
Abstract
Background:
An everyday clinical practice dilemma in the 20–30% of metastatic colorectal cancer (CRC) patients that have not been operated on their primary tumour, is, under which specific histopathology and molecular circumstances, an endoscopic biopsy could be considered adequate to provide a representative RAS/BRAF molecular status to guide treatment.
Methods:
A consecutive series of 193 paired biopsy and primary CRC tumour samples between August 2008 and 2010 available in the Department of Pathology archives, University Hospitals, KU Leuven were retrieved. For a pair to be included, in the endoscopic biopsy, 20% of invasive adenocarcinoma cells should be present and enough slides to yield an extracted DNA concentration of ⩾5 ng μl−1, and no <2 ng μl−1 should be available for cutting. Exons 2–4 KRAS/NRAS, BRAF, PIK3CA molecular evaluation was performed with RT–PCR and Sequenom.
Results:
From 165 deemed adequate by the pathologist pairs, 85 (51.5%) were concordantly mutated in at least one of the tested genes, 70 (42.5%) were wt and 10 (6%) were discordant, harbouring a mutation in the primary and not in the endoscopic biopsy. In the re-evaluation, when more slides were cut per discordant pair, mutational status changed in two of the six discordantly KRAS-mutated pairs. A strong strength of agreement for both runs was observed (Cohen's kappa, k=0.877, P<0.001 and k=0.901, P<0.001, respectively) between the surgically acquired and the endoscopic biopsy specimens’ evaluation.
Conclusions:
Based on our results, an endoscopic biopsy could provide an accurate mutational profile and become a justified alternative to a surgically removed primary tumour specimen, as long as specific histopathology criteria are met.
Similar content being viewed by others


Main
Molecular diagnostics is a key element in the upfront management of metastatic colorectal cancer (mCRC). The use of the two monoclonal antibodies targeting the epidermal growth factor receptor (anti-EGFR moAbs), cetuximab and panitumumab, is since 2013 restricted to patients whose tumours do not harbour a mutation in exons 2–4 of the KRAS and the NRAS genes (Cunningham et al, 2004; Saltz et al, 2004; Van Cutsem et al, 2007; De Roock et al, 2008, 2010, 2011; Allegra et al, 2009; Loupakis et al, 2009; Janakiraman et al, 2010; Peeters et al, 2013; Vakiani and Solit, 2011). Furthermore, mounting evidence from retrospective studies suggest that the BRAF V600E mutation confers resistant to anti-EGFR moAbs (Di Nicolantonio et al, 2008; Laurent-Puig et al, 2009; Souglakos et al, 2009; De Roock et al, 2010; Saridaki et al, 2010, 2011, 2013). In addition, PIK3CA mutations have been shown to confer resistance to anti-EGFR moAbs and have been shown to co-exist with the KRAS and the BRAF mutations (De Roock et al, 2010).
Besides RAS and BRAF, there is a fast growing list of molecular alterations, such as HER2 and CMET amplifications, which we may soon need to base treatment decisions upon (Cancer Genome Atlas Network, 2012). The correct assessment of all the above-mentioned molecular alterations remains challenging in archival pathology material, as is the quality and amount of DNA extracted, the choice of the analytical method to be used and its sensitivity and specificity (van Krieken et al, 2008, 2013; Bellon et al, 2011; Cree et al, 2014). Of outmost relevance for the clinician is to get for every cancer patient where this is needed, a correct result, from a reliable molecular test, performed in a reliable, quality assured laboratory in a short turnaround time, to determine his or her eligibility for treatment with specific, targeted and often very expensive drugs (van Krieken et al, 2013; Cree et al, 2014).
Approximately 20–30% of mCRC patients have not undergone resection of their primary tumour because of synchronous inoperable metastatic disease. Furthermore, some patients are in no shape to undergo major surgery owing to bad performance status. Nevertheless, in these patients also, the molecular characterisation of their tumour is ‘key’ for their clinical management, and to obtain tissue two options exist; either revert to a biopsy of a metastatic site or get endoscopic biopsies of the primary tumour, the latter being more often available as a usual first step of the diagnostic work-up. In daily clinical routine, we very often have to decide whether this endoscopic material can be used for correct molecular definition of a tumour or invasive procedures need to be planned. In a simplified hypothetical version, endoscopic biopsies could be considered adequate, as long as pathology assessment verifies that they contain adenocarcinoma cells. The question is if key molecular elements, such as mutations in RAS or BRAF, are homogenously distributed in all tumour cells. Regarding primary and metastatic site comparisons, it has been shown that mutations are detected concordantly in most pairs, if large pieces of tumour are used, suggesting tumour homogeneity (Vakiani et al, 2012). Nevertheless, doubt arises towards that, through the recent published work by Misale et al (2012), Gerlinger et al (2012) and Diaz et al (2012) who suggest that clinically relevant minor subclones may exist in the tumour and can be detected, of course with the use of extremely sophisticated and high-throughput techniques, not fitting for massive everyday usage. However, increasing documentation of tumour heterogeneity even for key oncogenic mutations cast doubt on the representativity of a small tumour surface area, as found in endoscopic biopsies, compared with a more comprehensive assessment that can be obtained by using a whole slide from a surgically resected tumour.
Since its incorporation into clinical practice guidelines, KRAS mutational testing has become routine in many centers; however, there is no clear consensus yet as to how such testing should be performed and which analytical method should be used (Cree et al, 2014). Needless to note, that such matters have only just started to be discussed in a more official manner in the international literature for some of the other previously mentioned mutations (van Krieken et al, 2013). KRAS optimal testing procedures, though, are not the only issue; optimal tissue material is as well a matter of debate and discussion. Based on current knowledge, the most appropriate material for KRAS mutation testing is the primary tumour tissue (van Krieken et al, 2008). Van Krieken et al (2008) concluded that this is because usually this type of material contains a sufficient amount of invasive carcinoma cells and is commonly archived, thus, is easily accessible. The central leading role in this procedure towards a specimen’s molecular characterisation is played by the pathologist. Even before the actual molecular testing, the pathologist is the one who is obliged to select the most appropriate tissue block and evaluate the tumour content and density, which preferably should exceed 70% of invasive carcinoma cells. If only an endoscopic biopsy exists, van Krieken et al (2008) conclude that the pathologist should ensure that malignant cells are present in the biopsy and indicate the most appropriate blocks and slides to be used, but no actual guidelines for selection are described (van Krieken et al, 2008).
Endoscopic biopsy, in our case colonoscopy, provides the first source of tissue for the vast majority of CRC cases, and thus, potentially is a very important reservoir of cancerous material suitable for molecular testing. Furthermore, in some cases, for example, the ∼20% of newly diagnosed already metastatic patients, or those with a poor performance status who cannot undergo surgery, or those with symptomatic and life-threatening disease in need of immediate systemic treatment, an endoscopic biopsy could be the sole tissue source.
Having in mind the above-mentioned every day, clinical practice dilemmas and the lack of published documentation and concrete guidelines we sought to address whether the molecular analyses, for a number of ‘hot clinically relevant’ genes and mutations, of specimens from an endoscopic biopsy would yield results similar to those from a surgically removed primary CRC tumour, and thus, could provide us with an acceptable justified alternative.
Materials and methods
Sample and patient selection
A consecutive series of patients with histologically confirmed CRC, diagnosed according to the WHO criteria, who had an endoscopic biopsy and a surgical resection specimen of the primary tumour available in the Department of Pathology between August 2008 and August 2010 were selected from the database of the University Hospital Gasthuisberg, Leuven. This corresponded to a series of 193 selected paired biopsy and primary CRC tumour samples, documented by paraffin-embedded material, which were retrieved from the archives of the Department of Pathology of the University Hospitals of KU Leuven, Leuven, Belgium. For an endoscopic biopsy sample to be retrieved and subsequently evaluated, at least one fragment should have been recorded to be available in the pathology report.
Patients' and tumors' characteristics' variables were obtained from the same database and included where available: age at diagnosis, gender, primary tumour location, TNM status, grade differentiation, mucinous elements representation, obstruction existence, number of fragments determination in each endoscopic biopsy and percentage of invasive tumour cells in the endoscopic biopsy. The project was approved by the ethical committee of the University Hospitals Leuven.
Pathologic review
For 193 pairs, all hematoxylin and eosin (H&E) slides were reviewed by an expert digestive pathologist (XS). For a pair to be included, diagnosis of an invasive adenocarcinoma would have to be confirmed according to the histopathological criteria as defined by the WHO. Furthermore, the bioptic tissue would have no artefact damage, the endoscopic biopsy material would have an adequate percentage (20%) of invasive adenocarcinoma cells in the endoscopic biopsy, and, at least four slides of 5-μm-thick sections would be available from the endoscopic biopsy to fulfill the goal of having an extracted DNA concentration of ideally >5 ng μl−1 and clearly no <2 μl−1.
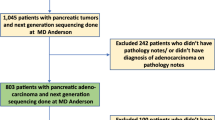
Based on the above-mentioned inclusion criteria, of the 193 paired cases, 28 were omitted from molecular testing (Figure 1). One pair was omitted because of artefact damage of the endoscopic biopsy, one pair because not enough slides (<4) were available for the endoscopic biopsy and 26 because of the presence of a limited amount of invasive cancer cells in the endoscopic biopsy (<20% of cancer cells) that would not give the amount of DNA needed for mutational analysis. The remaining 165 paired specimens were considered adequate by the pathologist, based on the above-mentioned criteria, to be subsequently forwarded to molecular testing (Supplementary Table S1).

Consort diagram of the matched endoscopic biopsy and primary tumour specimen pairs included in the analyses.
After discussions and careful considerations of the usual practice in most Pathology Laboratories (Cree et al, 2014) we decided, that for initial molecular testing, from the endoscopic biopsies' specimens, four 5 μm thickness' slides, and, for surgical resections' specimens, two 5 μm thickness' slides would be used. After the first round of analysis and in case discordances were observed, in those pairs, a re-evaluation by the pathologist would be performed. H&E-stained slides would again be assessed, and for the endoscopic biopsies additional slides would be cut, this time eight of a 5-μm-thick section, whereas, for the primary tumours, additional blocks would be obtained, to exclude heterogeneity within tumour, and, three of a 5-μm-thick section additional slides would be cut, as well.
Mutational analyses
Exon 2–4 KRAS and NRAS mutations and BRAF mutations were considered for our analyses as clinically relevant and influencing treatment decision making (De Roock et al, 2010; Douillard et al, 2013; Peeters et al, 2013; Saridaki et al, 2013). As the role of PIK3CA mutations regarding sensitivity to anti-EGFR moAbs treatment (Jacobs et al, 2009; Jhawer et al, 2008; Laurent-Puig et al, 2009; Ogino et al, 2009; Perrone et al, 2009; Sartore-Bianchi et al, 2009; De Roock et al, 2010; Saridaki et al, 2011) is not clear yet, in our series PIK3CA gene mutation status was purely exploratory.
The primary tumour specimen’s molecular status was set as the gold standard to test the endoscopic biopsies’ performance. In all cases, the pathologist demarcated the tumour area on every H&E slides, avoiding as much as possible, necrotic and haemorrhagic areas, stromal infiltration and extracellular mucous aggregates. The marked areas of the slides were subsequently macrodissected with a scalpel from de-paraffinized sections and molecular testing was performed in the Laboratory of Digestive Oncology, Department of Human Genetics at the University of Leuven. After macrodissection, DNA was extracted according to manufacturer’s protocol from the FFPE tissue slides using the QIAamp DNA FFPE Tissue Kit from Qiagen (Valencia, CA, USA; Cat. No. 56404). DNA concentration was measured using the Quant-iT PicoGreen dsDNA Reagent Kit from Invitrogen (Carlsbad, CA, USA; Cat. No. P11496).
For our study, two techniques were used, the Real Time Taqman PCR and the Sequenom MassARRAY System. Initially, for the first round, samples with a DNA concentration of ⩾5 ng μl−1 were analysed by Sequenom; samples with lower than acceptable for Sequenom analysis concentrations (as low as 2 ng μl−1) were analysed using Real Time Taqman PCR. With Sequenom, the mutational status of KRAS, NRAS, BRAF and PIK3CA genes was analysed (De Roock et al, 2010). With Real Time Taqman PCR specific primers were used to analyse the seven major KRAS mutation KRAS c.34G>T (p.G12C), KRAS c.34G>A (p.G12S), KRAS c.34G>C (p.G12R), KRAS c.35G>A (p.G12D), KRAS c.35G>T (p.G12V), KRAS c.35G>C (p.G12A), KRAS c.38G>T (p.G13D) and the major BRAF mutation BRAF c.1799T>A (p.V600E) (Gulley et al, 2007; Ogino et al, 2007; 2010; De Roock et al, 2010). For the second round of analyses, where only the discordant paired cases were included, both techniques were used for mutational re-evaluation. In this 2nd round, where applicable, no discordance between the two techniques was observed.
Statistical considerations
There was no predetermined sample size estimation in this study because of its observational nature. The primary end point of the study was to evaluate mutational concordance and discordance between tissues obtained from endoscopic biopsy and surgically removed primary tumours. Analysis included contingency tables and quantitative factors that were compared with the use of Continuity correction test or Fisher's exact test. The agreement between categorical variables was measured with the use of Cohen’s Kappa statistical method. All P-values <0.05 were considered statistically significant for all comparisons. Data were analysed using the SPSS program (version 22.0).
Results
KRAS, NRAS and BRAF genes
In the first run, from 165 paired cases, 85 (52%) were found to be concordantly mutated in at least one of all the tested (KRAS, NRAS, BRAF, PIK3CA) genes, 70 (43%) were found to be wt and 10 (6%) were found to be discordant, harbouring a mutation in the primary tumour’s surgical specimen but not in the endoscopic biopsy specimen. Eleven pairs harboured simultaneous mutations in two genes. The specific types of mutations can be seen in Supplementary Table S1.
Considering only the clinically relevant mutations, in the first run, 47 pairs (29%) harboured the same KRAS, 4 (3%) the same NRAS and 24 (15%) the same BRAF mutation in the endoscopic biopsy and the surgically acquired specimens. From the 10 discordantly detected mutations in the first run, 6 were in KRAS and 2 in BRAF (being clinically relevant), whereas the other 2 were in PIK3CA gene.
For the discordant cases a subsequent second run, with more slides cut per pair (three of 5-μm thickness from the primary tumour specimens and eight of 5 μm thickness from the endoscopic biopsies) and with the use of both Real Time Taqman PCR and Sequenom MassARRAY System, was performed. Every specimen was re-examined by the expert pathologist to check again for intratumoral hetereogeneity, and all samples with discordant pairs were found to be composed of a monomorphic pattern. The ‘discordant mutation status’ was changed in two of the six discordantly KRAS-mutated pairs, which were then found to harbour the same KRAS mutation in both biopsy and surgery specimens.
Correlations between concordantly and discordantly mutated pairs
Regarding the concordance of the surgically acquired and the endoscopic biopsy specimens in the first and second runs, Cohen’s kappa was run to determine whether there was agreement between the two methods. According to the result, there is an almost perfect agreement between the two methods in both the first and second run, k=0.877, P<0.001 and 0.901, P<0.001, respectively.
For all pairs, the average number of fragment in the endoscopy was six and the average percentage of adenocarcinoma cells present in endoscopy was 57%. For all concordantly mutated pairs and for all KRAS and BRAF concordant pairs, the average number of fragment was 6.3 and the average percentage of adenocarcinoma cells present in endoscopy was 60% in both cases. For all discordantly mutated pairs, the average number of fragment was 4.5 and the average percentage of adenocarcinoma cells was 53%, whereas for the KRAS and BRAF discordant pairs, the respective numbers were 3.5 and 53%.
Numbers are small in the discordant group, but nevertheless, no statistically significant differences were found between the pairs with concordant status (concordantly mutated and wt) and those with discordant status in relation to median age, gender, primary tumour location, T status, number of fragments and percentage of invasive tumour cells in the endoscopic biopsy, as well as tumour grade. However, a small trend (P=0.071, by continuity correction test), towards more tumours with poor differentiation grade being in the discordant group, was observed (Table 1).
PIK3CA gene
From the 85 concordantly mutated pairs 20 harboured a PIK3CA mutation. In 11 of these cases, the PIK3CA co-existed with a KRAS, an NRAS or a BRAF mutation. From the 10 discordantly mutated pairs, 2 were in the PIK3CA gene and the mutational status was not changed in the second run, where more slides were cut for each specimen (Supplementary Table S1).
Discussion
Almost the sum of published data in the international literature imply that patients with KRAS-, NRAS-, BRAF-mutant CRC tumours derive no or little benefit from treatment with anti-EGFR moAbs. In the era of inhibitors development of RAS and its key downstream effectors (BRAF and MEK inhibitors) it seems logical that a priori screening of mCRC tumours for the above-mentioned mutations would help stratify patients likely to benefit from such treatments (Souglakos et al, 2009; De Roock et al, 2010; Vakiani and Solit, 2011; Douillard et al, 2013; Saridaki et al, 2013). For this step of outmost importance to be realized though, and besides the need of prospectively collected information, additional elements of molecular analysis testing should be addressed.
In the process of the collection of our series of paired endoscopic and primary tumour specimens, the expert pathologists’ role was crucial. As far as primary tumours were concerned, to evaluate tumour content and of course prior to slides cutting, the pathologist selected the most appropriate paraffin block of the surgical specimen by assessing the corresponding H&E-stained sections of the tissue areas. The H&E section of the chosen paraffin block was then subjected to specific evaluation of the fixation characteristics, tumour content and tumour cell viability and was subsequently marked for macrodissection. As per existing guidelines, the tumour density of the tissue area was >70% of invasive carcinoma cells (van Krieken et al, 2008). For the endoscopic specimens, first, the pathologist had to be certain that malignant cells were present in the biopsy material. The pathologist’s opinion regarding adequacy of the biopsy to be submitted to molecular testing was then based on a number of elements, such as fixation characteristics, the quality of the biopsy, the viable tumour volume in the FFPE block (the latter being based on tumour surface on the H&E slide and the percentage of viable invasive carcinoma cells within the marked tumour area on that same H&E slide), as well slides to be cut availability. A minimum of 20% of invasive tumour cells per marked area per H&E slide was considered by the pathologist the lower limit of adequacy.
After the re-evaluation of discordantly mutated cases, where new slides were cut, and instead of four, from the endoscopic biopsy as in the first round, eight were cut in the second, we witnessed a clinically relevant discordant mutation status change in two out of the six KRAS discordant cases (33%) (or out of eight (25%), if we add the BRAF discordant ones). Noting that we used both techniques (sequenom and taqman) for this re-evaluation and the produced results were comparable, the above-mentioned genotyping alteration highlights the importance of the quantity of tumour DNA entering the analysis (more relative for the endoscopic biopsy specimens but applicable for the primary tumour ones, in line with the saying ‘the more the better’) but also of the representation of the more possible areas from the primary tumour in an attempt to minimise tumour heterogeneity. Indeed, in all discordant cases, of note also in the ‘unchanged’ ones, the pathologist concluded that the tumours that he re-evaluated were composed of a monomorphic pattern. This, of course does not mean that heterogeneity and other intratumoral clones could not be present in parts of the tumour that were not biopsied during endoscopy.
In a somewhat ‘older’ publication by Zauber et al (2003), the detection of KRAS gene mutations was assessed between colonic carcinomas and adenomas from the same 42 patients and it was found concordant in only 17 of the paired samples. This finding made the authors discuss that KRAS gene mutations may not necessarily be a consistent pancolonic or lesional feature (Zauber et al, 2003). In a much newer publication, Zauber et al (2013) analysed 134 adenomas, 84 in situ carcinomas and 171 carcinomas and concluded that tubulovillous and villous adenomas, as well as both the benign and malignant parts of in situ carcinomas, are statistically more likely to contain a somatic KRAS gene mutation than colorectal carcinomas (Zauber et al, 2013).
As we were able to show that the concordance of the surgically acquired and the endoscopic biopsy specimens’ evaluation in both runs was in strong agreement (Cohen's kappa, k=0.877, P<0.001 and k=0.901, P<0.001, respectively) and taking into consideration all our other observations regarding the level of detected mutational discordance in our series, we believe that it is safe to conclude that both endoscopic biopsy and primary tumour neoplastic material can be used for mutational analysis, as long as the laboratory performing the analysis is experienced and the standards of expert pathologists regarding the handling of a specimen are met (van Krieken et al, 2008, 2013; Cree et al, 2014). This includes, for the surgically removed primary tumour specimen, the already validated adequate tumour density of the tissue area (existence of >70% of invasive carcinoma cells) and for the endoscopic biopsy specimen, more than the usual quantity of 5-μm-thick section slides (at least eight), cutting deeper into tumour cell populations, and with at least 20% of invasive carcinoma cells present per marked area per slide. A pragmatic suggestion to surpass potential mutational discordances could be to use multiple endoscopic biopsies (with >6 fragments) to increase the chance of detecting a mutation. An approach that would need the endoscopist’s cooperation would be to take 10 biopsies (fragments) and fix them separately into two blocks, one for diagnosis and one for molecular studies (preferably with at least six). Furthermore, pathologists and medical oncologists could have in mind that in poorly differentiated tumours the endoscopy specimens may not be representative of the whole tumours’ mutational status, and if no surgicaly removed primary tumour is available an increased number of slides from the endoscopic biopsy could be needed.
Based on our results, mutational analysis of both endoscopic and primary tumour specimens from the same patient is neither justified, nor required. Probably in the future, liquid biopsies might emerge as a useful, non-invasive alternative to tissue biopsy for the molecular characterisation of a tumour. But for now, an endoscopic biopsy’s tumour material, given the circumstances, could be used as the means of providing an accurate and acceptable mutational tumour profile and become a justified alternative of a primary tumour specimen.
Change history
15 September 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
(2010) Conventional wisdom. Nat Genet 42: 363.
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL (2009) American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 27: 2091–2096.
Bellon E, Ligtenberg MJ, Tejpar S, Cox K, de Hertogh G, de Stricker K, Edsjo A, Gorgoulis V, Hofler G, Jung A, Kotsinas A, Laurent-Puig P, Lopez-Rios F, Hansen TP, Rouleau E, Vandenberghe P, van Krieken JJ, Dequeker E (2011) External quality assessment for KRAS testing is needed: setup of a European program and report of the first joined regional quality assessment rounds. Oncologist 16: 467–478.
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70.
Cree IA, Deans Z, Ligtenberg MJ, Normanno N, Edsjo A, Rouleau E, Sole F, Thunnissen E, Timens W, Schuuring E, Dequeker E, Murray S, Dietel M, Groenen P, Van Krieken JH (2014) Guidance for laboratories performing molecular pathology for cancer patients. J Clin Pathol 67: 923–931.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351: 337–345.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11: 753–762.
De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S (2011) KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 12: 594–603.
De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S (2008) KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol 19: 508–515.
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2008) Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 26: 5705–5712.
Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486: 537–540.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, Blasinska-Morawiec M, Smakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD (2013) Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369: 1023–1034.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892.
Gulley ML, Braziel RM, Halling KC, Hsi ED, Kant JA, Nikiforova MN, Nowak JA, Ogino S, Oliveira A, Polesky HF, Silverman L, Tubbs RR, Van Deerlin VM, Vance GH, Versalovic J (2007) Clinical laboratory reports in molecular pathology. Arch Pathol Lab Med 131: 852–863.
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, Humblet Y, Penault-Llorca F, De Hertogh G, Laurent-Puig P, Van Cutsem E, Tejpar S (2009) Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 27: 5068–5074.
Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, Halilovic E, Wilson M, Huberman K, Ricarte Filho JC, Persaud Y, Levine DA, Fagin JA, Jhanwar SC, Mariadason JM, Lash A, Ladanyi M, Saltz LB, Heguy A, Paty PB, Solit DB (2010) Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res 70: 5901–5911.
Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS, Nasser S, Arango D, Shin J, Klampfer L, Augenlicht LH, Perez-Soler R, Mariadason JM (2008) PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res 68: 1953–1961.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, Bouche O, Reid J, Stone S, Penault-Llorca F (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27: 5924–5930.
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, Floriani I, Bencardino K, Galluccio N, Catalano V, Tonini G, Magnani M, Fontanini G, Basolo F, Falcone A, Graziano F (2009) KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 101: 715–721.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486: 532–536.
Ogino S, Gulley ML, den Dunnen JT, Wilson RB (2007) Standard mutation nomenclature in molecular diagnostics: practical and educational challenges. J Mol Diagn 9: 1–6.
Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, Chan AT, Engelman JA, Kraft P, Cantley LC, Giovannucci EL, Fuchs CS (2009) PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol 27: 1477–1484.
Peeters M, Douillard JY, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J (2013) Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol 31: 759–765.
Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C, Andreola S, Bajetta E, Bertario L, Leo E, Pierotti MA, Pilotti S (2009) PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 20: 84–90.
Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, Lambrechts D, Van Cutsem E, Tejpar S (2009) PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res 15: 3184–3188.
Saltz LB, Meropol NJ, Loehrer PJ Sr., Needle MN, Kopit J, Mayer RJ (2004) Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol 22: 1201–1208.
Saridaki Z, Georgoulias V, Souglakos J (2010) Mechanisms of resistance to anti-EGFR monoclonal antibody treatment in metastatic colorectal cancer. World J Gastroenterol 16: 1177–1187.
Saridaki Z, Tzardi M, Papadaki C, Sfakianaki M, Pega F, Kalikaki A, Tsakalaki E, Trypaki M, Messaritakis I, Stathopoulos E, Mavroudis D, Georgoulias V, Souglakos J (2011) Impact of KRAS, BRAF, PIK3CA mutations, PTEN, AREG, EREG expression and skin rash in ⩾ 2 line cetuximab-based therapy of colorectal cancer patients. PloS One 6: e15980.
Saridaki Z, Tzardi M, Sfakianaki M, Papadaki C, Voutsina A, Kalykaki A, Messaritakis I, Mpananis K, Mavroudis D, Stathopoulos E, Georgoulias V, Souglakos J (2013) BRAFV600E mutation analysis in patients with metastatic colorectal cancer (mCRC) in daily clinical practice: correlations with clinical characteristics, and its impact on patients' outcome. PloS One 8: e84604.
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2009) PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 69: 1851–1857.
Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M, Silver J, Ogino S, Hooshmand S, Kwak E, Freed E, Meyerhardt JA, Saridaki Z, Georgoulias V, Finkelstein D, Fuchs CS, Kulke MH, Shivdasani RA (2009) Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 101: 465–472.
Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, Cercek A, Kemeny N, D'Angelica M, Viale A, Heguy A, Paty P, Chan TA, Saltz LB, Weiser M, Solit DB (2012) Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol 30: 2956–2962.
Vakiani E, Solit DB (2011) KRAS and BRAF: drug targets and predictive biomarkers. J Pathol 223: 219–229.
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25: 1658–1664.
van Krieken JH, Jung A, Kirchner T, Carneiro F, Seruca R, Bosman FT, Quirke P, Flejou JF, Plato Hansen T, de Hertogh G, Jares P, Langner C, Hoefler G, Ligtenberg M, Tiniakos D, Tejpar S, Bevilacqua G, Ensari A (2008) KRAS mutation testing for predicting response to anti-EGFR therapy for colorectal carcinoma: proposal for an European quality assurance program. Virchows Archiv 453: 417–431.
van Krieken JH, Normanno N, Blackhall F, Boone E, Botti G, Carneiro F, Celik I, Ciardiello F, Cree IA, Deans ZC, Edsjo A, Groenen PJ, Kamarainen O, Kreipe HH, Ligtenberg MJ, Marchetti A, Murray S, Opdam FJ, Patterson SD, Patton S, Pinto C, Rouleau E, Schuuring E, Sterck S, Taron M, Tejpar S, Timens W, Thunnissen E, van de Ven PM, Siebers AG, Dequeker E (2013) Guideline on the requirements of external quality assessment programs in molecular pathology. Virchows Archiv 462: 27–37.
Zauber P, Marotta S, Sabbath-Solitare M (2013) KRAS gene mutations are more common in colorectal villous adenomas and in situ carcinomas than in carcinomas. Int J Mol Epidemiol Genetics 4: 1–10.
Zauber P, Sabbath-Solitare M, Marotta SP, Bishop DT (2003) Molecular changes in the Ki-ras and APC genes in primary colorectal carcinoma and synchronous metastases compared with the findings in accompanying adenomas. Mol Pathol 56: 137–140.
Acknowledgements
ZS was a recipient of a research fellowship from the Hellenic Society of Medical Oncology (Hesmo).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Saridaki, Z., Saegart, X., De Vriendt, V. et al. KRAS, NRAS, BRAF mutation comparison of endoscopic and surgically removed primary CRC paired samples: is endoscopy biopsy material adequate for molecular evaluation?. Br J Cancer 113, 914–920 (2015). https://doi.org/10.1038/bjc.2015.307
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.307
Keywords
This article is cited by
-
Treatment with checkpoint inhibitors in a metastatic colorectal cancer patient with molecular and immunohistochemical heterogeneity in MSI/dMMR status
Journal for ImmunoTherapy of Cancer (2019)
