containing PKD1 gene using Lipofectamine 2000 (Thermo Fisher Scientific, Carlsbad, CA, USA). After 6 h of transfection, the media was replaced with 10% serum containing media. Transfected cells were propagated in the presence of a selection agent (G418) and used after 48 h of stable transfection.
Real-time PCR array analysis
RNA samples from C4-2-PKD1, C4-2-GFP, SW480-PKD1 and SW480-GFP cells were prepared and cDNA was synthesised using superscript II RNAase H (High capacity RNA to cDNA kit). The cDNA was amplified by Taqman real-time PCR using gene-specific primers. The PCR amplification was performed in the Roche Lightcycler 480 (Roche, Indianapolis, IN, USA).
Immunoprecipitation
Immunoprecipitation was performed in C4-2-PKD1 and C4-2-GFP-overexpressing cells using PKD1, lysine 48-specific polyubiquitin antibody and PKD1 substrate antibody, and immunoblotted using MTA1 antibody. PKD1 deletion mutants-overexpressing cells were also immunoprecipitated using PKD1 antibody and immunoblotted using MTA1 antibody. Equal amount of proteins (500 μg) were incubated with antibodies using protein A/G agarose beads (Santa Cruz Biotechnology) followed by incubation at 4 °C overnight. Immunoprecipitates were eluted using SDS sample buffer.
Proximity ligation assay
Proximity ligation assay was performed using Duolink Red starter kit according to the manufacturer’s guidelines. Briefly, oligonucleotide conjugated anti-mouse minus and anti-rabbit plus PLA secondary probes were added after overnight conjugation with mouse MTA1 and rabbit PKD1 antibody. The secondary probes were allowed to incubate in humidified chamber for 1 h at 37 °C followed by incubation with DNA ligase for 30 min at 37 °C and DNA polymerase for 100 min to amplify DNA by rolling cycle amplification which gives off red fluorescence. DAPI was used as a counterstain for nuclei and the cells were visualised using Zeiss 710 Confocal microscope.
Real-time cell migration and invasion assays through xCELLigence system
To validate the effects of PKD1 on motility and invasiveness, real-time migration and invasion assays were performed using the xCELLigence system (Khan et al, 2015). xCELLigence system is an electrical impedance-based method that allows for the measurement of cell migration, invasion and proliferation in real time. Briefly, cells (C4-2-GFP and C4-2-PKD1) were seeded in invasion and migration (7 × 104) plates and were analysed in xCELLigence instrument at 37 °C, 5% CO2 for real-time cell migration and invasion assays.
Animal studies
Athymic nude male mice were used for these experiments. The mice were maintained in a pathogen-free environment and all procedures were carried out as approved by the UTHSC Institutional Animal Care and Use Committee (UTHSC-IACUC). All the procedures and methods were carried out in accordance with the approved guidelines of UTHSC-IACUC.
Subcutaneous tumours
Athymic nude mice (Cancer Research Animal Core, UTHSC, Memphis, TN, USA) (seven per group) were injected subcutaneously with C4-2-PKD1 and C4-2-GFP cells. Briefly, C4-2-PKD1 and C4-2-GFP cells (2 × 106 cells per mouse) were dispersed in 100 μl 1 × PBS and 100 μl Matrigel (BD Biosciences, Bedford, MA, USA) and injected subcutaneously directly into the dorsal flank of nude mice. The animals were periodically monitored for tumour development and the tumour volume was measured using a digital Vernier caliper. The tumour volume was calculated using the ellipsoid volume formula: tumour volume (mm3) = π/6 × L × W × H, wherein L is length, W is width and H is height. The tumour volume was regularly monitored and allowed to grow until the tumour burden reached a maximum volume of 1000 mm3. At the time of killing, the mice tumours were removed, fixed in formalin, embedded in paraffin and sliced into 5 μm sections for further processing and analysis. Intra-tibial bone metastasis model: Athymic nude mice were used to generate an intra-tibial model of prostate cancer. Briefly, C4-2-PKD1 and C4-2-GFP cells (1 × 105 cells per mouse) were dispersed in 10 μl 1 × PBS and directly into injected intra-tibially in the mice. The animals were periodically monitored for tumour development for over 2 months. At the time of killing, the mice tibiae were removed, fixed in formalin, demineralised and embedded in paraffin, for further processing and analysis.
Immunohistochemistry
Prostate cancer mouse tissues (PTEN KO and WT, and transgenic adenocarcinoma of mouse prostate (TRAMP)) obtained from University of Wisconsin, Madison, from Dr. Bilal Hafeez, were immunostained using heat-induced antigen retrieval immunohistochemistry techniques with the help of Biocare kit (Biocare Medical, Concord, CA, USA) and analysed as previously described (Gupta et al, 2012). Human cancer tissue microarray was purchased from AccuMax (ISU Abxis Co., Ltd, Newark, CA, USA) for prostate cancer and US Biomax (Rockville, MD, USA) for breast and colon cancer. After deparaffinisation, rehydration and peroxidation, antigen retrieval was performed in Biocare Decloacking Chamber at 125 °C for 30 s, whereas the slides were immersed in 1 × Diva solution. Slides were then incubated in background sniper following incubation with rabbit PKD1 (C-20) (1:3500) or mouse MTA1 (1:50) antibody diluted in Da Vinci Green diluent (in case of double stain IHC). After washing, slides were incubated with mouse probe for 30 min and HRP-polymer probe for another 30 min and then incubated with 3,3’-diaminobenzidine reagent solution for 3 min. Haematoxylin was used as counterstain before dehydrating the slides and mounting with Ecomount mounting media. Slides treated with no primary antibody were used as negative controls. Composite Scoring for the staining intensity was calculated as (% of cell stained) × (staining intensity). The scoring criteria for % of cell stained was <25% cells stained – ‘1’, 25–50% cells stained – ‘2’, 50–75% cells stained – ‘3’ and 75–100% cells stained – ‘4’. Scoring criteria for staining intensity was as follows: ‘1’, ‘2’, ‘3’ and ‘4’. The maximum composite score for staining was 16 and minimum was 0.
Statistical analyses
Student’s t-test was used for analysis of statistical significance and the significance was determined using a paired t-test. A P-value of<0.05 was considered significant.
Results
An inverse PKD1–MTA1 relationship in human cancers
As the progression of human cancer and metastasis have been shown to be accompanied by downregulation of PKD1, as well as overexpression of MTA1 chromatin remodelling factor (Molli et al, 2008; Du et al, 2010) we sought to first interrogate publicly available human cancer data sets for an inherent relationship between the levels of PKD1 and MTA1. We observed a significant negative correlation between the levels of PKD1 and MTA1 in different human cancer data sets, at large (Supplementary Figure S1A). For example, downregulation of PKD1 mRNA expression in human prostate, breast and colon cancer tissues was accompanied by a substantial upregulation of MTA1 mRNA expression in the Oncomine data sets (Figure 1A and Supplementary Figure S1B). Prostate cancer data set comprised prostate adenocarcinoma (normal=20; cancer=69; n=89) (Wallace et al, 2008), prostate carcinoma (normal=4; cancer=8; n=12) (Magee et al, 2001) and (normal=28; cancer=59; n=87) (Grasso et al, 2012), and benign prostatic hyperplasia (normal=3; cancer=3; n=6) (Tomlins et al, 2007). Breast cancer, data set comprised invasive ductal breast adenocarcinoma (normal=9; cancer=30; n=39 and normal=5; cancer=19; n=24) (Radvanyi et al, 2005), tubular breast carcinoma (normal=144; cancer=67; n=211) (Curtis et al, 2012), invasive ductal breast carcinoma (normal=61; cancer=389; n=450) (TCGA Breast 2011, Oncomine), invasive breast carcinoma (normal=61; cancer=76; n=137) (TCGA Breast 2011, Oncomine), mucinous breast carcinoma (normal=61; cancer=4; n=65) (TCGA Breast 2011, Oncomine). Similarly, for colon cancer the data set comprised colon adenocarcinoma (normal=18; cancer=18; n=36) (Notterman et al, 2001), rectal adenocarcinoma (normal=3; cancer=60; n=63) (TCGA Colorectal 2011, Oncomine), caecum adenocarcinoma (normal=19; cancer=22; n=41) (TCGA Colorectal 2011, Oncomine) and colon adenocarcinoma (normal=19; cancer=101; n=120) (TCGA Colorectal 2011, Oncomine). These results suggest that the noticed negative correlation in the expression data sets of respective cancers for the levels of PKD1 and MTA1 might contribute to cancer progression.

PKD1 and MTA1 levels negatively correlate in human cancers. (A) mRNA expression pattern from Oncomine and TCGA data sets for prostate, breast and colon cancer tissues. For prostate cancer: red solid circle represents Magee et al, 2001, blue solid triangle represents Grasso et al, 2012, green solid square represents Tomlins et al, 2007 and solid orange hexagon represents Wallace et al, 2008 data sets respectively. For breast cancer: red solid circle represents Curtis et al, 2012, solid blue triangle represents TCGA study for invasive breast carcinoma, solid green square represents TCGA data set for invasive ductal breast carcinoma, solid orange hexagon represents TCGA data set for mucinous breast carcinoma and solid magenta star represents Radvanyi et al, 2005 data sets. Similarly, for colon cancer: solid red dot represents TCGA data set for rectal adenocarcinoma, solid blue triangle represents TCGA data set for colon adenocarcinoma, solid green square represents TCGA data set for cecum adenocarcinoma and solid magenta diamond represents Notterman et al, 2001 data set. (B) To define expression correlation between PKD1 and MTA1, western blot analysis was performed using protein extracts of prostate (LNCaP, C4-2) and colon (SW480, SW48) cancer cells. (C) Confocal microscopy images to show subcellular localisation of PKD1 (Green) and MTA1 (Red) in LNCaP, C4-2, SW480 and SW48 cells. DAPI (blue) is used for nuclear staining. Original magnifications × 400, scale bar=10 μm. (D) C4-2 and SW480 cells were stably transfected with either PKD1-overexpressing vector (C4-2-PKD1, SW480-PKD1) or GFP-tagged vector (C4-2-GFP, SW480-GFP) (control) and protein lysates were subjected to western blot analysis. (E) Quantitative PCR analysis of MTA1 mRNA levels in PKD1-overexpressing (C4-2-PKD1, SW480-PKD1) and vector control (C4-2-GFP, SW480-GFP) cells. (F) Western blot analysis of PKD1 and MTA1 in PKD1-expressing (PKD1-control siRNA) and PKD1 knockdown (PKD1-siRNA) LNCaP cells. LNCaP cells were transiently transfected with control and PKD1 siRNAs for 48 h. (G) Similarly, cells were transiently transfected with control and MTA1 siRNA and protein lysates were examined for MTA1 and PKD1 expression by western blot analysis. MTA1=metastasis-associated protein 1; PKD1=protein kinase D1. *P<0.01.
To investigate the functional significance of the newly recognised inverse PKD1–MTA1 relationship, we next evaluated the levels of PKD1 and MTA1 in the context of metastatic potentials of cancer cell models. We found an increased PKD1 expression in less-metastatic LNCaP prostate, SW480 colon and MCF-7 breast cancer cell lines as compared with metastatic C4-2 prostate, SW48 colon and MDA-MB231 breast cell lines, whereas the levels MTA1 were converse to the levels of PKD1 in these cells (Figure 1B and C, and Supplementary Figure S1C). To experimentally validate the presumed negative regulation of MTA1 by PKD1, we found that indeed PKD1 overexpression in C4-2 prostate and SW480 colon cancer cells inhibit the expression of MTA1 mRNA and MTA1 protein (Figure 1D and E). Consistent with the notion of a regulatory function of PKD1 upon MTA1’s expression, selective depletion of PKD1 in LNCaP prostate cancer cells or MTA1 in C4-2 cells leads to an increased MTA1 or PKD1 expression, respectively (Figure 1F and G). Collectively, these results suggest that PKD1 expression inversely correlates with MTA1 status and that PKD1 may be an important upstream regulator of MTA1 status in cancer cells.
PKD1 stimulates cytoplasmic accumulation of MTA1
To understand the basis of noted inverse relationship between the levels of PKD1 and MTA1 in human cancer, we performed confocal microscopic studies using PKD1-overexpressing cancer cells. We found a remarkably decreased expression of MTA1 in the nucleus of cells stably overexpressing PKD1 in C4-2 prostate and SW480 colon cancer cells (Figure 2A and B). Conversely, PKD1 knockdown by selective siRNA in LNCaP prostate cancer cells was accompanied by a subsequent increase in the level of MTA1 (Figure 1G), revealing that the level of PKD1 may control the expression and/or stability of MTA1 protein. In this context, results from CHX-chase studies demonstrated a relative faster degradation of MTA1 in PKD1-overexpressing cells as compared to the levels of MTA1 in the control cells (Figure 2C and Supplementary Figure S2A), suggesting a role of PKD1 in regulating the levels of MTA1 protein.

PKD1 induces nucleo-cytoplasmic translocation and degradation of MTA1. (A and B) Immunofluorescence analysis of PKD1 and MTA1 expression in vector control (C4-2-GFP and SW480-GFP) and PKD1-overexpressing C4-2 (C4-2-PKD1 and SW480-PKD1) cells. Control and PKD1 vectors were detected using GFP tag (Green), whereas MTA1 was detected using CY3-labelled secondary antibody (Red) and DAPI was used for nuclear staining. Original magnifications × 400. (C and D) Western blot analysis of MTA1 and pPKD1 expression in vector control and PKD1-overexpressing C4-2 cells that were treated with indicated concentration of cyclohexamide (CHX for 2, 2.5 and 3 h) or Bryostatin-1 (20 and 30 nM) for 24 h. (E–G) Confocal microscopy images Bryostatin-1 treated C4-2 cells at 3, 12 and 24 h. At 3 h localisation of MTA1 (Red) into Golgi as detected by GM130 (Green), a Golgi marker, at 12 h in trans-Golgi network (TGN) using P230 (Green) as TGN marker and at 24 h MTA1 (Green) in lysosomes using Red lysotracker (Red) as a marker for lysosome. Original magnifications × 400, scale bar=10 μm. MTA1=metastasis-associated protein 1; PKD1=protein kinase D1.
To elucidate the possible mechanism of PKD1-mediated noticed degradation of MTA1, time-course confocal microscopy experiments were performed using a known pharmacological activator of PKD1, Bryostatin-1, which stimulates PKD1-S916 phosphorylation (Figure 2D). In these studies, we first evaluated the effect of PKD1 activation by Bryostatin-1 on the sub-cellular distribution of MTA1 using distinct organelle markers as a function of PKD1 treatment. We found that at 3 h post-PKD1 activation, MTA1 predominantly localises in the Golgi apparatus (Figure 2E) followed by its accumulation in the trans-Golgi network (Figure 2F) at 12 h post-PKD1 activation, whereas MTA1 localises to the lysosome at 24 h post-PKD1 stimulation with Bryostatin-1 (Figure 2G).
Intracellular proteins are degraded via phosphorylation coupled ubiquitin–proteosome pathway (UPP) (Lecker et al, 2006). As the ubiquitin degradation machinery are cytoplasmic (Lecker et al, 2006) and the fact that MTA1 accumulates in the cytoplasm (this study), we sought to investigate whether PKD1 phosphorylates MTA1 and such modification serves as a prerequisite for its cytoplasmic translocation and ubiquitination. Therefore, we examined the impact of PKD1 phosphorylation of MTA1 on its stability. Using the pharmacological activator of PKD1, we observed a distinct co-localisation of MTA1 with a ubiquitin protein antibody (Figure 3A and Supplementary Figure S2B(i)), which was abrogated by a ubiquitin inhibitor MG132 (Figure 3B and Supplementary Figure S2B(ii)). Accordingly, Bryostatin-1 treatment of cells in presence of MG132 prevents Bryostatin-1-mediated reduction in the level of MTA1 (Figure 3C).

PKD1 degrades MTA1 protein via ubiquitin mechanism. (A) MTA1 (Red) co-localisation with Ubiquitin (UBX) marker (Green) in response to Bryostatin-1, a PKD1 activator treatment in confocal microscopy analysis. Bryostatin-1 treatment (24 h) translocates MTA1 to lysosome via ubiquitin-dependent mechanism. (B) MTA1 (Red) co-localisation with Ubiquitin (UBX) marker (Green) in cells that were treated with Bryostatin-1 and MG132, a proteasome inhibitor (10 μM) for 24 h. MG132, abrogated the co-localisation of MTA1 and UBX. Original magnifications × 400. (C). Western blot analysis for MTA1 analysis in lysates of C4-2 cells that were treated with Bryostatin-1, MG132 (10 μ M) and combination of Bryostatin-1 and MG132 for 24 h. β-Actin was used as a loading control. (D) Protein lysates of transiently transfected C4-2-PKD1 and C4-2-GFP cells were subjected to immunoprecipitation with K48-specific polyubiquitin antibody followed by western blotting to detect interaction of MTA1 with K48 protein. IgG was used as an internal control. Original magnifications × 400, scale bar=10 μm. IP=immunoprecipitation; MTA1=metastasis-associated protein 1; PKD1=protein kinase D1; WB=western blotting.
Different topologies of polyubiquitin chains mediate different cellular functions. For example, K48 polyubiquitin chain mediates proteosomal degradation, whereas K63 mediates proteasome-independent signal for endocytosis (Ohtake et al, 2016). To determine whether PKD1 activation triggered degradation of MTA1 is a K48-polyubiquitin-dependent mechanism, we immunoprecipitated MTA1 using K48-polyubiquitin-specific antibody and found PKD1 in the immunoprecipitated material (Figure 3D). These results suggest a role of PKD1 in the noted nucleo-cytoplasmic MTA1 translocation and polyubiquitin-mediated MTA1 degradation in cancer cells.
PKD1 interacts and phosphorylates MTA1
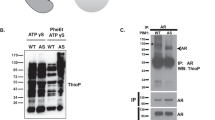
To examine whether PKD1 inhibits MTA1 expression through its direct interaction with MTA1, we performed co-immunoprecipitation studies. By transiently overexpressing PKD1 and its deletion mutant, we found that PKD1 physically interacts with MTA1 but not kinase-dead PKD1 (Figure 4A). To verify the physical interaction of PKD1 with MTA1, we performed PLA as illustrated in Figure 4B. In this assay, we observed that PKD1 and MTA1 interaction is predominantly localised in the extra-nuclear space in PKD1-overexpressing cells. In these studies, we used PKD1 only and MUC13/HER2 as negative and positive controls. Using a series of PKD1 deletion mutants, we next showed that N-terminal domain and kinase domain deletion attenuated interaction between PKD1 and MTA1 as compared to the wild-type PKD1 (Figure 4C), suggesting a crucial role of these domains in the noted MTA1-PKD1 interaction and, consequently, in the nucleus to cytoplasmic redistribution of MTA1 upon PKD1 activation. Consistent with these results, due to loss of interaction between the N-terminal deletion mutant PKD1 or kinase-dead PKD1, MTA1 localises in the nucleus (Figure 4D). As PKD1 is an important member of serine threonine kinase family, we next investigated whether PKD1 induces phosphorylation of MTA1. Using phospho-PKD substrate antibody in immunoprecipitation or western blotting assays, we confirmed that PKD1 indeed phosphorylates MTA1 only in PKD1-overexpressing cells (Figure 4D). In brief, PKD1 antagonises MTA1’s nuclear activity via its phosphorylation, cytoplasmic translocation and degradation.

PKD1 interacts and phosphorylates MTA1. (A) Protein lysates of C4-2-PKD1, C4-2-GFP and C4-2-PKD1-KD cells were subjected to immunoprecipitation (IP) with PKD1 antibody followed by western blot analysis with MTA1- and PKD1-specific antibodies, to detect the interaction between PKD1 and MTA1. (B) Proximity ligation assay (PLA) for interaction of PKD1 with MTA1 in C4-2-PKD1 cells, which is showing PKD1-MTA1 interaction in peri-nuclear space as indicated by red dots (i). Proximity ligation assay (PLA) image of PKD1 only and HER2/MUC13 in HPAF-II cells serves as negative (ii) and positive controls (iii), respectively. (iv) represent the mean fluorescence intensity of nuclear and cytoplasmic MTA1 in C4-2-PKD-overexpressing cells. *P<0.01. (iv). Original magnifications × 400, scale bar=10 μm. (C) To determine which domain of PKD1 is responsible for interaction MTA1, C4-2 cells were transiently transfected with different deletion mutants of PKD1 (ΔAC (acidic region deleted), ΔC1a (Cysteine-rich region a deleted), ΔC1b (Cysteine-rich region b deleted), ΔCRD (Cysteine-rich region a and b deleted) ΔPH (pleckstrin homology domain deletion), ΔN (N-terminal domain deletion), PKD1-KD (Kinase dead domain)) and full-length PKD1. Protein lysates were subjected to IP with PKD1 antibody followed by western blot analysis for MTA1 and PKD1 proteins. (D) To determine phosphorylation of MTA1 by PKD1, kinase assay was performed using protein lysates from C4-2-PKD1 and C4-2-GFP cells. Protein lysates were subjected to IP with phospho-PKD substrate antibody followed by western blot analysis with MTA1 and PKD1 antibodies. IgG was used as negative control. IP=immunoprecipitation; MTA1=metastasis-associated protein 1; PKD1=protein kinase D1; WB=western blotting.
PKD1 overexpression represses metastatic potential of cancer cells
To understand the relevance of PKD1 regulation of MTA1 axis in cancer cells, we performed functional metastasis assays. The overexpression of PKD1, which downregulates MTA1 expression, effectively inhibited invasion and migration of C4-2 cancer cells (Figure 5A and B). Consistent with these results, selective downregulation of MTA1 also inhibited cellular invasion and migration (Supplementary Figure S3). Next, we examined the impact on PKD1 overexpression on metastatic potential of C4-2 cells using ectopically transplanted xenograft and bone metastatic mouse models. We found that PKD1-overexpressing C4-2 cells is accompanied by a significant (P<0.05) reduction of tumour volume as compared with tumour volumes from the control cells (Figure 5C (i, ii)). As bone metastasis is commonly observed in prostate cancer patients (Simpson et al, 2012) to assess an anti-metastatic potential of PKD1 overexpression, we also used intra-tibial bone metastatic mouse model involving intra-tibially implantation of test cells in nude mice. We found distinct bone metastasis in mice with control C4-2 cells but not in PKD1-overexpressing C4-2 cells (Figure 5D i). In addition, mice implanted with control cells showed higher MTA1 and RANK (marker of osteoblast to osteoclast differentiation) expression as compared with PKD1-overexpressing cells (Figure 5D ii). These results suggest a role of PKD1 status in prostate tumour formation and distant metastasis.

PKD1 inhibits metastatic and growth phenotypes cancer cells. (A and B) Cellular invasion and migration of C4-2-PKD1 (red line) and C4-2-GFP (green line) were determined using real-time xCeLLigence system. Red bar indicates significantly less invasion and migration of C4-2-PKD1 cells compared with C4-2-GFP. (C) C4-2-PKD1 and C4-2-GFP cells (2 × 106) were ectopically transplanted into the dorsal flank of nude mice (n=4). Representative images of tumour bearing mice (i) and tumour volume (ii) of C4-2-GFP (n=2) and C4-2-PKD1 (n=0) implanted cells are shown at day 29 post injection. Asterisk represents statistical significance. (D) C4-2-GFP and C4-2-PKD1 cells (1 × 105) were intra-tibially transplanted in nude mice. Representative images of tumour growth and bone metastasis are shown in mice and in X-Ray images. C4-2-GFP implanted mice showed tumour growth and bone metastasis, whereas C4-2-PKD1 implanted mice failed to show growth and bone metastasis (i). Histopathological analysis of bone of C4-2-GFP and C4-2-PKD1-implanted mice for the expression of PKD1, MTA1 and RANK proteins as analysed by immunohistochemistry (ii). MTA1=metastasis-associated protein 1; PKD1=protein kinase D1. Scale bar=10 μm.
PKD1 and MTA1 in spontaneous prostate cancer models and human tumours
To validate the biological relevance of the PKD1-MTA1 axis in a whole animal model, we utilised spontaneous cancer mouse models. Transgenic adenocarcinoma of mouse prostate (TRAMP) is a commonly used prostate cancer (Suman et al, 2016) mouse model, spontaneously develop tumours at 9 weeks from the dorsal lobe (Hafeez et al, 2012) leading to well and moderately differentiated adenocarcinomas at 16 weeks and 24 weeks (Siddiqui et al, 2008), respectively. To determine the significance of PKD1-MTA1 in tumour progression and metastasis, we evaluated the expression of PKD1 and MTA1 in TRAMP model. Immunohistochemistry analysis of prostate tumour tissues showed a progressive downregulation of PKD1 expression and upregulation of MTA1 (Figure 6A). These findings were supported by protein and transcriptome analyses performed using different stage prostate tumour tissues (Figure 6B and C). In addition to TRAMP model, similar findings were observed in PTEN-knockout (PTEN-KO) prostate cancer mouse model which mimics human prostate cancer progression to metastatic stage (Hafeez et al, 2015) (Figure 6D and F). Collectively, these data suggest a high significance of PKD1 in MTA1 regulation in physiologically relevant models.

PKD1 negatively correlates with MTA1 in prostate tumour tissues of TRAMP and PTEN-KO mice. (A–C) PKD1 and MTA1 expression in the prostate tissues of WT and TRAMP mice at different weeks by immunohistochemical (A), immunoblotting (B) and qPCR (C) analyses. Result indicate decreased expression of PKD1 and subsequent increased of MTA1 in the prostate tumour of TRAMP mice compared to the WT control mice. (D–F). Similarly, the expression of PKD1 and MTA1 in the prostate tissues of wild-type (WT) and PTEN-KO mice was analysed at different weeks by immunohistochemical (D), immunoblotting (E) and qPCR (F) analyses. Values in bar graphs are shown as mean±s.d. obtained from three experimental replicates. MTA1=metastasis-associated protein 1; PKD1=protein kinase D1; PTEN-KO=PTEN-knockout; WT=wild type. *P<0.01; Student’s t-test, scale bar=10 μm.
To further understand the clinical relevance of PKD1 and MTA1 association, we performed immunohistochemistry of PKD1 and MTA1 in different grades of human prostate tumours. This analysis showed high PKD1 expression in adjacent normal but its expression was repressed while tumour progressed from Gleason Grade 7 to 9. Conversely, there was low or no expression of MTA1 in adjacent normal but it progressively increased from grade 7 to 9 (Figure 7A). Interestingly, similar results were obtained for different stages of colon and breast cancer tissues (Figure 7C and D). Composite scoring for the staining intensity for PKD1 and MTA1 staining in prostate, breast and colon cancer is shown as dot plot in Supplementary Figure S4.

Clinical significance of PKD1 and MTA1 negative correlation in human cancer. (A) Expression of PKD1 and MTA1 in prostate tissue microarray (TMA: total 96 spots; adjacent normal; n=32, grade 7; n=30, grade 8; n=10, grade 9; n=24), which suggest loss of PKD1 and gain of MTA1 with the progression of cancer (Gleason grade 7 to 9). Similar pattern of PKD1 and MTA1 was observed in colon cancer TMA (total of 65 spots; adjacent normal; n=30, stage 2A; n=8, stage 2B; n=15, stage 3B; n=12) (B) and breast cancer TMA (total 81 spots; adjacent normal; n=45, stage 2A; n=23, stage 2B; n=9, stage 3B; n=4). (C) Original magnifications × 400. (D) Schematic diagram depicting molecular mechanism of PKD1-mediated regulation of MTA1 expression in cancer cells. MTA1=metastasis-associated protein 1; PKD1=protein kinase D1.
Overall, these results suggest that PKD1 downregulation and MTA1 upregulation proffers aggressive and metastatic tumour phenotype. However, resurgence of PKD1 levels leading to subsequent downregulation of MTA1 that effectively abrogates aggressiveness and metastatic behaviour of tumour cells. These findings suggest that PKD1 is an important upstream modulator of MTA1 functionality, and that identification of PKD1-MTA1 axis may have clinical implications in future for reducing cancer metastasis by developing approaches targeting the PKD1-MTA1 axis.
Discussion
Several lines of evidence suggest a crucial role of MTA1 protein in cancer invasion and metastasis. MTA1 predominantly localises in the nucleus to exert its oncogenic functionality (Sen et al, 2014). This study elucidates a novel molecular mechanism related to repression of MTA1-mediated oncogenic signalling by PKD1 (Figure 1). PKD1 inhibits MTA1’s function by supporting its redistribution from the nucleus to the cytoplasm, where it under goes proteasomal degradation (Figures 3 and 4). We also found that MTA1 preferentially interests with PKD1 through its N-terminal and kinase domain. PKD1 overexpression and/or pharmacological PKD1 activation effectively inhibits MTA1-induced cancer metastatic phenotypes. Our studies demonstrate biological and clinical significance of the noticed novel phenomenon and an inverse relationship between the levels of PKD1 and MTA1 in spontaneous cancer mouse models and human cancer tissues. These findings establish crucial role of PKD1 in the suppression of cancer cell invasion and metastasis via influencing the levels of MTA1 and hence resulting functions. In brief, PKD1 might be a novel molecular target for biochemical intervention to diminish MTA1-mediated cancer invasion and metastasis. This line of investigation is likely to have a significant clinical implication as it activates the PKD1 pathway in cancer cells which might be developed as an effective strategy to inhibit cancer progression and metastasis.
Accumulating evidence suggest that PKD1 is an important modulator of signal transduction pathways (Radvanyi et al, 2005) and is downregulated in the prostate, breast and colon cancers (Borges et al, 2013; Li et al, 2013b; Deng et al, 2015). Protein kinase D1 functions as a regulator of growth factor receptors trafficking, as well as of a modifier of cell shape and tumour cell invasion (Radvanyi et al, 2005). MTA1 is an integral member of the nucleosome remodelling and histone deacetylase complex and multifunctional DNA-damage response protein (Curtis et al, 2012). MTA1 is a dual coregulator of gene expression, as it acts both as a coactivator or corepressor of target gene in a context-dependent manner (Notterman et al, 2001). MTA1 is highly overexpressed in the nucleus of cancer cells (Lecker et al, 2006) and its nuclear expression correlates with the cancer progression and metastasis (Ohtake et al, 2016) via enhancing EMT transformation (Simpson et al, 2012). In contrast, PKD1 expression in cancer inversely associates with metastasis and patient outcome, thus suggesting an opposing functional relationship between the PKD1 and MTA1. We found a direct PKD1 interaction and phosphorylation of MTA1 and its increased cytoplasmic accumulation and degradation. These findings raise the possibility that increased expression of MTA1 in a subset of human cancer might be the result of the loss of PKD1.
Emerging data suggest that MTA1 has been widely distributed to the cytoplasmic and other subcellular compartments in addition to the nucleus and that the functions of MTA1 are not limited to its nuclear localisation (Li et al, 2009; ). However, no study has yet shown a molecular mechanism pertaining to cytoplasmic translocation coupled degradation of MTA1 in cancer cells. As we have previously shown that PKD1 activation promotes subcellular distribution of β-catenin (Sen et al, 2014), we sought to analyse whether PKD1 activation facilitates MTA1’s intracellular redistribution of MTA1. We found that pharmacological activation of PKD1 by Bryostatin-1 induces MTA1 translocation from the nucleus to cytoplasmic organelles in a time-dependent manner, where it eventually targeted for degradation via UPP pathway (Liu et al, 2014b). We found that PKD1-mediated phosphorylation triggers MTA1 polyubiquitination and its degradation. Interestingly, this PKD1-mediated nucleo-cytoplasmic translocation of MTA1 was abrogated when PKD1 was mutated at N-terminal and kinase domains, suggesting a crucial function of these PKD1 domains in the noted redistribution of MTA1.
These novel findings however raised multiple interesting possibilities and questions that need to be addressed in future, to utilise PKD1 as potential therapeutic target for the inhibition of cancer metastasis. Having a clear understanding about the inverse relationship between MTA1 and PKD1 in cancer (Supplementary Figure S5), it will be utmost important to define role of environmental, genetic and epigenetic factors that are responsible for the repression of the PKD1 in cancer cells. It would be of high interest to investigate whether MTA1 activation could acetylate or methylate histone and non-histone substrates that are crucial for PKD1 expression in cells. This is a next logical question that needs to be answered in future, as we have seen immediate suppression of PKD1 upon onset of MTA1 expression in cancer cells, as well as in spontaneous cancer mouse models. Another central topic for future studies could be related to the screening and development of clinically translatable novel PKD1 activators/modulators which might be useful to conquer cancer invasion and metastasis. These findings broaden our horizon regarding the tumour suppressor functionality and underlying molecular mechanisms of PKD1 in cancer biology which would be a critical point for further investigations to find molecular mechanisms of its downregulation and how it can be rescued in cancer cells to block tumour progression and metastasis.
Change history
20 February 2018
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bastea LI, Doppler H, Balogun B, Storz P (2012) Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PLoS ONE 7 (1): e30459.
Borges S, Doppler H, Perez EA, Andorfer CA, Sun Z, Anastasiadis PZ, Thompson E, Geiger XJ, Storz P (2013) Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res 15 (2): R66.
Coxam B, Sabine A, Bower NI, Smith KA, Pichol-Thievend C, Skoczylas R, Astin JW, Frampton E, Jaquet M, Crosier PS, Parton RG, Harvey NL, Petrova TV, Schulte-Merker S, Francois M, Hogan BM (2014) Pkd1 regulates lymphatic vascular morphogenesis during development. Cell Rep 7 (3): 623–633.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Borresen-Dale AL, Brenton JD, Tavare S, Caldas C, Aparicio S (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486 (7403): 346–352.
Deng L, Yang H, Tang J, Lin Z, Yin A, Gao Y, Wang X, Jiang R, Sun B (2015) Inhibition of MTA1 by ERalpha contributes to protection hepatocellular carcinoma from tumor proliferation and metastasis. J Exp Clin Cancer Res 34: 128.
Du C, Jaggi M, Zhang C, Balaji KC (2009) Protein kinase D1-mediated phosphorylation and subcellular localization of beta-catenin. Cancer Res 69 (3): 1117–1124.
Du C, Zhang C, Hassan S, Biswas MH, Balaji KC (2010) Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res 70 (20): 7810–7819.
Du C, Zhang C, Li Z, Biswas MH, Balaji KC (2012) Beta-catenin phosphorylated at threonine 120 antagonizes generation of active beta-catenin by spatial localization in trans-Golgi network. PLoS One 7 (4): e33830.
Durand N, Borges S, Storz P (2016) Protein kinase D enzymes as regulators of EMT and cancer cell invasion. J Clin Med 5 (2).
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA (2012) The mutational landscape of lethal castration-resistant prostate cancer. Nature 487 (7406): 239–243.
Gupta BK, Maher DM, Ebeling MC, Sundram V, Koch MD, Lynch DW, Bohlmeyer T, Watanabe A, Aburatani H, Puumala SE, Jaggi M, Chauhan SC (2012) Increased expression and aberrant localization of mucin 13 in metastatic colon cancer. J Histochem Cytochem 60 (11): 822–831.
Hafeez BB, Fischer JW, Singh A, Zhong W, Mustafa A, Meske L, Sheikhani MO, Verma AK (2015) Plumbagin inhibits prostate carcinogenesis in intact and castrated PTEN knockout mice via targeting PKCepsilon, Stat3, and epithelial-to-mesenchymal transition markers. Cancer Prev Res (Philadelphia, PA) 8 (5): 375–386.
Hafeez BB, Zhong W, Mustafa A, Fischer JW, Witkowsky O, Verma AK (2012) Plumbagin inhibits prostate cancer development in TRAMP mice via targeting PKCepsilon, Stat3 and neuroendocrine markers. Carcinogenesis 33 (12): 2586–2592.
Ivanova P, Atanasova G, Poumay Y, Mitev V (2008) Knockdown of PKD1 in normal human epidermal keratinocytes increases mRNA expression of keratin 10 and involucrin: early markers of keratinocyte differentiation. Arch Dermatol Res 300 (3): 139–145.
Jaggi M, Rao PS, Smith DJ, Hemstreet GP, Balaji KC (2003) Protein kinase C mu is down-regulated in androgen-independent prostate cancer. Biochem Biophys Res Commun 307 (2): 254–260.
Kang H-B, Lee H-R, Jee DJ, Shin S-H, Nah S-S, Yoon SY, Kim JW (2016) PRDM1, a tumor-suppressor gene, is induced by Genkwadaphnin in human colon cancer SW620 cells. J Cell Biochem 117 (1): 172–179.
Khan S, Ebeling MC, Chauhan N, Thompson PA, Gara RK, Ganju A, Yallapu MM, Behrman SW, Zhao H, Zafar N, Singh MM, Jaggi M, Chauhan SC (2015) Ormeloxifene suppresses desmoplasia and enhances sensitivity of gemcitabine in pancreatic cancer. Cancer Res 75 (11): 2292–2304.
Lecker SH, Goldberg AL, Mitch WE (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17 (7): 1807–1819.
Li DQ, Pakala SB, Nair SS, Eswaran J, Kumar R (2012) Metastasis-associated protein 1/nucleosome remodeling and histone deacetylase complex in cancer. Cancer Res 72 (2): 387–394.
Li DQ, Pakala SB, Reddy SD, Peng S, Balasenthil S, Deng CX, Lee CC, Rea MA, Kumar R (2013a) Metastasis-associated protein 1 is an integral component of the circadian molecular machinery. Nat Commun 4: 2545.
Li S, Tian H, Yue W, Li L, Gao C, Si L, Li W, Hu W, Qi L, Lu M (2013b) Down-regulation of MTA1 protein leads to the inhibition of migration, invasion, and angiogenesis of non-small-cell lung cancer cell line. Acta Biochim Biophys Sinica 45 (2): 115–122.
Li W, Ma L, Zhao J, Liu X, Li Z, Zhang Y (2009) Expression profile of MTA1 in adult mouse tissues. Tissue Cell 41 (6): 390–399.
Li Z, Zhang C, Chen L, Li G, Qu L, Balaji KC, Du C (2016) E-cadherin facilitates protein kinase D1 activation and subcellular localization. J Cell Physiol 231 (12): 2741–2748.
Liu J, Wang H, Huang C, Qian H (2014a) Subcellular localization of MTA proteins in normal and cancer cells. Cancer Metastasis Rev 33 (4): 843–856.
Liu J, Xu D, Wang H, Zhang Y, Chang Y, Zhang J, Wang J, Li C, Liu H, Zhao M, Lin C, Zhan Q, Huang C, Qian H (2014b) The subcellular distribution and function of MTA1 in cancer differentiation. Oncotarget 5 (13): 5153–5164.
Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, Catalona WJ, Watson MA, Milbrandt J (2001) Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res 61 (15): 5692–5696.
Mak P, Jaggi M, Syed V, Chauhan SC, Hassan S, Biswas H, Balaji KC (2008) Protein kinase D1 (PKD1) influences androgen receptor (AR) function in prostate cancer cells. Biochem Biophys Res Commun 373 (4): 618–623.
Molli PR, Singh RR, Lee SW, Kumar R (2008) MTA1-mediated transcriptional repression of BRCA1 tumor suppressor gene. Oncogene 27 (14): 1971–1980.
Nguyen DX, Bos PD, Massague J (2009) Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9 (4): 274–284.
Notterman DA, Alon U, Sierk AJ, Levine AJ (2001) Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res 61 (7): 3124–3130.
Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K (2016) The K48-K63 branched ubiquitin chain regulates NF-kappaB signaling. Mol Cell 64 (2): 251–266.
Pakala SB, Singh K, Reddy SD, Ohshiro K, Li DQ, Mishra L, Kumar R (2011) TGF-beta1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells. Oncogene 30 (19): 2230–2241.
Qin XJ, Gao ZG, Huan JL, Pan XF, Zhu L (2015) Protein kinase D1 inhibits breast cancer cell invasion via regulating matrix metalloproteinase expression. Eur J Gynaecol Oncol 36 (6): 690–693.
Radvanyi L, Singh-Sandhu D, Gallichan S, Lovitt C, Pedyczak A, Mallo G, Gish K, Kwok K, Hanna W, Zubovits J, Armes J, Venter D, Hakimi J, Shortreed J, Donovan M, Parrington M, Dunn P, Oomen R, Tartaglia J, Berinstein NL (2005) The gene associated with trichorhinophalangeal syndrome in humans is overexpressed in breast cancer. Proc Natl Acad Sci USA 102 (31): 11005–11010.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM (2004) ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (New York, NY) 6 (1): 1–6.
Rozengurt E, Rey O, Waldron RT (2005) Protein kinase D signaling. J Biol Chem 280 (14): 13205–13208.
Sen N, Gui B, Kumar R (2014) Role of MTA1 in cancer progression and metastasis. Cancer Metastasis Rev 33 (4): 879–889.
Siddiqui IA, Shukla Y, Adhami VM, Sarfaraz S, Asim M, Hafeez BB, Mukhtar H (2008) Suppression of NFkappaB and its regulated gene products by oral administration of green tea polyphenols in an autochthonous mouse prostate cancer model. Pharmaceutical Res 25 (9): 2135–2142.
Simpson MA, Weigel JA, Weigel PH (2012) Systemic blockade of the hyaluronan receptor for endocytosis prevents lymph node metastasis of prostate cancer. Int J Cancer 131 (5): E836–E840.
Suman S, Das TP, Moselhy J, Pal D, Kolluru V, Alatassi H, Ankem MK, Damodaran C (2016) Oral administration of withaferin A inhibits carcinogenesis of prostate in TRAMP model. Oncotarget 7 (33): 53751–53761.
Sundram V, Chauhan SC, Ebeling M, Jaggi M (2012) Curcumin attenuates beta-catenin signaling in prostate cancer cells through activation of protein kinase D1. PLoS ONE 7 (4): e35368.
Sundram V, Chauhan SC, Jaggi M (2011) Emerging roles of protein kinase D1 in cancer. Mol Cancer Res 9 (8): 985–996.
Sundram V, Ganju A, Hughes JE, Khan S, Chauhan SC, Jaggi M (2014) Protein kinase D1 attenuates tumorigenesis in colon cancer by modulating beta-catenin/T cell factor activity. Oncotarget 5 (16): 6867–6884.
Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, Kalyana-Sundaram S, Wei JT, Rubin MA, Pienta KJ, Shah RB, Chinnaiyan AM (2007) Integrative molecular concept modeling of prostate cancer progression. Nat Genet 39 (1): 41–51.
Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, Stephens RM, Caporaso NE, Loffredo CA, Ambs S (2008) Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res 68 (3): 927–936.
Yu J, Lei R, Zhuang X, Li X, Li G, Lev S, Segura MF, Zhang X, Hu G (2016) MicroRNA-182 targets SMAD7 to potentiate TGFbeta-induced epithelial-mesenchymal transition and metastasis of cancer cells. Nat Commun 7: 13884.
Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, Ren G, Zhou T, Storz P, Wang HY, Kang Y (2014) PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer cell 26 (3): 358–373.
Acknowledgements
We thank Ms. Sarah Radel for ectopic xenograft study and Sonam Kumari for editorial assistance. We are also grateful to Dr. Angelika Hausser (Institute of Cell Biology and Immunology, University of Stuttgart, Germany) for providing PKD1 deletion mutants. Confocal microscopy studies were conducted at the imaging core in the Neuroscience Department at The University of Tennessee Health Science Center. The help provided by Pathology core is kindly acknowledged. This work was supported by Department of Defense (DOD) (PC130870 to SCC and MJ); NIH U01CA162106 to SCC and MJ, NIH R01CA142736, R01CA210192, R01CA204452 and R01CA2206069 to SCC and College of Pharmacy/University of Tennessee Health Science Center Seed Grant to MJ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ganju, A., Chauhan, S., Hafeez, B. et al. Protein kinase D1 regulates subcellular localisation and metastatic function of metastasis-associated protein 1. Br J Cancer 118, 587–599 (2018). https://doi.org/10.1038/bjc.2017.431
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.431

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}