
Abstract
CK2 is a protein kinase involved in several human diseases (ranging from neurological and cardiovascular diseases to autoimmune disorders, diabetes, and infections, including COVID-19), but its best-known implications are in cancer, where it is considered a pharmacological target. Several CK2 inhibitors are available and clinical trials are underway in different cancer types. Recently, the suitability of CK2 as a broad anticancer target has been questioned by the finding that a newly developed compound, named SGC-CK2-1, which is more selective than any other known CK2 inhibitor, is poorly effective in reducing cell growth in different cancer lines, prompting the conclusion that the anticancer efficacy of CX-4945, the commonly used clinical-grade CK2 inhibitor, is to be attributed to its off-target effects. Here we perform a detailed scrutiny of published studies on CK2 targeting and a more in-depth analysis of the available data on SGC-CK2-1 vs. CX-4945 efficacy, providing a different perspective about the actual reliance of cancer cells on CK2. Collectively taken, our arguments would indicate that the pretended dispensability of CK2 in cancer is far from having been proved and warn against premature conclusions, which could discourage ongoing investigations on a potentially valuable drug target.
Similar content being viewed by others
Introduction
CK2 is a protein kinase implicated in many human diseases [1]. Recently, it has been found relevant also in the viral infection by SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) and its exploitation as a COVID-19 target is under investigation [2, 3]. The best-known functions of CK2, however, are in cancer, where it is frequently overexpressed and currently considered a valuable pharmacological target [4,5,6,7]. Several CK2 inhibitors are available and two of them are in clinical trials [8, 9].
A recent publication [10] reported that a newly developed CK2 inhibitor, SGC-CK2-1, is much more selective than previously employed CK2 inhibitors, but surprisingly is poorly effective in reducing cell growth in different cancer lines. This prompted some investigators to question the suitability of CK2 as a broad anticancer target [11], attributing the efficacy of CX-4945, the commonly used clinical-grade CK2 inhibitor [9], to its off-target effects.
We think that such a conclusion is premature and eventually deceptive, as it unduly nullifies valuable efforts made so far by many researchers, discouraging investigations in a field that instead still appears quite promising. Our point of view is mainly based on three pieces of evidence, as described below: (1) a survey of data in the literature about the effects produced in tumor cells by targeting CK2 with non-pharmacological tools; (2) the abnormally high CK2 levels in many kinds of tumors; and (3) an in-depth analysis of the available data on SGC-CK2-1, which leads to an interpretation different from the one provided in ref. [11].
Non-pharmacological CK2 targeting produces antiproliferative and pro-apoptotic effects in cancer cells
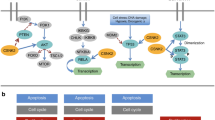
The functional relevance of CK2 in regulating cancer cell proliferation and survival has been suggested not only by observing the effect of different structurally unrelated CK2 inhibitors whose specificity may be questionable, but also by using a variety of non-pharmacological treatments to control kinase expression and, consequently, kinase activity (Fig. 1). Non-pharmacological approaches, ranging from antisense oligonucleotides or small interfering RNA (siRNA) to overexpression of kinase-death mutants, have been applied since the beginning of the investigation on the role of CK2 in cancer. Several independent reports proved that, when the kinase amount was transiently decreased by regulating the expression level of its subunits, cancer cell proliferation and survival were reduced, as well as their migration and invasiveness. Although the effects of downregulating an individual CK2 subunit on cell proliferation/viability are variable, according to the cell type, they have been observed in a wide variety of cancer cells. In glioblastoma cells, siRNAs treatment against both α- and β-subunit of CK2 decreases cell viability [12, 13]; in colorectal cancer cells, the CK2α targeting by siRNA inhibits cell proliferation, promoted cell senescence, and inhibit cell migration and invasion [14]; in breast cancer cells, knockdown of CK2α inhibited cell proliferation, migration, and invasion [15]; in hepatoma cells, knockdown of CK2α inhibits cell proliferation, cell migration, and invasion, and induces apoptosis [16]; in non-small cell lung cancers, CK2α targeting induces apoptosis [17]; siRNA against CK2α in a chronic lymphocytic leukemia cell line resulted in a significant decrease in cell viability [18]; in multiple myeloma cell lines, CK2α targeting induces apoptosis [19]; and in human osteosarcoma cells, knockdown of CK2α or CK2β inhibited the proliferation [20].

The left side shows the major pharmacological targeting strategies, represented by ATP-competitive small molecule inhibitors (as the clinical-grade inhibitor CX-4945, upper) or peptide-based inhibitors (as the clinical-grade compound CIGB-300, bottom). The right side schematically shows the most common non-pharmacological approaches to target CK2: the mRNA downregulation by antisense oligonucleotides or shRNA/siRNA (upper), the expression of kinase-dead mutants (bottom, left), and the gene knockout by CRISPR/Cas9 technology (bottom, right). All strategies reduce the survival and attenuate the oncological features of cancer cells.
Concomitantly, the downregulation of CK2 subunits increases the sensitivity to different chemotherapeutic agents [21,22,23,24,25,26,27,28,29,30,31,32].
To be mentioned, finally, the effects of CK2 downregulation in xenograft cancer models. The in vivo delivery to cancer cells of specific CK2-siRNA targeting both the catalytic subunits by nanocapsules has been found effective in inhibiting tumor growth and inducing cell death in xenograft models of head and neck squamous cell carcinoma [33, 34], prostate cancer [35, 36], and breast cancer [37]. Moreover, in a U87-MG-derived glioblastoma xenograft model, expressing an inducible short hairpin RNA targeting CK2α, a significant reduction in tumor size and growth was observed in parallel with the reduction of CK2α expression [38].
With the advent of the CRISPR/Cas9 tool, it has been possible to produce viable knockout cancer cells for each CK2 catalytic subunit. Even if the production of these cells proved that the single individual catalytic CK2 subunit is not strictly required for cancer cell survival, these cells generally show a reduction in cell proliferation, motility/invasiveness, and an increase in sensitivity against chemotherapeutic agents when compared with wild-type cells [39,40,41,42]. Again, these effects are variably pronounced, according to the cell type and the target subunit gene. Any attempt to produce tumor cells fully devoid of both CK2 catalytic subunits has failed so far, suggesting that a complete absence of CK2 activity is incompatible with cell life.
These data by themselves unambiguously demonstrate the reliance of cancer cells on CK2 for their survival by an approach fully independent of pharmacological treatment, thus circumventing the hypothesis that the effects of CX-4945 are mediated by its nonspecific targets.
Abnormally elevated CK2 in cancer cells: a neglected clue
An aspect of malignancy that has not been duly considered in refs. [10, 11] is the abnormally high CK2 expression/activity found in a wide variety of cancer cells [4, 43,44,45,46], consistent with the view that such a genetic trait is selected by transformed cells, because it maintains/potentiates the tumor phenotype. Although such an argument is per se coincidental in nature, a cause–effect relatedness between elevated CK2 and malignancy is disclosed by observing the pro-apoptotic efficacy of CK2 inhibitors, which is more accentuated in tumor cells as compared with non-tumor ones (see ref. [47]). Such a scenario is hardly explainable by purported off-target effects of CK2 inhibitors unless we assume the implausible theory that these effects are invariably more pronounced in cancer cells.
There is not sufficient evidence of a lower antitumor efficacy of SGC-CK2-1 compared to CX-4945
The claim that CK2 is dispensable in cancer cells [11] is mostly based on the comparison between the effects induced by a very specific inhibitor (SGC-CK2-1) [10] and those of the less selective and commonly used inhibitor, CX-4945 [9]: as “SGC-CK2-1 does not demonstrate significant antiproliferative activity against a panel of 140 different cancer cell lines,” [10] the authors conclude that the antitumor properties of CX-4945 (as reported by hundreds of studies, reviewed in ref. [48]) are mediated by unspecific targets.
However, it is noteworthy that when Wells et al. [10] compared SGC-CK2-1 and CX-4945 for their antiproliferative effects, they found very similar half maximal inhibitory concentration (IC50) values. The direct comparison has been performed only on HCT116 cells, showing IC50 > 10 μM for both compounds, calculated from a ProQuinase proliferation assay. Surprisingly, by a different method, the same work reports 1.9 and 2.2 μM IC50 values for SGC-CK2-1 and CX-4945, respectively. Therefore, there is a discrepancy between the efficacy of SGC-CK2-1 in the same cell line depending on the method, but the same discrepancy is mirrored by CX-4945. From these data, the assignment of the CX-4945 effects to its off-targets appears inconsistent.
Of course, the efficacy of an inhibitor in cells primarily depends on its inhibitory potency in vitro. The in vitro inhibition of CK2 (in terms of Ki or IC50) has never been reported for SGC-CK2-1, but a nanoBRET assays (for the compound binding to CK2) indicates IC50 in the nM range (36 nM for CK2α and 16 nM for CK2α’) [10]. These values are in line with those of CX-4945 (Ki = 0.38 nM, IC50 1 nM) [9], which is, in case, even more potent.
The inhibitory efficacies of SGC-CK2-1 and CX-4945 are similar also towards endogenous CK2 in cells. The concentrations of SGC-CK2-1, which are effective in cells, are in the sub-micromolar range [10]. Also for CX-4945, we found a very strong inhibition in cells at sub-micromolar concentrations [49], at variance with what was reported by Wells et al. [10], who found CX-4945 effects at higher concentrations, probably due to the artificial system employed, with CK2 ectopically overexpressed in cells. Direct experimental comparison on endogenous CK2 in HCT116 cells is not possible, as in ref. [10] a single, very high concentration of CX-4945 was used (10 μM, completely inhibitory).
It can be argued that, given the inhibitory values, a cellular response could have been expected at lower concentrations and this has probably driven the conclusions on the poor antiproliferative effects of SGC-CK2-1 [10, 11]. However, as discussed in ref. [49], this discrepancy in the concentration range required for CK2 inhibition vs. antiproliferative efficacy is also observed for CX-4945 (Ki towards recombinant CK2 of 0.38 nM [9] and antiproliferative effect in the μM range [49]).
In summary, our scrutiny suggests that the cellular efficacy and the antiproliferative effects of SGC-CK2-1 roughly correspond with what is expected in comparison with CX-4945, thus challenging the conclusion that the effects induced by CX-4945 must be ascribed to its unspecific targets.
It should be also noted that very recently, a chemical modification of CX-4945 has given rise to a CK2 inhibitor more specific than CX-4945, but with improved, rather than impaired, antitumor properties [50].
It is also worth noting that, in some cases, the specificity of the cellular effects induced by a chemical inhibitor has been confirmed by transfecting a CK2 mutant insensitive to the inhibitor (because bearing mutations of sites crucial for the compound binding) and showing that this prevented the cell responses to the inhibitor (e.g., see ref. [19]).
We agree that SGC-CK2-1 is a very powerful tool, probably representing the most selective CK2 inhibitor available, which means the first-choice compound for dissecting the CK2 cellular functions. However, further investigation is necessary to disclose its potential effects in cancer. CK2 controls practically all cancer hallmarks [51], whereas the efficacy of SGC-CK2-1 has been evaluated only on proliferation, so far. Especially important, the induction of apoptosis in response to SGC-CK2-1 has not been thoroughly evaluated. Moreover, individual substrates of CK2 have been associated with specific features of cancer cells. Wells et al. [10] evaluated the endogenous CK2 residual activity by Akt phospho-Ser129. Although this site is widely considered a reliable reporter of cellular CK2 activity [52], there are individual substrates that mediate CK2-specific functions and whose phosphorylation state responds to a variable extent to the same inhibitor concentration, due to different reasons. We have already observed and discussed this phenomenon [49, 53]. Therefore, the phosphorylation level of CK2 physiological targets other than Akt Ser129 should be evaluated in response to SGC-CK2-1.
In summary, on one hand, additional tumor features, besides cell proliferation, should be analyzed before drawing conclusions about the antitumor properties of a given CK2 inhibitor. On the other hand, the inhibitor concentrations used for cell treatment are not sufficient to completely abolish CK2 activity and to prevent the phosphorylation of a subset of critical CK2 substrates. Pertinent to this may be the observation that C2C12 myoblasts fully deprived of both CK2 catalytic subunits survive, thanks to the presence of minimal traces of CK2 activity, attributable to a deleted form of the α’-subunit still able to generate a number of phospho-sites unambiguously attributable to CK2 [53]. It is noteworthy that these phospho-sites differentiate from those of wild-type cells for their different susceptibility to two CK2 inhibitors (manuscript in preparation).
Concerning the variable sensitivity of different cell lines to CK2 inhibition, as reported by Wells et al. [10] in the case of SGC-CK2-1, it should be mentioned that the same phenomenon has been observed in several studies also with other CK2 inhibitors (e.g., see ref. [54]) and we can speculate about peculiar features that cancer cells must acquire in order to become more resistant to the reduction of CK2 activity/amount. In general, blood cancer cells seem to display a higher sensitivity to CK2 inhibitors (with the possible speculation, however, about the higher capacity for the compounds to reach intracellular CK2 in cells growing in suspension, compared to adherent cells). It could be speculated that there might be a genetic background, which is shared by all very sensitive (or very resistant) cells. A p53-dependent induction of apoptosis by CK2 inhibition has been already proposed in acute myeloid leukemia [24] and glioblastoma [12, 55], suggesting that p53-null cells are less sensitive to CK2 inhibitors. However, conflicting data have been reported on this point [56, 57]. We suppose that other hidden genetic traits might be shared by cells with similar sensitivity to CK2 targeting. Their identification will be important to plan the most appropriate clinical studies and precision medicine approaches based on CK2 targeting.
Conclusions
To sum up, we strongly believe that the broad cancer essentiality of CK2 is far from having been disproved. Hopefully, newly discovered compounds, such as SGC-CK2-1 [10], the CX-4945 derivative 1c [50], and GO289 [58], will give new impetus to the investigations on this multipurpose anticancer target. In the meantime, premature conclusions about the dispensability of CK2 in cancer should not be considered consistent with the whole array of data available in the literature.
In this connection, it may be worthy to note that the purported dispensability of CK2 in non-cancer cells, grounded on the generation of viable C2C12 clones deprived of both CK2 catalytic subunits [59] has been subsequently challenged by the discovery in these cells of a CK2α’ C-terminally deleted mutant still able, despite its very low activity, to surrogate at least some of the CK2 functions [53]. This observation, in conjunction with the above mentioned, amply documented, abnormally high CK2 expression/activity found in a wide range of cancer cells, provide a strong, albeit indirect, argument (entirely independent of pharmacological implications) supporting the view that reliance on CK2 for survival is a general feature becoming especially stringent in tumor cells, possibly due to the well-known non-oncogene addiction phenomenon [5]. According to this scenario, a moderate decrease of endogenous CK2, still compatible with viability of non-cancer cells, may be lethal to tumor cells if it compromises functions that are essential for malignancy. Such a lethal/antiproliferative outcome, however, will also depend on a number of additional factors, besides the extent of CK2 decrement. One is the background of the tumors under scrutiny, which may rely on different mechanisms for their survival. Another, in the case of pharmacological treatments, is the nature of the CK2 inhibitor, which may affect to variable extents the phospho-proteome generated by CK2 [60]. This means that, coming back to the SGC-CK2-1 vs. CX-4945 issue, a comparative phospho-proteomics analysis with the two inhibitors is urgently needed in order to shed light on the controversial points dealt with in this perspective contribution.
Data availability
Data sharing is not applicable to this article, as no data sets were generated or analyzed during the current study.
References
Borgo C, D’Amore C, Sarno S, Salvi M, Ruzzene M. Protein kinase CK2: a potential therapeutic target for diverse human diseases. Signal Transduct Target Ther. 2021;6:183.
Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, et al. The Global phosphorylation landscape of SARS-CoV-2 infection. Cell 2020;182:685–712.e19.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–68.
Ortega CE, Seidner Y, Dominguez I. Mining CK2 in cancer. PLoS ONE. 2014;9:e115609.
Ruzzene M, Pinna LA. Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta. 2010;1804:499–504.
St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66:1817–29.
Trembley JH, Chen Z, Unger G, Slaton J, Kren BT, Van Waes C, et al. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 2010;36:187–95.
Perea SE, Baladrón I, Valenzuela C, Perera Y. CIGB-300: a peptide-based drug that impairs the protein kinase CK2-mediated phosphorylation. Semin Oncol. 2018;45:58–67.
Pierre F, Chua PC, O’Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J Med Chem. 2011;54:635–54.
Wells CI, Drewry DH, Pickett JE, Tjaden A, Krämer A, Müller S, et al. Development of a potent and selective chemical probe for the pleiotropic kinase CK2. Cell Chem Biol. 2021;28:546–58.
Licciardello MP, Workman P. A new chemical probe challenges the broad cancer essentiality of CK2. Trends Pharmacol Sci. 2021;42:313–5.
Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012;3:e271.
Rowse AL, Gibson SA, Meares GP, Rajbhandari R, Nozell SE, Dees KJ, et al. Protein kinase CK2 is important for the function of glioblastoma brain tumor initiating cells. J. Neurooncol. 2017;132:219–29.
Zou J, Luo H, Zeng Q, Dong Z, Wu D, Liu L. Protein kinase CK2α is overexpressed in colorectal cancer and modulates cell proliferation and invasion via regulating EMT-related genes. J Transl Med. 2011;9:97.
Bae JS, Park S-H, Jamiyandorj U, Kim KM, Noh SJ, Kim JR, et al. CK2α/CSNK2A1 phosphorylates SIRT6 and is involved in the progression of breast carcinoma and predicts shorter survival of diagnosed patients. Am J Pathol. 2016;186:3297–315.
Zhang H-X, Jiang S-S, Zhang X-F, Zhou Z-Q, Pan Q-Z, Chen C-L, et al. Protein kinase CK2α catalytic subunit is overexpressed and serves as an unfavorable prognostic marker in primary hepatocellular carcinoma. Oncotarget 2015;6:34800–17.
So KS, Rho JK, Choi YJ, Kim SY, Choi CM, Chun YJ, et al. AKT/mTOR down-regulation by CX-4945, a CK2 inhibitor, promotes apoptosis in chemorefractory non-small cell lung cancer cells. Anticancer Res. 2015;35:1537–42.
Martins LR, Lúcio P, Silva MC, Anderes KL, Gameiro P, Silva MG, et al. Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood 2010;116:2724–31.
Piazza FA, Ruzzene M, Gurrieri C, Montini B, Bonanni L, Chioetto G, et al. Multiple myeloma cell survival relies on high activity of protein kinase CK2. Blood 2006;108:1698–707.
Takahashi K, Setoguchi T, Tsuru A, Saitoh Y, Nagano S, Ishidou Y, et al. Inhibition of casein kinase 2 prevents growth of human osteosarcoma. Oncol Rep. 2017;37:1141–7.
Jung M, Park KH, Kim HM, Kim TS, Zhang X, Park S-M, et al. Inhibiting casein kinase 2 overcomes paclitaxel resistance in gastric cancer. Gastric Cancer. 2019;22:1153–63.
Trembley JH, Li B, Kren BT, Gravely AA, Caicedo-Granados E, Klein MA, et al. CX-4945 and siRNA-mediated knockdown of CK2 improves cisplatin response in HPV(+) and HPV(−) HNSCC cell lines. Biomedicines 2021;9:571.
Borgo C, Cesaro L, Salizzato V, Ruzzene M, Massimino ML, Pinna LA, et al. Aberrant signalling by protein kinase CK2 in imatinib-resistant chronic myeloid leukaemia cells: biochemical evidence and therapeutic perspectives. Mol Oncol. 2013;7:1103–15.
Quotti Tubi L, Gurrieri C, Brancalion A, Bonaldi L, Bertorelle R, Manni S, et al. Inhibition of protein kinase CK2 with the clinical-grade small ATP-competitive compound CX-4945 or by RNA interference unveils its role in acute myeloid leukemia cell survival, p53-dependent apoptosis and daunorubicin-induced cytotoxicity. J Hematol Oncol. 2013;6:78.
Li Q, Li K, Yang T, Zhang S, Zhou Y, Li Z, et al. Association of protein kinase CK2 inhibition with cellular radiosensitivity of non-small cell lung cancer. Sci Rep. 2017;7:16134.
Yang B, Yao J, Li B, Shao G, Cui Y. Inhibition of protein kinase CK2 sensitizes non-small cell lung cancer cells to cisplatin via upregulation of PML. Mol Cell Biochem. 2017;436:87–97.
Pathak HB, Zhou Y, Sethi G, Hirst J, Schilder RJ, Golemis EA, et al. A synthetic lethality screen using a focused siRNA library to identify sensitizers to dasatinib therapy for the treatment of epithelial ovarian cancer. PLoS ONE. 2015;10:e0144126.
Zhao T, Jia H, Li L, Zhang G, Zhao M, Cheng Q, et al. Inhibition of CK2 enhances UV-triggered apoptotic cell death in lung cancer cell lines. Oncol Rep. 2013;30:377–84.
Luo W, Yu W-D, Ma Y, Chernov M, Trump DL, Johnson CS. Inhibition of protein kinase CK2 reduces Cyp24a1 expression and enhances 1,25-dihydroxyvitamin D(3) antitumor activity in human prostate cancer cells. Cancer Res. 2013;73:2289–97.
Kreutzer JN, Ruzzene M, Guerra B. Enhancing chemosensitivity to gemcitabine via RNA interference targeting the catalytic subunits of protein kinase CK2 in human pancreatic cancer cells. BMC Cancer. 2010;10:440.
Di Maira G, Brustolon F, Bertacchini J, Tosoni K, Marmiroli S, Pinna LA, et al. Pharmacological inhibition of protein kinase CK2 reverts the multidrug resistance phenotype of a CEM cell line characterized by high CK2 level. Oncogene 2007;26:6915–26.
Zhou B, Ritt DA, Morrison DK, Der CJ, Cox AD. Protein kinase CK2α maintains extracellular signal-regulated kinase (ERK) activity in a CK2α kinase-independent manner to promote resistance to inhibitors of RAF and MEK but not ERK in BRAF mutant melanoma. J Biol Chem. 2016;291:17804–15.
Brown MS, Diallo OT, Hu M, Ehsanian R, Yang X, Arun P, et al. CK2 modulation of NF-kappaB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin Cancer Res. 2010;16:2295–307.
Unger GM, Kren BT, Korman VL, Kimbrough TG, Vogel RI, Ondrey FG, et al. Mechanism and efficacy of sub-50-nm tenfibgen nanocapsules for cancer cell-directed delivery of anti-CK2 RNAi to primary and metastatic squamous cell carcinoma. Mol Cancer Ther. 2014;13:2018–29.
Trembley JH, Unger GM, Tobolt DK, Korman VL, Wang G, Ahmad KA, et al. Systemic administration of antisense oligonucleotides simultaneously targeting CK2α and α’ subunits reduces orthotopic xenograft prostate tumors in mice. Mol Cell Biochem. 2011;356:21–35.
Ahmed K, Kren BT, Abedin MJ, Vogel RI, Shaughnessy DP, Nacusi L, et al. CK2 targeted RNAi therapeutic delivered via malignant cell-directed tenfibgen nanocapsule: dose and molecular mechanisms of response in xenograft prostate tumors. Oncotarget 2016;7:61789–805.
Kren BT, Unger GM, Abedin MJ, Vogel RI, Henzler CM, Ahmed K, et al. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference targets for triple negative breast cancer therapy. Breast Cancer Res. 2015;17:19.
Nitta RT, Gholamin S, Feroze AH, Agarwal M, Cheshier SH, Mitra SS, et al. Casein kinase 2α regulates glioblastoma brain tumor-initiating cell growth through the β-catenin pathway. Oncogene 2015;34:3688–99.
Di Maira G, Gentilini A, Pastore M, Caligiuri A, Piombanti B, Raggi C, et al. The protein kinase CK2 contributes to the malignant phenotype of cholangiocarcinoma cells. Oncogenesis 2019;8:61.
Lettieri A, Borgo C, Zanieri L, D’Amore C, Oleari R, Paganoni A, et al. Protein Kinase CK2 Subunits Differentially Perturb the Adhesion and Migration of GN11 Cells: A Model of Immature Migrating Neurons. Int J Mol Sci. 2019. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6928770/.
Zonta F, Borgo C, Quezada Meza CP, Masgras I, Rasola A, Salvi M, et al. Contribution of the CK2 catalytic isoforms α and α’ to the glycolytic phenotype of tumor cells. Cells 2021 ;10:181.
Schmitt BM, Boewe AS, Götz C, Philipp SE, Urbschat S, Oertel J, et al. CK2 activity mediates the aggressive molecular signature of glioblastoma multiforme by inducing nerve/glial antigen (NG)2 expression. Cancers (Basel). 2021;13:1678.
Gapany M, Faust RA, Tawfic S, Davis A, Adams GL, Ahmed K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med. 1995;1:659–66.
Faust RA, Gapany M, Tristani P, Davis A, Adams GL, Ahmed K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett. 1996;101:31–5.
Trembley JH, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell Mol Life Sci. 2009;66:1858–67.
Chua MMJ, Lee M, Dominguez I. Cancer-type dependent expression of CK2 transcripts. PLoS ONE. 2017;12:e0188854.
Cozza G, Girardi C, Ranchio A, Lolli G, Sarno S, Orzeszko A, et al. Cell-permeable dual inhibitors of protein kinases CK2 and PIM-1: structural features and pharmacological potential. Cell Mol Life Sci. 2014;71:3173–85.
D’Amore C, Borgo C, Sarno S, Salvi M. Role of CK2 inhibitor CX-4945 in anti-cancer combination therapy - potential clinical relevance. Cell Oncol (Dordr). 2020;43:1003–16.
Zanin S, Borgo C, Girardi C, O’Brien SE, Miyata Y, Pinna LA, et al. Effects of the CK2 inhibitors CX-4945 and CX-5011 on drug-resistant cells. PLoS ONE. 2012;7:e49193.
Wang Y, Lv Z, Chen F, Wang X, Gou S. Discovery of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine derivatives as highly selective CK2 inhibitors with potent cancer cell stemness inhibition. J Med Chem. 2021;64:5082–98.
Borgo C, Ruzzene M. Role of protein kinase CK2 in antitumor drug resistance. J Exp Clin Cancer Res. 2019;38:287.
Ruzzene M, Di Maira G, Tosoni K, Pinna LA. Assessment of CK2 constitutive activity in cancer cells. Meth Enzymol. 2010;484:495–514.
Borgo C, D’Amore C, Cesaro L, Itami K, Hirota T, Salvi M, et al. A N-terminally deleted form of the CK2α’ catalytic subunit is sufficient to support cell viability. Biochem Biophys Res Commun. 2020;531:409–15.
Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010;70:10288–98.
Kang Y-J, Balter B, Csizmadia E, Haas B, Sharma H, Bronson R, et al. Contribution of classical end-joining to PTEN inactivation in p53-mediated glioblastoma formation and drug-resistant survival. Nat Commun. 2017;8:14013.
Dubois N, Willems M, Nguyen-Khac M-T, Kroonen J, Goffart N, Deprez M, et al. Constitutive activation of casein kinase 2 in glioblastomas: absence of class restriction and broad therapeutic potential. Int J Oncol. 2016;48:2445–52.
Schneider CC, Hessenauer A, Montenarh M, Götz C. p53 is dispensable for the induction of apoptosis after inhibition of protein kinase CK2. Prostate 2010;70:126–34.
Oshima T, Niwa Y, Kuwata K, Srivastava A, Hyoda T, Tsuchiya Y, et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci Adv. 2019;5:eaau9060.
Borgo C, Franchin C, Scalco S, Bosello-Travain V, Donella-Deana A, Arrigoni G, et al. Generation and quantitative proteomics analysis of CK2α/α’(-/-) cells. Sci Rep. 2017;7:42409.
Borgo C, Cesaro L, Hirota T, Kuwata K, D’Amore C, Ruppert T, et al. Comparing the efficacy and selectivity of Ck2 inhibitors. A phosphoproteomics approach. Eur J Med Chem. 2021;214:113217.
Acknowledgements
We thank all the members of our laboratories for their contributions.
Funding
The work performed in our laboratory was supported by grants from Associazione. Italiana per la Ricerca sul Cancro (AIRC), grant IG 18756 to LAP; AFM-Telethon, grant 22974 to MR; Fondazione per la Ricerca sulla Fibrosi Cistica (grants FFC#12/2017 and FFC#11/2019 adopted by Delegazione FFC di Fabriano Ancona con la Delegazione FFC di Umbertide Città di Castello) to MS; and University of Padova, SID Project BORG_BIRD2121_01 to CB.
Author information
Authors and Affiliations
Contributions
MS conceived the study, collected the literature, and drafted the manuscript. CB collected the literature, prepared the figure, and drafted the manuscript. LAP conceived the study, collected the literature, drafted and revised the manuscript. MR conceived the study, collected the literature, drafted and revised the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salvi, M., Borgo, C., Pinna, L.A. et al. Targeting CK2 in cancer: a valuable strategy or a waste of time?. Cell Death Discov. 7, 325 (2021). https://doi.org/10.1038/s41420-021-00717-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-021-00717-4
This article is cited by
-
Hyperphosphorylated PTEN exerts oncogenic properties
Nature Communications (2023)
-
Targeting CK2-mediated phosphorylation of p53R2 sensitizes BRCA-proficient cancer cells to PARP inhibitors
Oncogene (2023)
-
Protein kinase CK2 – diverse roles in cancer cell biology and therapeutic promise
Molecular and Cellular Biochemistry (2023)
