
Abstract
The PI3K/Akt signalling pathway is one of the most frequently altered signalling networks in human cancers and has become an attractive target in anticancer therapy. Several drugs targeting this pathway are currently in different phases of clinical trials. However, accumulating reports suggest that adverse effects such as hyperglycaemia and hyperinsulinaemia accompany treatment with pan-PI3K and pan-Akt inhibitors. Thus, understanding the consequences of the systemic deletion or inhibition of Akt activity in vivo is imperative. Three Akt isoforms may individually affect different cancer cells in culture to varying degrees that could suggest specific targeting of different Akt isoforms for different types of cancer. However, the results obtained in cell culture do not address the consequences of Akt isoform inhibition at the organismal level and consequently fail to predict the feasibility of targeting these isoforms for cancer therapy. This review summarises and discusses the consequences of genetic deletions of Akt isoforms in adult mice and their implications for cancer therapy. Whereas combined Akt1 and Akt2 rapidly induced mortality, hepatic Akt inhibition induced liver injury that promotes hepatocellular carcinoma. These findings may explain some of the side effects exerted by pan-PI3K and pan-Akt inhibitors and suggest that close attention must be paid when targeting all Akt isoforms as a therapeutic intervention.
Similar content being viewed by others

Main
Before addressing the feasibility of targeting PI3K and Akt for cancer therapy, the evolution of this pathway should be understood. The PI3K/Akt signalling and its downstream effectors, the FOXO transcription factors and mTORC1, constitute an evolutionarily conserved pathway that executes the metabolic response to insulin at the organismal and cellular levels (Kandel and Hay, 1999; Bhaskar and Hay, 2007; Robey and Hay, 2009; Hay, 2011a, b). In mammalian cells, the pathway was first implicated in growth factor-mediated cell survival, and it was proposed as target for cancer therapy long before it was identified to be frequently hyperactivated in cancer (Kennedy et al, 1997; Kandel and Hay, 1999). The activation of the PI3K/Akt signalling pathway in cancer cells evolved, at least in part, to employ its conserved function in metabolism to drive cancer cell metabolism and fulfil anabolic demands. As a kinase Akt also affects multiple processes in the cell that contribute to tumourigenesis.
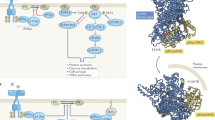
For the past two decades, the PI3K/Akt pathway has been one of the most intensively investigated signalling networks in cancer research. Akt is hyperactivated in cancer cells by multiple mechanisms, including the loss of PTEN, mutations that activate the catalytic subunit of PI3K, p110α, mutations that activate Akt isoforms, the activation of RAS and growth factor receptors and amplification of the genes encoding the catalytic subunit of PI3K and Akt (Figure 1). When PI3K is activated by various signalling events, phosphatidylinositol 3,4,5-trisphosphate (PIP3) is generated from the substrate phosphatidylinositol 3,4-bisphosphate (PIP2). The PIP3 then binds the pleckstrin homology (PH) domain of Akt and translocates Akt to the cell membrane. The recruitment of Akt to the plasma membrane, or other membranes, drives the phosphorylation of Akt at Ser473 within the hydrophobic C-terminal domain by mTORC2, and by the phosphoinositide-dependent kinase 1 (PDK1) at Thr308 within the catalytic domain for full activation (Figure 1). Akt is then able to translocate to other cellular compartments, such as the cytoplasm and nucleus, and phosphorylate various downstream substrates at serine or threonine residues (Manning and Cantley, 2007).

The PI3K/Akt signalling and its activation in cancer. The catalytic subunit of PI3K, p110, is activated by the binding of the regulatory subunit, p85, either to insulin receptor substrate (IRS) proteins downstream of IGF1 or insulin receptor or to the intracellular domain of tyrosine kinase receptors. Activated Ras activates p110 through physical interaction. Lesions that activate the pathway in cancer are indicated (see text for details).
In mammalian cells, the Akt kinase family comprises three members that share a high degree of amino acid identity, Akt1, Akt2 and Akt3, that are encoded by distinct genes. Akt1 is highly expressed in the majority of tissues; Akt2 is the predominant isoform in insulin-sensitive tissues, including the liver, skeletal muscle and adipose tissue; and Akt3 expression is highest in the brain and testes. The phenotypes of individual Akt isoform knockout mice correlate with the relative Akt expression levels in the affected tissues or organs. For instance, Akt2−/− mice exhibit hyperinsulinaemia and insulin resistance (Cho et al, 2001a; Garofalo et al, 2003), consistent with the maximised Akt2 expression in insulin-responsive tissues. Moreover, Akt3−/− mice exhibit smaller brains (Tschopp et al, 2005), consistent with the maximised Akt3 expression observed in the brain. However, the germline deletion of Akt1 did not result in a severe phenotype, and Akt1−/− mice live longer than wild-type mice (Chen et al, 2001, 2006a; Cho et al, 2001b). The phenotypes of combined deletions of different Akt isoforms (reviewed in Hay, 2011a) showed that the deletion of two isoforms could exacerbate the phenotype exerted by the deletion of a single isoform, especially with respect to diabetes. For instance, haploinsufficiency of Akt1 in Akt2−/− mice converts hyperinsulinaemia to hyperglycaemia and hyperactivation of Akt1 in Akt2−/− and in Akt1+/−; Akt2−/− mice decreased hyperinsulinaemia and hyperglycaemia respectively (Chen et al, 2009), suggesting synergetic and overlapping activities of the different Akt isoforms. The diabetic phenotypes of Akt2−/− and Akt1+/−; Akt2−/− mice are due in part to lipodystrophy and leptin deficiency (Chen et al, 2009). These results exemplify the relevance of studies in mice to humans, as it was reported that a family with inherited mutation in Akt2, which exerts dominant negative activity, developed type II diabetes associated with lipodystrophy (George et al, 2004; Tan et al, 2007).
Together, these results suggest that the functions of the different Akt isoforms at the organismal level may not be determined by their different substrate specificities but rather by their relative levels of expression. Indeed, the three Akt isoforms exhibited similar substrate specificity towards a generic substrate in a kinase assay in vitro (Walker et al, 1998). However, multiple reports showed that the different Akt isoforms exhibited specific activities and substrate specificity at the cellular level (Iliopoulos et al, 2009; Toker, 2012) that were, at least in part, attributed to differences in the intracellular localisation of the different isoforms (Gonzalez and McGraw, 2009; Cariaga-Martinez et al, 2013; Toker and Marmiroli, 2014).
Currently, pan-Akt inhibitors are being used in clinical trials for cancer therapy. Specifically, two types of inhibitors, ATP-competitive inhibitors (GSK690693, GDC0068, AZD5363) and an allosteric inhibitor, MK-2206, are being examined. Some clinical trials have shown promising results, either by treatment with pan-Akt inhibitors alone or in combination with other therapeutic regimens, but several toxicities were also observed. In particular, diarrhoea and hyperglycaemia were observed in many clinical trials (Hudis et al, 2013; Yap et al, 2014; Ma et al, 2015; Ramanathan et al, 2015; Jansen et al, 2016; Saura et al, 2017). Notably, results reported in clinical trials also showed a certain percentage of patients with elevated levels of liver enzymes, suggesting liver injury (https://clinicaltrials.gov/ct2/results?term=akt+inhibitor&show_down=Y).
The effect of akt gene deletion on tumourigenesis in mouse models of cancer
Several mouse models have been used to characterise the function of the different Akt isoforms in tumourigenesis. Akt1 deficiency, and even the haploinsufficiency of Akt1, is sufficient to dramatically inhibit the incidence and development of tumours in Pten+/− mice in all tissues tested, including the prostate, endometrium and small intestine (Chen et al, 2006b). In contrast, Akt2 deficiency is not sufficient to significantly inhibit the incidence of tumours in these mice, except in the thyroid, where Akt2 is the predominantly expressed isoform (Xu et al, 2012). The inability of Akt2 deletion to inhibit tumour development in Pten+/− mice was attributed to the high circulating level of insulin as a consequence of Akt2 deletion (Xu et al, 2012) that hyperactivates the other Akt isoforms and possibly other oncogenic signalling pathways and may consequently curb the effect of Akt2 deletion on the tumours. Another group also showed that Akt1, but not Akt2, germline deletion prevents lung tumourigenesis in carcinogen-induced or oncogenic K-Ras mouse models, whereas Akt3 deletion increased tumour incidence in the carcinogen-induced model and tumour size in the genetic model (Hollander et al, 2011). In a different mouse model of lung cancer, Akt1 germline deletion inhibited tumourigenesis, whereas Akt2 deletion increased it (Linnerth-Petrik et al, 2014). In an ErbB2-induced mammary tumourigenesis model, Akt1 deficiency delayed tumour growth and reduced lung metastases (Ju et al, 2007). In a different report in both polyoma middle T (PyMT) and ErbB2-driven mammary adenocarcinoma mouse models, the deletion of Akt1 inhibited, whereas the deletion of Akt2 accelerated, tumour induction (Maroulakou et al, 2007).
The consequences of systemic akt gene deletion in adult mice
The studies described above employed mice with germline deletions of Akt isoforms and can consequently only address the requirement of Akt isoforms for tumour initiation and development. To emulate drug therapy, we began to systemically delete Akt isoforms in adult mice after tumour detection. We showed that the systemic deletion of Akt1 in p53−/− mice after tumour onset regressed thymic lymphoma and substantially increased the lifespan of the mice without adverse physiological consequences (Yu et al, 2015). The effect exerted by systemic Akt1 deletion on p53−/− thymic lymphoma phenocopies the effect of p53 restoration on p53−/− thymic lymphoma (Ventura et al, 2007), and the allosteric pan-Akt inhibitor MK2206 recapitulated this effect of systemic Akt1 deletion. However, MK2206 inhibits Akt1 and Akt2 with similar IC50 values.
The germline deletion of both Akt1 and Akt2 or both Akt1 and Akt3 in mice is neonatal lethal and embryonic lethal, respectively (Peng et al, 2003; Yang et al, 2005). To determine the consequences of their deletion in adult mice, we first systemically deleted Akt1 in either adult Akt2−/− or Akt3−/− mice. Interestingly, unlike the germline deletion, the systemic deletion of Akt1 in Akt3−/− mice was tolerated in adult mice, whereas the systemic deletion of Akt1 in Akt2−/− mice rapidly elicited mortality (Wang et al, 2016). Similar results were obtained after the systemic deletion of both Akt1 and Akt2 or after treating the mice with MK2206 at a dose only double the commonly used dose (Wang et al, 2016). Mortality was preceded by an increase in circulating glucose and insulin levels, followed by a decrease in glucose to a hypoglycaemic level. The mice lost body weight and body fat, the intestinal villi in the mice were disrupted and crypt cell proliferation was diminished. The intestinal damage observed in mice may explain the high incidence of diarrhoea after treatment with pan-Akt inhibitors in clinical trials. We also observed severe inflammation, as measured by the high level of IL-6 in the blood. This high level of inflammation may be due, at least in part, to infiltrating bacteria resulting from the disrupted intestinal barrier. We speculate that the mice could not absorb food because of the disrupted villi and consequently consumed body fat instead until exhausted, leading to hypoglycaemia and death. It should be noted that systemic deletion of the individual Akt isoforms did not elicit the intestinal phenotype. However, the ability of specifically deleting both Akt1 and Akt2 in the crypt cells to cause the same phenotype remains to be seen. Although these experiments were conducted in mice, their results raise concerns regarding the potential toxicity associated with the use of pan-Akt or pan-PI3K in clinical trials at doses that markedly ablate total Akt activity.
Although the systemic deletion of Akt1 and Akt2 is not tolerated in adult mice, the hepatic deletion of Akt1 in Akt2−/− mice is tolerated. However, unexpectedly, these mice develop early-onset aggressive hepatocellular carcinoma (HCC) (Figure 2). Adult mice in which hepatic deletion of both Akt1 and Akt2 is induced also develop HCC, but with much longer latency period. The loss of Akt1 and Akt2 in hepatocytes resulted in cell apoptosis and consequently elevated the serum level of liver enzymes, resulting in macrophage infiltration and inflammation, as measured by high levels of IL-6 and TNFα. Then, IL-6 activated STAT3 and induced the proliferation of surviving hepatocytes. In our study, we used Ki67 to evaluate cell proliferation and found that most Ki67-positive cells were located inside the tumour, whereas apoptotic cells were located around the tumour, as detected by caspase-3 staining. Notably, liver injury and inflammation is due to the activation FOXO1 in the absence of Akt activity. Activated FOXO1 upregulates some pro-apoptotic genes, such as Fasl and Bcl2l11 (Bid), that are responsible for cell death. The HCC that developed in the absence of Akt1 and Akt2 exhibited the gene signature of aggressive human HCC. Moreover, the dramatic induction of Igf2BP3, which is strongly associated with advanced tumour stage and has been considered a predictor of poor prognosis among patients with HCC (Jeng et al, 2008), is especially interesting. Although these results seem counterintuitive, it should be noted that obesity and fatty liver, which inhibit hepatic Akt activity, are also risk factors for HCC (Sun and Karin, 2012). Finally, in diethylnitrosamine (DEN)-treated mice, a model of HCC, the incidence of lung metastasis was markedly increased in Akt2−/− but not Akt1−/− mice. Again, this phenomenon could be attributed to the very high level of insulin in Akt2-deficient mice (Wang et al, 2016).

Schematic depicting the stages of HCC development after the ablation of hepatic Akt activity. Deletion of Akt1 and Akt2 in hepatocytes results in cell death, liver damage and inflammation in a FoxO1-dependent manner. Consequently, macrophages (Kupffer cells) are recruited as well as plasma cells that induce inflammatory cytokines such as IL-6. In turn, IL-6 activates STAT3 in the survived hepatocytes and induces proliferation and survival. Proliferating hepatocytes accumulate mutations that eventually results in HCC.
Notably, the hyperactivation of Akt due to the hepatic deletion of PTEN also induces HCC, but with a much longer latency period than that observed in the absence of Akt activity (Horie et al, 2004). Interestingly, it was reported the hepatic PTEN deletion also increased liver injury that is attenuated by hepatic deletion of Akt2 (Galicia et al, 2010). However, it is likely that total hepatic Akt activity was not markedly decreased because PTEN deficiency hyperactivates Akt1 (hepatocytes do not express Akt3) and the mice likely do not have hyperinsulinaemia. Finally, there are other precedents in which the ablation of pro-oncogenic and survival signalling have been shown to accelerate hepatocarcinogenesis in several examples (Feng, 2012).
Concluding remarks
The results obtained in mice suggest the following. First, the complete inhibition of Akt activity in the liver by treatment with pan-PI3K or pan-Akt inhibitors may increase liver injury and inflammation that are prerequisites for liver cancer. Second, these results suggest that treating obese patients or patients who experienced liver damage with pan-PI3K/Akt inhibitors may exacerbate liver damage and inflammation as well as the risk for liver cancer. Third, close attention should be paid to inflammation and liver injury when pan-PI3K/Akt inhibitors are being used, particularly for HCC therapy.
The effects of pan-PI3K/Akt inhibitors may not have been fully manifested in clinical trials because of the influence of these inhibitors on glucose homeostasis. The systemic inhibition by pan-PI3K/Akt inhibitors may induce hyperinsulinaemia and consequently attenuate the efficacy of the inhibitors. However, it cannot be excluded that a certain dose of the pan-inhibitor could be effective without having a marked effect on glucose homeostasis and insulin level. The side effects on glucose homeostasis and insulin levels may be overcome by combining the treatment with a diabetes drug, such as metformin, that may decrease insulin levels following pan-PI3K/Akt inhibition. As metformin has also been considered for cancer therapy (Chae et al, 2016), the combination of metformin and pan-PI3K/Akt inhibitors may be highly beneficial. Alternatively, Akt isoform-specific inhibitors may be employed because early studies that led to the development of MK2206 identified compounds that more selectively inhibit individual Akt isoforms (DeFeo-Jones et al, 2005). The use of isoform-specific inhibitors may be more effective if the inhibitors are tailored to the cancer in which the specific Akt isoform is highly expressed or activated. However, one drawback to the use of isoform-specific inhibitors is a potential compensatory response that may lead to the hyperactivation of other Akt isoforms. The results obtained in mice indicate that Akt2-specific inhibition should be avoided if possible because it is the major cause of hyperinsulinaemia and hyperglycaemia. This effect is also observed in humans – a missense mutation in the Akt2 gene has been implicated in insulin resistance and diabetes that phenocopies Akt2 deletion in mice (George et al, 2004; Tan et al, 2007; Chen et al, 2009). Finally, other approaches that exploit the metabolic consequence of Akt activation could be developed to selectively eradicate cancer cells exhibiting Akt hyperactivation (Nogueira et al, 2008).
References
Bhaskar PT, Hay N (2007) The two TORCs and Akt. Dev Cell 12: 487–502.
Cariaga-Martinez AE, Lopez-Ruiz P, Nombela-Blanco MP, Motino O, Gonzalez-Corpas A, Rodriguez-Ubreva J, Lobo MV, Cortes MA, Colas B (2013) Distinct and specific roles of AKT1 and AKT2 in androgen-sensitive and androgen-independent prostate cancer cells. Cell Signal 25: 1586–1597.
Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, Kaplan J, Kalyan A, Altman JK, Platanias L, Giles F (2016) Repurposing metformin for cancer treatment: current clinical studies. Oncotarget 7: 40767–40780.
Chen ML, Xu PZ, Peng X, Chen WS, Guzman G, Yang X, Di Cristofano A, Pandolfi PP, Hay N (2006a) The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev 20: 1569–1574.
Chen ML, Xu PZ, Peng XD, Chen WS, Guzman G, Yang X, Di Cristofano A, Pandolfi PP, Hay N (2006b) The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev 20: 1569–1574.
Chen WS, Peng XD, Wang Y, Xu PZ, Chen ML, Luo Y, Jeon SM, Coleman K, Haschek WM, Bass J, Philipson LH, Hay N (2009) Leptin deficiency and beta-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol Cell Biol 29: 3151–3162.
Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N (2001) Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev 15: 2203–2208.
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ (2001a) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292: 1728–1731.
Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ (2001b) Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276: 38349–38352.
DeFeo-Jones D, Barnett SF, Fu S, Hancock PJ, Haskell KM, Leander KR, Mcavoy E, Robinson RG, Duggan ME, Lindsley CW, Zhao Z, Huber HE, Jones RE (2005) Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol Cancer Ther 4: 271–279.
Feng GS (2012) Conflicting roles of molecules in hepatocarcinogenesis: paradigm or paradox. Cancer Cell 21: 150–154.
Galicia VA, He L, Dang H, Kanel G, Vendryes C, French BA, Zeng N, Bayan JA, Ding W, Wang KS, French S, Birnbaum MJ, Rountree CB, Stiles BL (2010) Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 139: 2170–2182.
Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, Mcneish JD, Coleman KG (2003) Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest 112: 197–208.
George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, Dunger DB, Barford D, Umpleby AM, Wareham NJ, Davies HA, Schafer AJ, Stoffel M, O'Rahilly S, Barroso I (2004) A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 304: 1325–1328.
Gonzalez E, McGraw TE (2009) Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci USA 106: 7004–7009.
Hay N (2011a) Akt isoforms and glucose homeostasis - the leptin connection. Trends Endocrinol Metab 22: 66–73.
Hay N (2011b) Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta 1813: 1965–1970.
Hollander MC, Maier CR, Hobbs EA, Ashmore AR, Linnoila RI, Dennis PA (2011) Akt1 deletion prevents lung tumorigenesis by mutant K-ras. Oncogene 30: 1812–1821.
Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, Mizuno K, Hasegawa G, Kishimoto H, Iizuka M, Naito M, Enomoto K, Watanabe S, Mak TW, Nakano T (2004) Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest 113: 1774–1783.
Hudis C, Swanton C, Janjigian YY, Lee R, Sutherland S, Lehman R, Chandarlapaty S, Hamilton N, Gajria D, Knowles J, Shah J, Shannon K, Tetteh E, Sullivan DM, Moreno C, Yan L, Han HS (2013) A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res 15: R110.
Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN (2009) MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal 2: ra62.
Jansen VM, Mayer IA, Arteaga CL (2016) Is there a future for AKT inhibitors in the treatment of cancer? Clin Cancer Res 22: 2599–2601.
Jeng YM, Chang CC, Hu FC, Chou HY, Kao HL, Wang TH, Hsu HC (2008) RNA-binding protein insulin-like growth factor II mRNA-binding protein 3 expression promotes tumor invasion and predicts early recurrence and poor prognosis in hepatocellular carcinoma. Hepatology 48: 1118–1127.
Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, Zhou J, Turner J, Lisanti MP, Russell RG, Mueller SC, Ojeifo J, Chen WS, Hay N, Pestell RG (2007) Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci USA 104: 7438–7443.
Kandel ES, Hay N (1999) The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res 253: 210–229.
Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev 11: 701–713.
Linnerth-Petrik NM, Santry LA, Petrik JJ, Wootton SK (2014) Opposing functions of Akt isoforms in lung tumor initiation and progression. PLoS ONE 9: e94595.
Ma BB, Goh BC, Lim WT, Hui EP, Tan EH, Lopes Gde L, Lo KW, Li L, Loong H, Foster NR, Erlichman C, King AD, Kam MK, Leung SF, Chan KC, Chan AT (2015) Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Invest New Drugs 33: 985–991.
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274.
Maroulakou IG, Oemler W, Naber SP, Tsichlis PN (2007) Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res 67: 167–177.
Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N (2008) Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14: 458–470.
Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, Sundararajan D, Chen WS, Crawford SE, Coleman KG, Hay N (2003) Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev 17: 1352–1365.
Ramanathan RK, Mcdonough SL, Kennecke HF, Iqbal S, Baranda JC, Seery TE, Lim HJ, Hezel AF, Vaccaro GM, Blanke CD (2015) Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: A SWOG cooperative group trial (S1005). Cancer 121: 2193–2197.
Robey RB, Hay N (2009) Is Akt the ‘Warburg kinase’?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol 19: 25–31.
Saura C, Roda D, Rosello S, Oliveira M, Macarulla T, Perez-Fidalgo JA, Morales-Barrera R, Sanchis-Garcia JM, Musib L, Budha N, Zhu J, Nannini M, Chan WY, Sanabria Bohorquez SM, Meng RD, Lin K, Yan Y, Patel P, Baselga J, Tabernero J, Cervantes A (2017) A first-in-human phase I study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov 7: 102–113.
Sun B, Karin M (2012) Obesity, inflammation, and liver cancer. J Hepatol 56: 704–713.
Tan K, Kimber WA, Luan J, Soos MA, Semple RK, Wareham NJ, O'Rahilly S, Barroso I (2007) Analysis of genetic variation in Akt2/PKB-beta in severe insulin resistance, lipodystrophy, type 2 diabetes, and related metabolic phenotypes. Diabetes 56: 714–719.
Toker A (2012) Achieving specificity in Akt signaling in cancer. Adv Biol Regul 52: 78–87.
Toker A, Marmiroli S (2014) Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul 55: 28–38.
Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA (2005) Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 132: 2943–2954.
Ventura A, Kirsch DG, Mclaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T (2007) Restoration of p53 function leads to tumour regression in vivo. Nature 445: 661–665.
Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR (1998) Activation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alpha. Biochem J 331 (Pt 1): 299–308.
Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ, Guzman G, Hay N (2016) Spontaneous hepatocellular carcinoma after the combined deletion of Akt isoforms. Cancer Cell 29: 523–535.
Xu PZ, Chen ML, Jeon SM, Peng XD, Hay N (2012) The effect Akt2 deletion on tumor development in Pten(+/−) mice. Oncogene 31: 518–526.
Yang ZZ, Tschopp O, Di-Poi N, Bruder E, Baudry A, Dummler B, Wahli W, Hemmings BA (2005) Dosage-dependent effects of Akt1/protein kinase Balpha (PKBalpha) and Akt3/PKBgamma on thymus, skin, and cardiovascular and nervous system development in mice. Mol Cell Biol 25: 10407–10418.
Yap TA, Yan L, Patnaik A, Tunariu N, Biondo A, Fearen I, Papadopoulos KP, Olmos D, Baird R, Delgado L, Tetteh E, Beckman RA, Lupinacci L, Riisnaes R, Decordova S, Heaton SP, Swales K, Desouza NM, Leach MO, Garrett MD, Sullivan DM, De Bono JS, Tolcher AW (2014) Interrogating two schedules of the AKT inhibitor MK-2206 in patients with advanced solid tumors incorporating novel pharmacodynamic and functional imaging biomarkers. Clin Cancer Res 20: 5672–5685.
Yu WN, Nogueira V, Sobhakumari A, Patra KC, Bhaskar PT, Hay N (2015) Systemic Akt1 deletion after tumor onset in p53(−/−) mice increases lifespan and regresses thymic lymphoma emulating p53 restoration. Cell Rep 12: 610–621.
Acknowledgements
Work in NH laboratory is supported by NIH Grants R01AG016927, R01 CA090764 and R01 CA206167, and by VA merit award BX000733.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Wang, Q., Chen, X. & Hay, N. Akt as a target for cancer therapy: more is not always better (lessons from studies in mice). Br J Cancer 117, 159–163 (2017). https://doi.org/10.1038/bjc.2017.153
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.153
