
Abstract
Background:
Tumour-initiating cells (TICs) account for chemoresistance, tumour recurrence and metastasis, and therefore represent a major problem in tumour therapy. However, strategies to address TICs are limited. Recent studies indicate Cdk5 as a promising target for anti-cancer therapy and Cdk5 has recently been associated with epithelial–mesenchymal transition (EMT). However, a role of Cdk5 in TICs has not been described yet.
Methods:
Expression of Cdk5 in human cancer tissue was analysed by staining of a human tissue microarray (TMA). Functional effects of Cdk5 overexpression, genetic knockdown by siRNA and shRNA, and pharmacologic inhibition by the small molecule roscovitine were tested in migration, invasion, cell death, and tumorsphere assays and in tumour establishment in vivo. For mechanistic studies, molecular biology methods were applied.
Results:
In fact, here we pin down a novel function of Cdk5 in TICs: knockdown and pharmacological inhibition of Cdk5 impaired tumorsphere formation and reduced tumour establishment in vivo. Conversely, Cdk5 overexpression promoted tumorsphere formation which was in line with increased expression of Cdk5 in human breast cancer tissues as shown by staining of a human TMA. In order to understand how Cdk5 inhibition affects tumorsphere formation, we identify a role of Cdk5 in detachment-induced cell death: Cdk5 inhibition induced apoptosis in tumorspheres by stabilizing the transcription factor Foxo1 which results in increased levels of the pro-apoptotic protein Bim.
Conclusions:
In summary, our study elucidates a Cdk5-Foxo1-Bim pathway in cell death in tumorspheres and suggests Cdk5 as a potential target to address TICs.
Similar content being viewed by others


Main
Tumour-initiating cells (TICs) limit therapeutic success of anti-cancer chemotherapy as they show high tumour-initiating potential, cause the establishment of metastases and are resistant to standard therapy (Mani et al, 2008; Scheel and Weinberg, 2012; Pinto et al, 2013; Pattabiraman and Weinberg, 2014). Consequently, therapeutic strategies that address TICs may substantially improve anti-cancer treatment and patient prognosis.
In order to find new approaches for targeting TICs, research on TICs has been strongly intensified during recent years. Various mechanisms that contribute to TIC generation and survival have been elucidated (Scheel and Weinberg, 2012); TIC generation has been associated with epithelial–mesenchymal transition (EMT), a process of cellular plasticity that confers mesenchymal properties to epithelial cells. During EMT, epithelial cells change their morphology, acquire highly migratory and invasive mesenchymal traits and gain stem-like properties (Mani et al, 2008; Scheel et al, 2011; Schmidt et al, 2015). Mechanisms responsible for the increased survival of TICs include increased DNA damage repair, altered cell cycle checkpoint control, overexpression of drug efflux proteins like multidrug resistance transporters as well as impaired apoptosis (Signore et al, 2013). Nevertheless, our understanding of the mechanisms and the regulation of signalling components contributing to TIC survival is still limited.
Cdk5 is a serine/threonine kinase pivotal for neuronal development, synaptic plasticity and neurotransmission and implicated in neurodegenerative diseases like Alzheimer’s and Parkinson’s disease (Dhavan and Tsai, 2001). During the last years, important non-neuronal functions of Cdk5 have been elucidated (Liebl et al, 2011a). Amongst others, our own studies demonstrated indispensable functions of endothelial Cdk5 in lymphatic vessel development (Liebl et al, 2015) and in tumour angiogenesis (Merk et al, 2016). Moreover, recent studies elucidated functions of Cdk5 in cancer, for example, we elucidated a role of Cdk5 in DNA damage response in hepatocellular carcinoma (HCC; Ehrlich et al, 2015). Importantly, Cdk5 was associated with tumour initiation: aberrant Cdk5 activity induced neuroendocrine medullary thyroid carcinoma (Pozo et al, 2013) and ASCL1-driven upregulation of Cdk5 contributed to the establishment of neuroendocrine and small-cell cancers (Meder et al, 2016). Notably, Cdk5 has recently been linked with EMT (Liang et al, 2013; Ren et al, 2015; Sun et al, 2015). However, a function of Cdk5 in TICs has not been described yet.
Along this line, we hypothesised that Cdk5 might contribute to tumour initiation. According, the aim of the present study was to evaluate a potential role and the molecular signalling of Cdk5 in TICs in order to judge Cdk5 as a potential target to address TICs.
Materials and methods
Cells
Cells were cultured under constant humidity at 37 °C and 5% CO2. MCF10A cells were from ATCC (Manassas, VA, USA) and cultured with DMEM/Ham’s F12 containing Horse serum 5%, EGF 20 ng ml−1, Insulin 5 μg ml−1, Hydrocortisone 0.5 μg ml−1, Choleratoxin 0.1 μg ml−1 and penicillin/streptomycin (P/S). MCF7 cells were from DSMZ (Braunschweig, Germany) and cultured with RPMI 1640 containing FCS 10%, Insulin 5 μg ml−1, Pyruvate 1 mM, NEAA 1% and P/S. The human urinary carcinoma cell line T24 was kindly provided by Dr B Mayer (Surgical Clinic, LMU, Munich, Germany). T24 were cultured with McCoyś medium containing FCS 10%, glutamine (1.5 mM) and for lentiviral-transduced T24 cells, puromycin (1 μg ml−1) was used. Immortalised human mammary epithelial (HMLE) cells stably transfected with Twist-ER were described previously (Scheel et al, 2011). In brief, HMLE cells were cultivated in mammary epithelial cell growth medium (PromoCell GmbH, Heidelberg, Germany) supplemented with P/S (PAA Laboratories, Pasching, Austria) and 10 μg ml−1 blasticidin (Gibco, Germering, Germany). To induce EMT, HMLE Twist-ER cells were treated with 20 nM 4-Hydroxytamoxifen (4-OH-TX; Sigma-Aldrich, Taufkirchen, Germany) for 10 days, whereby cells were split and supplied with fresh medium and stimulation reagents every 3 days.
Transfection of cells
Overexpression experiments were performed using FuGENE HD reagent (Promega, Madison, WI, USA) according to the manufacturers protocol with a reagent:DNA ratio of 3 : 1. The following plasmids were used: Cdk5 (addgene 1871), p35 (addgene 1347).
Silencing experiments were performed with DharmaFECT 1 Transfection Reagent (Thermo Scientific, Waltham, MA, USA) corresponding to the manufacturers recommendation. The following siRNAs were used: non-targeting (nt) siRNA: D-001810-01; Cdk5 siRNA: J-003239-09 and J-003239-10 (Thermo Scientific/Dharmacon, Lafayette, CO, USA).
For stable downregulation of Cdk5, lentiviral transduction with non-targeting (nt) and Cdk5 shRNA was performed using MISSION shRNA Lentiviral Transduction Particles (Sigma-Aldrich) according to the manufacturer’s protocol.
Cell viability assay
Cells were seeded in 96-well plates and incubated for 24 h. Cells were treated as indicated for further 24 h. Cell viability was measured using CellTiter-Blue reagent (Promega) according to the manufacturers protocol.
Proliferation assay
Cells were seeded in 96-well plates and incubated for 24 h. The initial cell number was determined. Cells were treated as indicated for further 72 h. Cell viability was measured using CellTiter-Blue reagent (Promega) according to the manufacturers protocol. For proliferation assay cells were stained with crystal violet (0.5% in methanol 20%), lysed with sodium citrate (0.05 M in ethanol 50%) and absorbance (550 nm) was measured in a plate-reading photometer (SpectraFluor PlusTM, Tecan, Crailsheim, Germany).
Apoptosis analysis
Apoptosis was performed according to Nicoletti et al (1991). Briefly, tumorspheres were collected, washed and stained with HFS-solution containing PI (50 μg ml−1, overnight, 4 °C) followed by flow cytometry (FACSCantoII, BD Biosciences, Franklin Lakes, NJ, USA) the next day. SubG1 cells were analysed using the FlowJo 7.6 analysis software (Tree Star Inc., Ashland, OR, USA). Percentage of specific apoptosis was calculated using the formula 100 × ((experimental apoptosis (%)−spontaneous apoptosis (%))/(100%−spontaneous apoptosis (%)); Fulda et al, 2001).
Clonogenic growth
Cells were treated as indicated for 24 h. Afterwards, cells were trypsinized and freshly seeded (5000 cells per 6-well) and incubated for 7 days. Colonies were stained with crystal violet (0.5% in methanol 20%), lysed with sodium citrate (0.05 M in ethanol 50%) and the absorbance was measured in a plate-reading photometer (SpectraFluor PlusTM, Tecan, Crailsheim, Germany).
Migration and invasion
After indicated treatments (24 h), cells were labelled with CellTrackerGreen CMFDA (Life Technologies, Carlsbad, CA, USA), resuspended in medium without serum and added to the upper compartment of the transwell chamber (6.5 mm, polycarbonate membrane, 8.0 μm pore size, Corning Incorporated, Tewksbury, MA, USA). For invasion assays, the transwell inserts were coated with Matrigel. Culture medium containing 10% serum was added to the lower chamber, medium without serum served as negative control. After migration, cells were fixed in 4% PFA (10 min) and cells in the upper chamber were removed using cotton buds. Four pictures per well were taken by means of an inverted microscope (Axiovert 25/200, Zeiss, Jena, Germany) at 10-fold magnification. Cells were counted using ImageJ software.
Sphere formation assay
Sphere formation assays were performed as described (Dontu et al, 2003). Cells were treated with Roscovitine (24 h) or transfected as indicated (48 h). Cells were resuspended in sphere-formation medium containing 1% methyl cellulose (Sigma-Aldrich) and seeded into poly-(2-hydroxyethyl methacrylate; pHEMA, Sigma-Aldrich)-coated 12-well plates at 4 × 104 cells per well. After incubation for 10 days, 12 pictures per well were taken by using a LSM 510 Meta (Zeiss, Jena, Germany) microscope. Number of spheres bigger than 50 μm was evaluated using ImageJ.
Immunoblotting
Immunoblotting was performed as described (Ehrlich et al, 2015). The following primary antibodies were used: AKT (9272, Cell Signaling), AKT phospho (Ser 473; 4051, Cell Signaling), β-actin (MAB1501, Millipore), β-tubulin (2146, Cell Signaling), Bim (2819, Cell Signaling), Cdk5 (AHZ0492, Life Technologies), COX IV (4844, Cell Signaling), CREB (9104, Cell Signaling), ERK (9102, Cell Signaling), ERK phospho (9106, Cell Signaling), Foxo1 (2880, Cell Signaling), GAPDH (sc-69778, Santa Cruz), HIF1α (610958, BD Biosciences), phospho-histone H2aX (γH2aX, 2577, Cell Signaling), matrix metalloproteinase-9 (MMP-9; 3852 Cell Signaling), MMP-2 (4022 Cell Signaling), NICD-1 (4147, Cell Signaling), NICD-4 (sc-5594, Santa Cruz), Notch 1 (3608, Cell Signaling), Retinoblastoma protein phospho (Ser807/811; 8516, Cell Signaling), Retinoblastoma protein (554136, BD Biosciences), Stat3 phospho (Ser 727; 9134, Cell Signaling), Stat3 (9132, Cell Signaling). Anti-vimentin, -snail, -β-catenin, -claudin-1, N-cadherin were from Cell Signaling (EMT Sampler Kit, 9782), c-Myc (sc-788, Santa Cruz). Loading control by stain-free gels was performed by adding 0.5% TCE (2,2,2-Trichloroethanol, Sigma-Aldrich) to the PAGE gels according to the TGX Stain-Free Gels system (BioRad, Hercules, CA, USA).
Kinase activity assay
Cdk5 was immunoprecipitated using anti-Cdk5 antibody (sc-173, Santa Cruz Biotechnology) and Protein G Agarose beads (Sigma-Aldrich). Beads were resuspended in 50 μl kinase buffer (50 mM HEPES pH 7.0, 10 mM MgCl2, 1 mM DTT, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, 3 mM β-glycerophosphate, 4 mM Complete EDTAfree). An aliquot of 2 μ M ATP, 10 μCi 32P-γ-ATP (Hartmann Analytic, Braunschweig, Germany) and 0.05 μg μl−1 histone H1 (Type III from calf thymus, Sigma-Aldrich) were added and samples were incubated at 30 °C for 20 min before termination by boiling with 5 × SDS sample buffer (5 min, 95 °C). Electrophoresis and autoradiography was performed.
Immunostaining
Cells were seeded in 8-well slides (ibidi GmbH, μ-Slide 8-Well ibiTreat, 80826), washed, fixed with 4% PFA and permeabilized using 0.1% TritonX-100. After blocking, cells were incubated with anti-Foxo1 antibody and a secondary antibody (Alexa Fluor 488, A-11008, Invitrogen (Carlsbad, CA, USA); 1 h each). Nuclei were visualised using 5 μg ml−1 Hoechst33342. Microscopy was conducted using LSM 510 Meta (Zeiss, Jena, Germany) microscope.
Mitochondrial fractionation
Cells were collected, incubated with permeabilization buffer for 20 min on ice (210 mM mannitol, 200 mM sucrose, 10 mM HEPES, pH 7.2, 0.2 mM Na2EGTA, 5 mM succinate, bovine serum albumin 0.15%, 80 μg ml−1 digitonin) and centrifuged (1300 r.p.m., 4 °C, 10 min). The supernatant was collected (cytosolic fraction) and the pellet permeabilized with 0.1% TritonX-100 (15 min, on ice; mitochondrial fraction).
Cytosol-nuclei fractionation
Cells were collected, washed and pelleted (1500 r.p.m., 4 °C, 5 min). The pellet was incubated with BufferA (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0,5 mM PMSF, Complete 1 : 100) for 15 min. After addition of Nonidet P-40 10% cells were again pelleted (12 000 r.p.m., 4 °C, 1 min). The supernatant (cytosolic fraction) was removed and the pellet was resuspended in hypertonic BufferB (20 mM HEPES, pH 7.9, 0,4 mM NaCl, 1 mM EDTA, 1 mM EGTA, 25% Glycerol, 1 mM DTT, 1 mM PMSF, Complete 1 : 100; 15 min, 4 °C shaker). After centrifugation (12 000 r.p.m., 4 °C, 5 min) the supernatant was taken as nuclei fraction.
Quantitative real-time PCR
Cells were collected and RNA was isolated using RNeasy Mini Kit (74106, Qiagen, Hilden, Germany) according to the manufacturers protocol. Reverse transcription was conducted using High Capacity cDNA Reverse Transcription Kit (4368814, Life technologies). Bim primers were purchased from Life technologies (4453320). E-cadherin, N-cadherin and vimentin primers were purchased from Metabion international AG. (E-Cadherin fw 5′ CAG CAC GTA CAC AGC CCT AA 3′, rv 5′ AAG ATA CCG GGG GAC ACT CA 3′; N-Cadherin fw 5′ ACA GTG GCC ACC TAC AAA GG 3′, rv 5′ CCG AGA TGG GGT TGA TAA TG 3′; Vimentin fw 5′ CGG CGG GAC AGC AGG 3′, rv 5′ TCG TTG GTT AGC TGG TCC AC 3′). GAPDH was used as control.
CD44 staining and FACS analysis
For CD44 surface protein quantification, a FITC labelled CD44 antibody was used (560977, BD Pharmingen). CD44 staining was carried out according to the manufacturers protocol. FITC Mouse IgG2b κ antibody was used as isotype control (555742, BD Pharmingen). Surface expression was measured by a flow cytometer (FACS Canto, BD Pharmingen). For gating and population analysis FlowJo 7.6 software (Tree Star Unc.) was used.
Tumour xenograft model
Mouse experiments were performed with approval by the District Government of Upper Bavaria in accordance with the German animal welfare and institutional guidelines. T24 cells stably transfected with non-targeting shRNA and Cdk5 shRNA (1 × 105 cells in 100 μl PBS) were subcutaneously injected into the flanks of 6-week-old female Balb/c nude mice (Harlan Envigo). Five mice per group were used. The time of tumour establishment and the number of established tumours was determined.
Immunohistochemistry
For staining of tumour sections, tumours were removed, fixed with PFA 4% for 24 h, left in PFA 1%, embedded into paraffin and 10 μm sections were prepared. The slides were deparaffinized in xylene (15 min) and rehydrated through descending concentrations of ethanol (20 min in 100% and 20 min in 95%). For antigen retrieval sections were boiled in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 20 min. Endogenous peroxidase was blocked by incubation in 7.5% hydrogen peroxide for 10 min. For staining of the human tissue microarray (TMA), anti-Cdk5 antibody (Thermo Fisher Scientific, AHZ0492), diluted 1 : 100 in PBS, was applied as primary antibodies for 1 h at room temperature. For antibody detection Vectastain Universal Elite ABC Kit (Vector Laboratories, Burlingame, CA, USA) was taken according to the manual and AEC (Vector Laboratories) was used as a chromogen. Slides were counterstained with hematoxylin for 1 min and mounted using FluorSave Reagent mounting medium (Merck, Darmstadt, Germany). Images were obtained with an Olympus BX41 microscope and an Olympus DP25 camera (Olympus, Tokyo, Japan). Cdk5 staining of the TMA section was assessed using the (Remmele and Stegner, 1987) immunoreactive score as described 0 absent; 1–4 weak; 5–8 moderate; 9–12 strong expression.
Statistical analysis
All experiments were performed at least three times unless otherwise indicated in the figure legend. Data are expressed as mean±s.e.m. Statistical analysis was performed with GraphPad Prism (version 5.04, GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was assumed if P⩽0.05.
Results
Expression of Cdk5 in human cancer tissue
Analysis of Cdk5 expression revealed increased levels of Cdk5 in the cancer cell lines MCF7 and T24, and MDA-MB-231 in comparison to the non-cancerous epithelial cell line MCF10A (Figure 1A). This was in line with the expression of Cdk5 in human cancer tissue: staining of a human TMA from 198 female breast cancer patients revealed that Cdk5 expression seemed to be stronger in cancer tissue compared to healthy breast tissue. The staining was evaluated according to the IRS score showing that human tumour tissues expressed Cdk5 in 22.5% at low IRS score, in 46.1% at medium IRS score and in 31.4% at high-IRS score, whereas healthy tissues expressed Cdk5 only at low (28.6%) or median (71.4%) intensity (Figure 1B). A strong staining-intensity was not detected in healthy breast tissue. Moreover, Cdk5 expression and kinase activity (Histone H1 phosphorylation) were increased in cells with an EMT-induced mesenchymal phenotype in comparison to epithelial cells (Figure 1C and D). This set of data points to a role of Cdk5 in mesenchymal tumour cells.

Expression of Cdk5 in human cell lines and cancer tissue.(A) The immunoblots show levels of Cdk5 in breast epithelial cells MCF10A, and cancer cell lines MDA-MB-231, MCF7 and T24. Actin indicates equal loading (n=3). (B) Representative immunostainings from a human breast cancer TMA show Cdk5 expression in healthy mamma tissue and breast cancer tissues. The table displays the evaluation of Cdk5 expression in tumour tissues (n=198) and healthy tissues (controls, n=7) according to the IRS score (intensity × positivity). (C) Immunoblots show Cdk5 expression in HMLE cells that have (+) or have not (−) undergone EMT. Tubulin indicates equal loading (n=3). (D) Cdk5 activity in HMLE cells that have (+) or have not (−) undergone EMT is shown by phosphorylated histone H1 (p-H1; n=3).
Cdk5 regulates growth and motility of cancer cells
Next, the impact of genetic knockdown of Cdk5 with transient silencing by siRNA and stable lentiviral shRNA transduction as well as pharmacological inhibition of Cdk5 with the small molecule roscovitine on cancer cell functions was analysed. Cdk5 inhibition impaired cancer cell growth as the proliferation of Cdk5 shRNA cells was reduced (Figure 2A). In line, pharmacological inhibition of Cdk5 with roscovitine concentration dependently reduced proliferation and cell viability of various breast and bladder cancer cell lines (Figure 2B and C). Although Cdk5 expression and kinase activity was increased in cells with a mesenchymal phenotype, EC50 of roscovitine was similar in epithelial (MCF10A, MCF7, T24), as well as in mesenchymal (MDA-MB-231, EMT-induced HMLE) cell lines. This might be due to the expression of different Cdks by the cell lines which are inhibited by roscovitine and therefore contribute to its anti-proliferative effect. Moreover, both Cdk5 downregulation and inhibition reduced long-term colony formation (Figure 2D and E).

Cdk5 inhibition impairs cancer cell growth.(A) Proliferation of non-targeting (nt) or Cdk5 shRNA-transduced cells is shown (mean±s.e.m., *P<0.001, n=3). Immunoblots of non-targeting (nt) or Cdk5 shRNA-transduced T24 cells for Cdk5 and β-actin (loading control) proof Cdk5 knockdown. (B) Proliferation of MCF10A, MCF7, T24, MDA-MB-231, epithelial and mesenchymal HMLE cells treated with roscovitine for 72 h at indicated concentrations is shown. EC50 values for the various cell lines are indicated. (C) Viability of MCF10A, MCF7, T24, MDA-MB-231, epithelial and mesenchymal HMLE cells treated with roscovitine for 24 h at indicated concentrations is shown. (D) Colony formation of non-targeting (nt) and Cdk5 shRNA T24 cells after 7 days is shown. Bar graph shows quantification (mean±s.e.m., *P<0.001, n=3). (E) Colony formation of T24 cells treated with roscovitine for 24 h before freshly seeding at low density and cultivation for further 7 days is shown. Bar graph shows quantification (mean±s.e.m., *P<0.001, n=3).
Besides abrogating cancer cell growth, Cdk5 knockdown reduced cancer cell migration and invasion (Figure 3A and B Supplementary Figure 1). A concentration-dependent decrease of migration by Cdk5 inhibition with roscovitine again demonstrates pharmacologic accessibility of Cdk5 (Figure 3C).

Cdk5 inhibition reduces cancer cell motility.(A) FCS-induced migration of non-targeting (nt) or Cdk5 shRNA-transduced T24 cells is shown (mean±s.e.m., *P<0.001, n=3). (B) FCS-induced invasion of T24 cells transduced with non-targeting (nt) or Cdk5 shRNA is shown (mean±s.e.m., *P<0.001, n=3). (C) FCS-induced migration of T24 cells with/without treatment with roscovitine is shown (mean±s.e.m., *P<0.001, n=3).
Cdk5 regulates sphere formation
To analyse a potential function of Cdk5 in cancer stem cell (TIC) formation, we performed tumorsphere assays with cultivation of cells under specific conditions in suspension and with serum starvation which exclusively allows clonal expansion of TICs. In fact, Cdk5 knockdown with shRNA (Figure 4A) as well as inhibition with roscovitine (Figure 4B and C and Supplementary Figure 2) reduced tumorsphere formation. Conversely, Cdk5 overexpression in non-cancerous epithelial cells led to increased sphere formation (Figure 4D). Furthermore, time of establishment and number of established tumours was reduced by Cdk5 knockdown in vivo (Figure 4E). In sum, this set of data suggests a potential contribution of Cdk5 to tumour initiation.

Cdk5 regulates sphere formation and tumour establishment.(A) Tumorsphere formation of non-targeting (nt) and Cdk5 shRNA T24 cells is shown (mean±s.e.m., *P<0.05, n=3). Immunoblots of non-targeting (nt) or Cdk5 shRNA-transduced T24 cells for Cdk5 and β-tubulin (loading control) proof Cdk5 knockdown. (B) Tumorsphere formation after pretreatment of T24 cells with roscovitine for 24 h before resuspension in fresh sphere-formation medium and cultivation for further 10 days in presence of roscovitine is shown (mean±s.e.m., *P<0.001, n=3). (C) Tumorsphere formation after pretreatment of MCF7 cells with roscovitine for 24 h before resuspension in fresh sphere-formation medium and cultivation for further 10 days in presence of roscovitine is shown (mean±s.e.m., *P<0.05, n=3). (D) Sphere formation of non-tumorous MCF10A cells overexpressing empty vector (ev) or Cdk5/p35 (Cdk5) is shown (mean±s.e.m., *P<0.05, n=3). The immunoblot of MCF10A cells overexpressing either empty vector (ev) or Cdk5/p35 (Cdk5) for Cdk5 and β-actin (loading control) proofs Cdk5 overexpression. (E) Cdk5 inhibition impairs tumor establishment in vivo. The tables indicate the time of tumour establishment and the number of established tumours of mice injected with non-targeting (nt) shRNA and Cdk5 shRNA tumour cells (1 × 105 cells).
Cdk5 does not regulate tumorsphere formation by modulating EMT and cell survival
To understand how Cdk5 contributes to tumorsphere formation, we first focused on EMT as Cdk5 was recently described to contribute to TGFβ-induced EMT (Liang et al, 2013) and as our data showed that Cdk5 was activated in cells that have undergone EMT (Figure 1C and D). However, common EMT markers like the epithelial marker E-cadherin and the mesenchymal markers N-cadherin and vimentin were not changed by Cdk5 knockdown (Figure 5A and B). The generation of TICs as well as the EMT process itself are described to be influenced by Notch signalling pathways (Harrison et al, 2010a) and we recently elucidated that Cdk5 interferes with Notch signalling in tumour angiogenesis (Merk et al, 2016). Nevertheless, Cdk5 neither influenced the generation of active Notch1 and Notch4 intracellular domains N1-ICD and N4-ICD nor the Notch downstream target c-Myc that have been associated with TICs (Cho et al, 2010; Harrison et al, 2010b; Lombardo et al, 2012; Figure 5C). Moreover, the expression of CD44, a transmembrane adhesion receptor that directs the migration of mesenchymal stem cells and represents a characteristic surface molecule of TICs (Zoller, 2011) was not changed in Cdk5 knockdown cells (Figure 5D). Finally, keeping in mind that Cdk5 affects cell migration and invasion we further focused on MMPs which were also described to drive cancer progression via the EMT process (Radisky and Radisky, 2010). However, MMP2 and MMP9 were not influenced by Cdk5 (Figure 5E). This set of data suggests that the observed impact of Cdk5 on sphere formation is not due to a regulation of the EMT process. Next, we investigated whether pathways that have been associated with cancer cell survival are influenced by Cdk5. AKT, ERK, Hypoxia-inducible factor HIF1α or Stat3 can contribute to detachment-induced survival (Lin et al, 2007; Courapied et al, 2010; Hsu et al, 2013; Buchheit et al, 2014; Hu et al, 2015b), but were not modulated by Cdk5 knockdown in tumorspheres (Figure 5F). Additionally, the phosphorylation of Retinoblastoma protein (Rb), a cell cycle regulator and target of Cdk5 in neuroendocrine thyroid cancer (Pozo et al, 2013) was not affected by Cdk5 knockdown (Figure 5F). We previously elucidated that Cdk5 regulates DNA damage response in HCC (Ehrlich et al, 2015). However, phosphorylation of histone H2aX which is caused by DNA damage was not modulated by Cdk5 knockdown in T24 cells (Figure 5G).

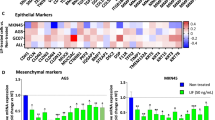
Cdk5 knockdown does neither affect EMT related signalling nor common cell survival pathways or DNA damage.(A) Bar graphs show mRNA levels of the EMT markers E-cadherin, N-cadherin and vimentin in non-targeting (nt) and Cdk5 shRNA-transduced cells (n=3). (B) Immunoblots show protein levels of the EMT markers vimentin, snail, β-catenin, N-cadherin and claudin-1 from non-targeting (nt) and Cdk5 shRNA cells. β-actin and β-tubulin indicate equal loading. (n=3). (C) Immunoblots for Notch1 and Notch4 intracellular domain (N1-ICD, N4-ICD) and the Notch downstream target c-Myc in non-targeting (nt) and Cdk5 knockdown cells are shown. β-actin and β-tubulin indicate equal loading. (n=3). (D) Histogram plot from FACS analysis from CD44 surface expression is shown. Bars represent quantification of CD44-positive cells in non-targeting (nt) and Cdk5 shRNA cells (mean±s.e.m., NS=not significant, n=3). (E) MMP-2 and MMP-9 immunoblots are shown. β-actin and β-tubulin indicate equal loading. (n=3). (F) Immunoblots show different proteins related to cell survival in non-targeting (nt) and Cdk5 shRNA cells. HIF1α, total and phosphorylated Stat3 (S727), ERK1/2, AKT (S473) and Retinoblastoma protein (807/811) are shown. β-actin indicates equal loading (n=3). (G) The immunoblots show phospho-histone H2aX (γH2aX) in non-targeting (nt) and Cdk5 shRNA cells. β-actin indicates equal loading (n=3).
Cdk5 inhibition mediates cell death in tumorspheres by increasing the pro-apoptotic protein Bim
TICs are often resistant to apoptotic cell death by altering proteins of the apoptosis machinery (Keitel et al, 2014). In fact, cell death was increased in tumorspheres from Cdk5 knockdown cells (Figure 6A) or in cells with pharmacologic Cdk5 inhibition (Figure 6B, Supplementary Figure 3). In addition, the pro-apoptotic BH3-only protein Bim that is a known mediator of detachment-induced cell death (Maamer-Azzabi et al, 2013; Buchheit et al, 2015) was increased in Cdk5 knockdown cells (Figure 6C). As the intrinsic apoptosis machinery is activated by Bim translocation to mitochondria (Buchheit et al, 2015), we performed mitochondria fractionation at different time points of cell detachment which revealed that Bim protein was increased in mitochondria from Cdk5 knockdown cells (Figure 6D). By investigating how Cdk5 knockdown affects Bim, we observed an increase in Bim mRNA in Cdk5 knockdown cells (Figure 6E). The transcription of Bim is regulated by forkhead transcription factor type O (Foxo) proteins and Cdk5 has previously been linked with Foxo1 in neurons (Zhou et al, 2015). In fact, we observed that Cdk5 knockdown and inhibition increased Foxo1 (Figure 6F and G) both in the cytoplasm as well as in the nucleus (Figure 6H and I), whereas mRNA transcription was not affected (Supplementary Figure 4). In sum, this set of data suggests that Cdk5 regulates cell death in tumorspheres by mediating Bim transcription via Foxo1.

Cdk5 knockdown induces apoptosis in tumorspheres by increasing the pro-apoptotic protein Bim.(A) Cdk5 knockdown induces apoptosis in tumorspheres. Specific apoptosis in sphere-forming T24 cells is shown (mean±s.e.m., *P<0.01, n=3). (B) Cdk5 inhibition induces apoptosis in tumorspheres. Specific apoptosis of MCF7 tumorspheres after pretreatment of cells with roscovitine for 24 h before resuspension in fresh sphere-formation medium and cultivation for further 10 days in presence of roscovitine is shown (mean±s.e.m., *P<0.05, n=3). (C) The immunoblot shows levels of the pro-apoptotic protein Bim in non-targeting (nt) and Cdk5 shRNA cells. Equal loading is indicated (n=3). (D) Immunoblots show Bim protein in mitochondrial fractions of non-targeting (nt) and Cdk5 shRNA T24 cells at detachment of 3 h and 24 h. The mitochondrial marker COX IV indicates equal loading (n=3). (E) The bar graph displays Bim mRNA levels of non-targeting shRNA (nt) and Cdk5 shRNA cells (mean±s.e.m., *P<0.05, n=3). (F) Immunoblots indicate increased Foxo1 protein levels in Cdk5 knockdown T24 cells. Whole-protein bands indicate equal loading (n=3). (G) Immunoblots indicate increased Foxo1 protein levels in mesenchymal HMLE cells treated with roscovitine (24 h). Whole-protein bands indicate equal loading (n=3). (H) Cdk5 knockdown leads to an increase of Foxo1. Immunoblots show Foxo1 protein of cytosolic and nuclear fractionation of T24 cells. cAMP response element-binding protein (CREB) serves as marker for the nuclei fraction. Whole-protein bands indicate equal loading (n=3). (I) Immunostainings show Foxo1 protein (green) and nucleus (Hoechst33342, blue) of non-targeting (nt) and Cdk5 shRNA T24 cells (n=3).
Discussion
Tumor-initiating cells represent a major problem in cancer therapy as they account for tumour recurrence, establishment of metastasis and resistance towards therapeutics. Thus, recent research strongly focused on investigating TIC properties and mechanisms that are involved in TIC formation and maintenance in order to drive the development of anti-TIC therapies (Pattabiraman and Weinberg, 2014): Signalling pathways which contribute to the self-renewing properties of stem cells like TGFβ, WNT, Notch and Hedgehog are supposed as potential attractive targets to address TICs. In fact, various compounds addressing components of these pathways have shown anti-TIC effects and are currently investigated in clinical trials. In order to identify targeted therapies that address TICs, compound library screens have been carried out and, amongst others, identified the compound salinomycine as anti-TIC drug. Moreover, anti-TIC approaches include targeting of the tumour microenvironment which often triggers EMT induction and tumour initiation. Differentiation therapy by all-trans retinoic acid promotes the differentiation of promyelocytic leukaemic cells into mature granulocytes and is successfully used in therapy of promyelocytic leukaemia. Likewise, also other forms of cancers associated with TICs might be effectively treated by differentiation inducing therapeutics. In addition, immunotherapy and the targeting of TIC metabolism were supposed as promising potential anti-TIC strategies. All in all, there are some promising drugs and strategies, but there is still a craving need for more candidates to address TICs.
In fact, here, we elucidate Cdk5 as a novel player in TICs which might point to Cdk5 as a potential target to address TICs. Cdk5 can be addressed pharmacologically and Cdk-inhibiting small molecules are currently investigated as potential anti-cancer drugs (Liebl et al, 2011b; Weitensteiner et al, 2013; Ehrlich et al, 2015). Two Cdk inhibitors are currently evaluated in clinical trials: Roscovitine represents a well-established inhibitor of Cdks that has been developed in the late 1990 (Meijer et al, 1997). It has shown modest anti-cancer activity in xenograft tumour models in vivo and has been tested in a number of Phase I and II clinical trials where it has shown some anti-cancer activity in around half of the patients (Khalil et al, 2015). Dinaciclib, a newer Cdk inhibitor, has demonstrated significant clinical activity in patients with lymphocytic leukaemia and multiple myeloma (Flynn et al, 2015; Kumar et al, 2015). Moreover, dinaciclib in combination with an AKT-inhibitor showed therapeutic efficiency in patient-derived human pancreatic cancer xenograft models and will be followed by clinical trial evaluation (Hu et al, 2015a). These results are very promising, however, in contrast, a phase I trial with patients suffering from triple-negative breast cancer has demonstrated severe toxic effects and failure of treatment response of a combination treatment of dinaciclib and epirubicin (Mitri et al, 2015). Thus, further trials are required to evaluate the potential of dinaciclib as anti-cancer agents.
In order to investigate the underlying mechanism of Cdk5 in TICs, we first focused on EMT as recent studies demonstrated an involvement of Cdk5 in EMT (Liang et al, 2013; Ren et al, 2015; Sun et al, 2015). Moreover, the forkhead transcription factor Foxc2 was identified as a critical regulator of EMT and TICs in breast cancer (Hollier et al, 2013) and we recently elucidated a relationship between Cdk5 and Foxc2 in the lymphatic endothelium (Liebl et al, 2015). In line, our results revealed that Cdk5 expression was increased in cells that have undergone EMT and in human cancer tissues. Nevertheless, Cdk5 did not regulate tumorsphere formation by EMT, suggesting a specific function of Cdk5 in TICs. Recently, Cdk5 was shown to contribute to the initiation of small-cell lung cancer: overexpression of the NOTCH target ASCL1-induced activation of Cdk5 that phosphorylated and inactivated Rb1 (Meder et al, 2016). In line, aberrant Cdk5 activity was shown to promote tumorigenesis of medullary thyroid cancer by phosphorylation of the retinoblastoma protein (Rb1; Pozo et al, 2013). Nevertheless, Cdk5 did not modulate Notch or Rb1 in Cdk5 knockdown cells. In fact, our work proposed a role of Cdk5 in cell death of tumorspheres by regulating the pro-apoptotic protein Bim. This is in line with previous studies showing that pro-apoptotic proteins like Bim were diminished in cells that have undergone EMT which contributed to apoptosis resistance of TICs (Keitel et al, 2014). As Bim suppressed the survival of disseminated tumour cells (Merino et al, 2015) and induced apoptosis in leukaemia stem/progenitor cells (Pan et al, 2015), induction of Bim might represent a potential anti-TIC strategy. As mechanism of Cdk5 to control Bim, we found that Cdk5 knockdown increased Bim at the transcriptional level by increasing the Forkhead box Type O transcription factor 1 (Foxo1). This is in line with findings in neurons, as neuronal Cdk5 regulates Foxo1 by phosphorylation at its S249 site, favoring its nuclear export and inhibiting its transcriptional activity (Zhou et al, 2015). Foxos play a pivotal role in tumour suppression and are regulated by PI3K/AKT; nuclear Foxos induce the expression of pro-apoptotic genes such as Bim, leading to cell death, whereas AKT-mediated phosphorylation induces cytoplasmic translocation and inhibits Foxo target gene transcription (Calnan and Brunet, 2008). In line with our study, the Akt/Foxo3/Bim pathway has been previously shown to be associated with cancer stem cell survival (Gargini et al, 2015).
In summary, our results demonstrate a Cdk5-Foxo1-Bim pathway in cell death in tumorspheres. As Cdk5 is pharmacologically accessible, it is suggested as a potential target to address TICs.
Change history
28 March 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Buchheit CL, Angarola BL, Steiner A, Weigel KJ, Schafer ZT (2015) Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ 22 (8): 1275–1286.
Buchheit CL, Weigel KJ, Schafer ZT (2014) Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer 14 (9): 632–641.
Calnan DR, Brunet A (2008) The FoxO code. Oncogene 27 (16): 2276–2288.
Cho KB, Cho MK, Lee WY, Kang KW (2010) Overexpression of c-myc induces epithelial mesenchymal transition in mammary epithelial cells. Cancer Lett 293 (2): 230–239.
Courapied S, Sellier H, de Carne Trecesson S, Vigneron A, Bernard AC, Gamelin E, Barre B, Coqueret O (2010) The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J Biol Chem 285 (35): 26765–26778.
Dhavan R, Tsai LH (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2 (10): 749–759.
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 17 (10): 1253–1270.
Ehrlich SM, Liebl J, Ardelt MA, Lehr T, De Toni EN, Mayr D, Brandl L, Kirchner T, Zahler S, Gerbes AL, Vollmar AM (2015) Targeting cyclin dependent kinase 5 in hepatocellular carcinoma—a novel therapeutic approach. J Hepatol 63 (1): 102–113.
Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S, Hessler J, Grever MR, Im E, Zhou H, Zhu Y, Zhang D, Small K, Bannerji R, Byrd JC (2015) Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 29 (7): 1524–1529.
Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM (2001) Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 20 (41): 5865–5877.
Gargini R, Cerliani JP, Escoll M, Anton IM, Wandosell F (2015) Cancer stem cell-like phenotype and survival are coordinately regulated by Akt/FoxO/Bim pathway. Stem Cells 33 (3): 646–660.
Harrison H, Farnie G, Brennan KR, Clarke RB (2010a) Breast cancer stem cells: something out of notching? Cancer Res 70 (22): 8973–8976.
Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB (2010b) Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res 70 (2): 709–718.
Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, Herschkowitz JI, Guerra R, Chang JT, Miura N, Rosen JM, Mani SA (2013) FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res 73 (6): 1981–1992.
Hsu FN, Chen MC, Lin KC, Peng YT, Li PC, Lin E, Chiang MC, Hsieh JT, Lin H (2013) Cyclin-dependent kinase 5 modulates STAT3 and androgen receptor activation through phosphorylation of Ser727 on STAT3 in prostate cancer cells. Am J Physiol Endocrinol Metab 305 (8): E975–E986.
Hu C, Dadon T, Chenna V, Yabuuchi S, Bannerji R, Booher R, Strack P, Azad N, Nelkin BD, Maitra A (2015a) Combined inhibition of cyclin-dependent kinases (dinaciclib) and AKT (MK-2206) blocks pancreatic tumor growth and metastases in patient-derived xenograft models. Mol Cancer Ther 14 (7): 1532–1539.
Hu M, Peng S, He Y, Qin M, Cong X, Xing Y, Liu M, Yi Z (2015b) Lycorine is a novel inhibitor of the growth and metastasis of hormone-refractory prostate cancer. Oncotarget 6 (17): 15348–15361.
Keitel U, Scheel A, Thomale J, Halpape R, Kaulfuss S, Scheel C, Dobbelstein M (2014) Bcl-xL mediates therapeutic resistance of a mesenchymal breast cancer cell subpopulation. Oncotarget 5 (23): 11778–11791.
Khalil HS, Mitev V, Vlaykova T, Cavicchi L, Zhelev N (2015) Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. J Biotechnol 202: 40–49.
Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, Fruth B, Roy V, Erlichman C, Stewart AK, Mayo Phase C (2015) Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 125 (3): 443–448.
Liang Q, Li L, Zhang J, Lei Y, Wang L, Liu DX, Feng J, Hou P, Yao R, Zhang Y, Huang B, Lu J (2013) CDK5 is essential for TGF-beta1-induced epithelial-mesenchymal transition and breast cancer progression. Sci Rep 3: 2932.
Liebl J, Furst R, Vollmar AM, Zahler S (2011a) Twice switched at birth: cell cycle-independent roles of the ‘neuron-specific’ cyclin-dependent kinase 5 (Cdk5) in non-neuronal cells. Cell Signal 23 (11): 1698–1707.
Liebl J, Krystof V, Vereb G, Takacs L, Strnad M, Pechan P, Havlicek L, Zatloukal M, Furst R, Vollmar AM, Zahler S (2011b) Anti-angiogenic effects of purine inhibitors of cyclin dependent kinases. Angiogenesis 14 (3): 281–291.
Liebl J, Zhang S, Moser M, Agalarov Y, Demir CS, Hager B, Bibb JA, Adams RH, Kiefer F, Miura N, Petrova TV, Vollmar AM, Zahler S (2015) Cdk5 controls lymphatic vessel development and function by phosphorylation of Foxc2. Nat Commun 6: 7274.
Lin H, Chen MC, Chiu CY, Song YM, Lin SY (2007) Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. J Biol Chem 282 (5): 2776–2784.
Lombardo Y, Filipovic A, Molyneux G, Periyasamy M, Giamas G, Hu Y, Trivedi PS, Wang J, Yague E, Michel L, Coombes RC (2012) Nicastrin regulates breast cancer stem cell properties and tumor growth in vitro and in vivo. Proc Natl Acad Sci USA 109 (41): 16558–16563.
Maamer-Azzabi A, Ndozangue-Touriguine O, Breard J (2013) Metastatic SW620 colon cancer cells are primed for death when detached and can be sensitized to anoikis by the BH3-mimetic ABT-737. Cell Death Dis 4: e801.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133 (4): 704–715.
Meder L, Konig K, Ozretic L, Schultheis AM, Ueckeroth F, Ade CP, Albus K, Boehm D, Rommerscheidt-Fuss U, Florin A, Buhl T, Hartmann W, Wolf J, Merkelbach-Bruse S, Eilers M, Perner S, Heukamp LC, Buettner R (2016) NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int J Cancer 138 (4): 927–938.
Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 243 (1-2): 527–536.
Merino D, Best SA, Asselin-Labat ML, Vaillant F, Pal B, Dickins RA, Anderson RL, Strasser A, Bouillet P, Lindeman GJ, Visvader JE (2015) Pro-apoptotic Bim suppresses breast tumor cell metastasis and is a target gene of SNAI2. Oncogene 34 (30): 3926–3934.
Merk H, Zhang S, Lehr T, Muller C, Ulrich M, Bibb JA, Adams RH, Bracher F, Zahler S, Vollmar AM, Liebl J (2016a) Inhibition of endothelial Cdk5 reduces tumor growth by promoting non-productive angiogenesis. Oncotarget 7 (5): 6088–6104.
Mitri Z, Karakas C, Wei C, Briones B, Simmons H, Ibrahim N, Alvarez R, Murray JL, Keyomarsi K, Moulder S (2015) A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer. Invest New Drugs 33 (4): 890–894.
Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C (1991) A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods 139 (2): 271–279.
Pan R, Ruvolo VR, Wei J, Konopleva M, Reed JC, Pellecchia M, Andreeff M, Ruvolo PP (2015) Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (−)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood 126 (3): 363–372.
Pattabiraman DR, Weinberg RA (2014) Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov 13 (7): 497–512.
Pinto CA, Widodo E, Waltham M, Thompson EW (2013) Breast cancer stem cells and epithelial mesenchymal plasticity—implications for chemoresistance. Cancer Lett 341 (1): 56–62.
Pozo K, Castro-Rivera E, Tan C, Plattner F, Schwach G, Siegl V, Meyer D, Guo A, Gundara J, Mettlach G, Richer E, Guevara JA, Ning L, Gupta A, Hao G, Tsai LH, Sun X, Antich P, Sidhu S, Robinson BG, Chen H, Nwariaku FE, Pfragner R, Richardson JA, Bibb JA (2013) The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 24 (4): 499–511.
Radisky ES, Radisky DC (2010) Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia 15 (2): 201–212.
Remmele W, Stegner HE (1987) Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe 8 (3): 138–140.
Ren Y, Zhou X, Yang JJ, Liu X, Zhao XH, Wang QX, Han L, Song X, Zhu ZY, Tian WP, Zhang L, Mei M, Kang CS (2015) AC1MMYR2 impairs high dose paclitaxel-induced tumor metastasis by targeting miR-21/CDK5 axis. Cancer Lett 362 (2): 174–182.
Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA (2011) Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145 (6): 926–940.
Scheel C, Weinberg RA (2012) Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol 22 (5-6): 396–403.
Schmidt JM, Panzilius E, Bartsch HS, Irmler M, Beckers J, Kari V, Linnemann JR, Dragoi D, Hirschi B, Kloos UJ, Sass S, Theis F, Kahlert S, Johnsen SA, Sotlar K, Scheel CH (2015) Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep 10 (2): 131–139.
Signore M, Ricci-Vitiani L, De Maria R (2013) Targeting apoptosis pathways in cancer stem cells. Cancer Lett 332 (2): 374–382.
Sun SS, Zhou X, Huang YY, Kong LP, Mei M, Guo WY, Zhao MH, Ren Y, Shen Q, Zhang L (2015) Targeting STAT3/miR-21 axis inhibits epithelial-mesenchymal transition via regulating CDK5 in head and neck squamous cell carcinoma. Mol Cancer 14: 213.
Weitensteiner SB, Liebl J, Krystof V, Havlicek L, Gucky T, Strnad M, Furst R, Vollmar AM, Zahler S (2013) Trisubstituted pyrazolopyrimidines as novel angiogenesis inhibitors. PLoS One 8 (1): e54607.
Zhou J, Li H, Li X, Zhang G, Niu Y, Yuan Z, Herrup K, Zhang YW, Bu G, Xu H, Zhang J (2015) The roles of Cdk5-mediated subcellular localization of FOXO1 in neuronal death. J Neurosci 35 (6): 2624–2635.
Zoller M (2011) CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer 11 (4): 254–267.
Acknowledgements
We thank Dr Christina Scheel (Helmholtz Zentrum München, Institute of Stem Cell Research, Neuherberg, Germany) for providing HMLE cells. We thank Kerstin Loske, Rita Socher, Silvia Schnegg and Julia Blenninger for their help with the experiments. The animal facility of the Department of Pharmacy, Ludwig-Maximilians-University Munich is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Mandl, M., Zhang, S., Ulrich, M. et al. Inhibition of Cdk5 induces cell death of tumor-initiating cells. Br J Cancer 116, 912–922 (2017). https://doi.org/10.1038/bjc.2017.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.39
Keywords
This article is cited by
-
CDK5: an oncogene or an anti-oncogene: location location location
Molecular Cancer (2023)
-
Cyclin-dependent kinase inhibitors exert distinct effects on patient-derived 2D and 3D glioblastoma cell culture models
Cell Death Discovery (2021)
-
Cyclin-dependent kinase 5 mediates pleiotrophin-induced endothelial cell migration
Scientific Reports (2018)

{kind=link}
{kind=link}
{kind=link}
{kind=link}