
Abstract
The relevance of KRAS mutation alleles to clinical outcome remains inconclusive in pancreatic adenocarcinoma (PDAC). We conducted a retrospective study of 803 patients with PDAC (42% with metastatic disease) at MD Anderson Cancer Center. Overall survival (OS) analysis demonstrated that KRAS mutation status and subtypes were prognostic (p < 0.001). Relative to patients with KRAS wildtype tumors (median OS 38 months), patients with KRASG12R had a similar OS (median 34 months), while patients with KRASQ61 and KRASG12D mutated tumors had shorter OS (median 20 months [HR: 1.9, 95% CI 1.2–3.0, p = 0.006] and 22 months [HR: 1.7, 95% CI 1.3–2.3, p < 0.001], respectively). There was enrichment of KRASG12D mutation in metastatic tumors (34% vs 24%, OR: 1.7, 95% CI 1.2–2.4, p = 0.001) and enrichment of KRASG12R in well and moderately differentiated tumors (14% vs 9%, OR: 1.7, 95% CI 1.05–2.99, p = 0.04). Similar findings were observed in the external validation cohort (PanCAN’s Know Your Tumor® dataset, n = 408).
Similar content being viewed by others


Introduction
Pancreatic ductal adenocarcinoma (PDAC) is projected to be the second leading cause of cancer death in the US by 2040; with limited available treatment options for metastatic PDAC, the 5-year survival rate is <5%1,2. The median overall survival (OS) for the current standard of care chemotherapy (oxaliplatin, irinotecan, fluorouracil, and leucovorin [FOLFIRINOX]) is 11.1 months in the first-line treatment of metastatic disease, with an objective response rate (ORR) of 31.6% and median progression-free survival (PFS) of 6.4 months3,4. The median OS for the other available first-line chemotherapy regimen, gemcitabine/nab-paclitaxel, is 8.5 months with an ORR of 23% and median PFS of 5.5 months5. In the setting of second-line treatment, the median OS with chemotherapy (liposomal irinotecan, fluorouracil and leucovorin) is only 6.1 months, with an ORR of 16% and median PFS of 3.1 months6. Better therapy for PDAC is urgently needed.
Among the identified genomic alterations (GAs) in PDAC, oncogenic KRAS mutations are the most common, occurring in close to 90% of patients, followed by TP53, CDKN2A, and SMAD47,8. The majority of KRAS mutations are at codon 12, with the highest prevalence of G12D mutation (35%), followed by G12V (20–30%), G12R (10–20%), Q61 (~5%), G12C (1–2%), and other rare muations9,10,11,12. Targeting KRAS has been challenging for decades until allosteric KRASG12C mutant-specific inhibition by covalent binding to the mutant cysteine beneath the switch-II region, which locks it in the inactive GDP bound form was discovered13. Exciting results from clinical trials of the KRASG12C inhibitors sotorasib (AMG510) and adagrasib (MRTX849) have been reported, and both have been approved by the US FDA for previously treated KRASG12C-mutated advanced lung cancer. Moreover, efficacy of both sotorasib and adagrasib against PDAC has also been observed14,15,16,17,18. Sotorasib had a 21% ORR with a median PFS of 4.0 months in patients with pancreatic cancer who had received chemotherapy previously19. Adagrasib monotherapy had an ORR of 33.3% with a median PFS of 5.4 months (95% CI 3.9–8.2) and a median OS of 8.0 months (95% CI 5.2–11.8) in patients with pancreatic cancer refractory to chemotherapy (n = 21)20. Furthermore, preclinical development of a KRASG12D inhibitor (MRTX 1133) has shown promising results and MRTX 1133 is currently in phase 1 clinical trial21. Most recently, pan-KRAS inhibitor RMC-6236, which binds to the chaperone protein cyclophilin A and active GTP-bound RAS (RAS ON inhibitor), is also being tested in patients (NCT05379985). Finally, T cell therapy with KRASG12D-targeting T cell receptors (TCRs) caused tumor regression in a patient with pancreatic cancer, and T cells with TCRs targeting other KRAS mutations, such KRASG12V, are under development22,23.
We are at a breakthrough point in attempts to target KRAS in pancreatic cancer. The remaining challenges include the short duration of response and primary/secondary resistance to KRAS inhibition. Additionally, while multiple genomic and non-genomic factors have been associated with resistance to KRAS inhibitors, such as co-mutations of KEAP1/STK11 with KRAS as observed in patients with lung cancer, KEAP1/STK11 co-mutations are rare in pancreatic cancer, and little is known about the landscape of KRAS mutations and co-mutations in pancreatic cancer or their impact on clinical outcomes12,24,25.
KRAS-mutated cancers are heterogeneous with different mutation allele subtypes and co-mutations26,27,28. Each KRAS mutation allele subtype has unique biochemical and clinicopathological features, and the differences between the mutation subtypes and co-mutations in pancreatic cancer have not been well studied26,27,28,29. The KRASG12D mutation has an intrinsic wildtype and SOS1 guanine exchange activities while the KRASQ61 mutation has deficiencies in GTP hydrolysis27,30. The KRASG12R mutation, which accounts for approximately 15% of the KRAS mutations in pancreatic cancer but less than 1% of the KRAS mutations in lung cancer, was reported to be associated with different downstream signaling pathways relative to other KRAS mutations27. The KRASG12D mutation was reported to be more immune suppressive with shorter survival in lung cancer and pancreatic cancer31,32. Moreover, it has been reported that genes most frequently co-mutated with KRAS vary with the KRAS mutation alleles in patients with lung cancer, and these different patterns of co-mutation with KRAS differentially affect clinical outcomes33. For example, co-mutation of KEAP1/STK11 was more common in patients with KRASG13-mutated lung cancer than in patients with KRASG12D-mutated lung cancer, and co-mutation of KEAP1/STK11 with KRASG13 was associated with poor prognosis and treatment resistance28.
Research to date on the impact of KRAS allele subtypes and co-mutations on PDAC clinical outcomes has been limited, and the conclusions remain controversial. Compared to KRASG12R-mutated PDAC, KRASG12D-mutated PDAC was reported to be associated with worse OS in a single institutional study (n = 126); however, within the KRASG12R-mutated PDAC group, those with PI3K pathway co-mutations experienced worse OS34. Meanwhile, another study found no statistically significant difference in OS between different KRAS mutation alleles12. Our institution has collaborated with the data science firm Syntropy to deploy the Palantir Foundry software platform for extraction and analysis of merged clinical and laboratory data across a variety of platforms, including the Electronic Health Record (EHR), molecular testing/next generation sequencing (NGS), pathology and radiology results, and tumor registry data35,36,37. Together with the development of data science tools such as natural language processing (NLP) and the increased use of NGS in pancreatic cancer, the Foundry platform now gives us the ability to analyze large datasets comprising real-world clinical and molecular information to dissect the heterogeneity of KRAS-mutated pancreatic cancer. In this study, we illustrate the co-mutation landscape of KRAS mutations and the allele-specific associations of KRAS-mutated pancreatic cancer with clinical outcome in our institution. In addition, we validated our findings in an external cohort from the Pancreatic Cancer Action Network (PanCAN)’s Know Your Tumor® (KYT) Dataset38.
Results
Patient characteristics
A total of 803 patients with PDAC who had tumor tissue somatic mutation testing performed at MD Anderson were identified (Fig. 1); the demographic and clinical characteristics of this cohort are summarized in Table 1. The median age was 63 years (range 26–86), 43% were female, and 29.3% had a smoking history (current or former). A total of 336 (42%) patients had documented stage IV disease at the time of their initial diagnosis, and 321 (40%) had poorly differentiated tumors. KRAS gene mutation status was tested in 703 patients, including 302 with stage IV disease; 578 (82%) were positive for mutated KRAS. In addition to KRAS, TP53 was tested in 604 patients, 418 (69%) of whom were positive; CDKN2A was tested in 509 patients, 102 (20%) of whom were positive; and SMAD4 was tested in 536 patients, 68 (13%) of whom were positive. The median follow-up time from the initial diagnosis was 41 months. Median OS of the entire cohort of 803 patients was 19 months (range 0.07–348).

The flowchart shows cohort patient selection. Abbreviations include MD Anderson (MD Anderson Cancer Center).
KRAS mutation status and allele subtype association with OS
Among the 578 patients whose tumors tested positive for a KRAS mutation, 227 had KRASG12D (39%), 182 had KRASG12V (31%), 81 had KRASG12R (14%), 35 had KRASQ61 (6%), and 53 had other uncommon KRAS variants (9%) (Fig. 2d). The Kaplan–Meier (KM) analysis of OS in all 703 patients with known KRAS mutation status (all stages included) demonstrated that KRAS mutation status and subtype was prognostic of OS (p < 0.001) (Fig. 2a); patients with KRAS wildtype tumors had a median OS of 38 months, patients with KRASG12R tumors had a median OS of 34 months (HR: 1, 95% CI 0.71–1.5, p = 0.88), patients with KRASQ61 tumors had a median OS of 20 months (HR: 1.9, 95% CI 1.2–3.0, p = 0.006), and patients with KRASG12D tumors had a median OS of 22 months (HR: 1.7, 95% CI 1.3–2.3, p < 0.001) (Fig. 2c). When limited to patients with stage IV metastatic disease (n = 302), KRAS mutation remained significantly associated with OS (p = 0.034) (Fig. 2b). Again, patients with KRASQ61 and KRASG12D mutations had shorter median OS (15 and 11 months, respectively) relative to those with KRASG12R-mutated and KRAS wildtype tumors (median OS of 25 and 24 months respectively). KRASG12D-mutated tumors (HR = 1.7, 95% CI 1.1–2.6, p = 0.009) were associated with significantly worse OS relative to KRAS wildtype tumors.

a KM OS curves of all patients, and stage IV patients only b with KRAS-mutated PDAC c Univariate analysis of OS with KRAS mutation subtypes and d Frequencies of different KRAS mutations in patients with KRAS-mutant PDAC (n = 578).
KRAS mutation allele subtype association with stage and tumor differentiation
Advanced disease stage was associated with decreased OS (p < 0.001). Patients with stage IV PDAC had median OS of 16 months (HR: 3.3, 95% CI 2.4–4.4) while patients with stage I PDAC had a median OS of 48 months (Fig. 3a). In the full cohort of patients with known KRAS mutation status (all stages included), tumor histopathology was also prognostic of OS (p < 0.001); poorly differentiated/anaplastic tumors had shorter overall survival (median OS = 21 months; HR: 2.3, 95% CI 1.4-3.9) than patients with well-differentiated tumors (median OS = 62 months) (Fig. 3b). We also found a greater prevalence of KRASG12D mutations in patients with metastatic disease (stage IV) than in patients with localized disease (stage I–III) (34% vs. 24%, OR:1.7, 95% CI:1.2–2.4, p = 0.001) (Fig. 3c) and an increased prevalence of KRASG12R mutations in well and moderately differentiated tumors than in poorly differentiated/anaplastic tumors (14% vs. 9%, OR:1.7, 95% CI: 1.05–2.99, p = 0.04) (Fig. 3d).

a KM OS curves for tumor stage of our cohort. b KM OS curves for tumor histopathological grade of our cohort. c Bar plot showing enrichment of KRASG12D mutation in metastatic disease. d Bar plot showing enrichment of KRASG12R in well and moderately differentiated tumors.
KRAS Co-mutations and OS
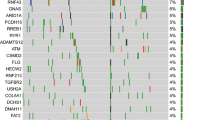
The top detected GAs (Fig. 4a) were sorted by the detected positivity rate among tested patients (the number of tested patients for each gene varied due to different gene panels in the testing platforms). KRAS (82%, N = 578 of 703), TP53 (69%, N = 418 of 608), CDKN2A (20%, N = 102 of 509), SMAD4 (13%, N = 68 of 536), and ARID1A (7%, N = 34 of 482) were the most commonly mutated genes in the MD Anderson cohort (Fig. 4c). TP53 was the most frequently detected co-mutation with KRAS, with a 67% TP53 co-positive rate, followed by CDKN2A (17%), SMAD4 (11%), and ARID1A (6%) (Fig. 4b). In the co-mutation analysis, KRAS was found to be frequently co-mutated with TP53 (OR: 1.77, 95% CI 0.85–3.6, false discovery rate (FDR)-corrected p = 0.29), and CDKN2A (OR: 2.05, 95% CI 0.71–8.13, FDR-corrected p = 0.47). Interestingly, KRAS and GNAS were mutually exclusive (OR: 0.23, 95% CI 0.07–1.05, FDR-corrected p = 0.14) while TP53 and ATM were mutually exclusive (OR: 0.31, 95% CI 0.12–0.81, FDR-corrected p = 0.095). (Fig. 5a). Moreover, TP53 and CDKN2A were frequently co-mutated (OR: 2.11, 95% CI 1.17–4.04, FDR-corrected p = 0.095). Also, ARID1A was found to be significantly co-mutated with CDKN2A (OR: 2.7, 95% CI 1.18–6.02, FDR-corrected p = 0.095) and SMARCA4 (OR: 5.17, 95% CI 1.15–8.44, FDR-corrected p = 0.1).

a Oncoplot showing the distribution of different KRAS mutational subtypes with the different genes in our cohort. b Heatmap showing the co-mutation landscape of the different KRAS mutation subtypes with the different genes and their frequencies. c Bar plot showing the most frequently mutated genes in our cohort.

a Co-mutation analysis of the MD Anderson cohort. Associations between prevalent driver mutations were assessed using Fisher’s exact method and a significant FDR-corrected p indicated by asterixis (*FDR-corrected p < 0.1). b Forest plot showing HR for death (from a univariable analysis) for driver mutations in our cohort, wildtype of each gene was used as reference. c KM OS analysis in patients with metastatic PDAC stratified by molecular subtype.
In univariate Cox proportional hazards analysis for the most commonly mutated genes in the MD Anderson cohort, ARID1A mutation was associated with poor OS, with median OS of 18 months in patients with ARID1A-mutated tumors vs 31 months in patients with ARID1A wildtype tumors (HR: 1.6, 95% CI 0.99–2.6, p = 0.05). However, SMAD4 mutant tumors had better OS than SMAD4 wildtype tumors (median OS 35 and 27 months, respectively, HR: 0.67, 95% CI 0.46–0.99, p = 0.046) (Fig. 5b). Interestingly, while none of the patients with KRASG12R-mutated tumors had ARID1A co-mutation, ARID1A co-mutation was observed in 8% of KRASG12D mutated tumors (p = 0.02). Conversely, SMAD4 co-mutation was observed in 15% of the patients with KRASG12R-mutated tumors compared with 10% in patients with KRASG12D-mutated tumors (p = 0.22) (Fig. 4b).
In patients with metastatic disease and known KRAS, TP53, and CDKN2A mutation status (n = 232), we classified four distinct molecular subtypes of metastatic PDAC (Fig. 5c): (1) KRAS mutant predominant (KRAS mutant, TP53 wildtype/CDKN2A wildtype) (n = 46/232), (2) TP53 mutant predominant (TP53 mutant, KRAS mutant or wildtype/CDKN2A wildtype) (n = 127/232), (3) CDKN2A mutant predominant (CDKN2A mutant, KRAS mutant or wildtype/TP53 mutant or wildtype) (n = 41/232), and (4) triple negative (all KRAS, TP53, and CDKN2A wildtype) (n = 18/232). Patients with triple negative (KRAS-/TP53-/CDKN2A-) tumors had the longest median OS (28 months), while the CDKN2A predominant group had the worst OS (median 12 months); the TP53 predominant group (median 17 months) and KRAS predominant group (median 14 months) had intermediate OS (p = 0.014) (Fig. 5c). PanCAN’s Know Your Tumor® (KYT) Dataset.
To validate our findings, an external cohort from PanCAN’s KYT dataset (n = 408) was analyzed. Baseline characteristics of patients in the KYT cohort are summarized in Table 2. The median age at the time of diagnosis was 65 years (range 36–88); 46% were female and 54% were male. The median follow-up time from diagnosis was 15 months. While disease staging information was not available for the majority of the patients in this cohort (59.8%); among those with known stage, 23.8% (n = 97) had documented stage IV disease at the time of diagnosis. Median overall survival in all the patients was 22 months (range 0.2–93 months). KRAS (92%), TP53 (77%), SMAD4 (24%), CDKN2A (21%), and ARID1A (5%) were the most commonly mutated genes in the PanCAN cohort (Fig. 6a).

a Oncoplot showing the somatic mutation distribution across the KYT cohort. b Heatmap showing the co-mutation landscape of the different KRAS mutation subtypes with the different genes and their frequencies in KYT cohort.
Similar to the MD Anderson cohort, TP53 was the most frequently detected co-mutation with KRAS, (73% positive rate), followed by CDKN2A (20%), SMAD4 (22%), and ARID1A (5%) (Fig. 6b). In the co-mutation analysis (Fig. 7a), KRAS was found to be frequently co-mutated with TP53 (OR: 2.6, FDR-corrected p = 0.18), and CDKN2A (OR: 2.6, FDR-corrected p = 0.84). TP53 and CDKN2A were frequently co-mutated (OR: 3.54, FDR-corrected p = 0.009). TP53 mutation was mutually exclusive with both ATM (OR: 0.04, FDR-corrected p = 9.8E-07) and GNAS (OR: 0.05, FDR-corrected p = 6.65E-05) mutations. KRAS and GNAS mutations were also mutually exclusive (OR: 0.17, FDR-corrected p = 0.18) (Fig. 7a). KRASG12R was associated with significantly longer median OS (32 months) than KRASQ61 (16 months, HR: 2.6, 95% CI 0.88–7.8, p = 0.02) and KRASG12D (23 months, HR: 1.68, 95% CI 1.06–2.65, p = 0.04) (Fig. 7b).

a Co-mutation analysis of KYT cohort, associations between prevalent driver mutations were assessed using the Fisher’s exact method and a significant FDR-corrected p indicated by asterixis (*FDR-corrected p < 0.1). b Bar plot showing the difference in median overall survival between different KRAS mutation subtypes. * indicate p < 0.05 using log-rank test for survival. ^Wildtype: indicate no pathogenic mutations were detected.
Discussion
In this study, we analyzed the impact of KRAS mutation status, KRAS allele subtypes, and co-occurring mutations on clinical outcome of patients with PDAC in two real-world datasets. The study included 803 patients who had been tested for somatic tumor mutations at MD Anderson Cancer Center and an external cohort (n = 408) of patients with pancreatic cancer from the PanCAN KYT® dataset. We found that KRAS mutation status and allele subtypes were associated with OS; median OS was longer in patients with KRAS wildtype and KRASG12R-mutated tumors compared to median OS in patients with KRASG12D or KRASQ61-mutated tumors. We illustrated the co-mutation landscape with KRAS mutation. We also found that ARID1A mutation was associated with worse OS and SMAD4 was associated with better OS. We found TP53 and ATM mutations were mutually exclusive. There was a higher rate of ARID1A mutation in KRASG12D compared with KRASG12R patients. We also found enrichment of KRASG12D in metastatic disease and enrichment of KRASG12R in well to moderately differentiated tumors.
Among the 803 patients with PDAC tested for somatic tumor mutations at MD Anderson, 703 were tested for KRAS mutations (Fig. 1). The overall positive rate for KRAS mutation was 82% (n = 578); the most common mutation was KRASG12D (39%), followed by KRASG12V (31%), KRASG12R (14%), KRASQ61 (6%), and other uncommon KRAS variants (9%) (Fig. 2d). There were differences in OS with KRAS mutation status and allele subtypes in both the overall population (all stages, Fig. 2a) and in the subset of patients with stage IV disease (n = 302) (Fig. 2b). Compared to patients with KRAS wildtype tumors, regardless of disease stages, patients with KRASG12D (median OS 22 months, HR: 1.7, 95% CI 1.3–2.3, p < 0.001) or KRASQ61 (median OS 20 months, HR: 1.9, 95% CI 1.2–3.0, p = 0.006) mutated tumors had worse survival. KRASG12R mutated patients (median OS 34 months, HR: 1, 95% CI 0.71–1.5, p = 0.88) had similar OS as wildtype patients (median OS 38 months, reference) (Fig. 2c). The external cohort from the PanCAN KYT® dataset (n = 408) validated the finding that KRASG12R mutation was associated with the longest median OS (32 months), while KRASQ61 (16 months, HR: 2.6, 95% CI 0.88–7.8, p = 0.02) and KRASG12D mutations (23 months, HR: 1.68, 95% CI 1.06–2.65, p = 0.04) were associated with shorter median OS (Fig. 7b). Our results were consistent with the previous report of significantly longer OS (HR 0.55) in patients with KRASG12R-mutated PDAC (n = 23) compared with those with non-KRASG12R PDAC (n = 88)34. Another study comparing KRASG12C (n = 30) and other KRAS mutations reported longer median OS (starting from the first line therapy, p = 0.03) for KRAS wildtype tumors (n = 91) in patients with metastatic PDAC, which was consistent with our findings of better survival in KRAS wildtype patients12; however, the authors did not show statistically significant difference between other KRAS alleles while compared against KRASG12C patients12. Due to the low frequency of KRASG12C mutation, we grouped the patients with KRASG12C mutations with patients that had other uncommon mutations. In our cohort, OS was defined from initial diagnosis and there was enrichment of KRASG12D mutation in metastatic disease (stage IV) (OR: 1.7, 95% CI 1.2–2.4, p = 0.001) (Fig. 3c). Our data suggested worse outcomes in patients with KRASG12D tumors. This is consistent with a previous study of 356 patients with resected PDAC, which reported that those with KRAS mutations had worse disease-free survival (DFS) (median 12.3 months) and OS (median 20.3 months) compared with those with wildtype KRAS (DFS 16.2 months and OS 38.6 months), and particularly poor outcomes were observed in patients with KRASG12D mutation (median OS 15.3 months)39.
The mechanisms of why KRASG12D is associated with poor prognosis relative to the other subtypes is not fully understood beyond the co-mutation with ARID1A and enrichment in metastatic disease. A more immunosuppressive tumor microenvironment (TME) in KRASG12D lung cancer tumors has been reported28,31. In a KRASG12D mutation driven PDAC mice model, immune suppressive cytokines IL-4 and IL-13 and remodeling of the myeloid cell composition in TME have been demonstrated40,41. In PDAC mouse models treated with the KRASG12D inhibitor MRTX 1133, increased macrophages (CD11b and F4/80+) in the TME and decreased total myeloid cells was observed42. Correlative tissue and blood samples for potential KRAS mutation allele-specific immune features were not included in this project and could be a future research direction in patients with PDAC.
KRASG12R was more common in PDAC (~15%) than in other cancer types12. It has distinct biochemical features from KRASG12D/V including an altered switch-II structure that cannot activate p110α/PI3K directly43. We found the median OS of patients with a KRASG12R mutation was comparable to that in patients with wildtype KRAS and longer than that in patients with KRASG12D or KRASQ61 mutations. There was enrichment of KRASG12R mutation in well and moderately differentiated tumors vs poorly differentiated/anaplastic tumors (OR: 1.7, 95% CI 1.05–2.99, p = 0.04) (Fig. 3d), which suggested less aggressive biology and better outcome for the KRASG12R-mutated tumors. On the other hand, KRASQ61 mutant tumors had a decreased GTP hydrolysis rate with high RAF-dependent MEK phosphorylation, and they did not response to SOS1 inhibition29,44. While KRASQ61 mutants had shorter median OS in our cohort, little is known about the clinical features of this KRAS mutation subtype. To our knowledge, this is the first study to report worse OS with KRASQ61, which could be consistent with its biochemical features. Due to the rarity of KRASQ61 mutations, we grouped different KRASQ61 mutations together, though the association with OS may be mutant-specific45. The clinical and molecular features of KRASG12R- and KRASQ61-mutated PDAC warrant further investigation; additional research in larger populations could help the development of KRAS allele-specific inhibitors such as the KRASG12R inhibitor46.
Co-mutations with KRAS could be one of the contributing factors for the allele specific clinical outcomes in PDAC. KEAP1 co-mutation with KRAS in lung cancer was associated with early progression on the KRASG12C inhibitor sotorasib25. Co-occurrence of other mutations were common in PDAC, and the disease progression model proposed based on observed co-mutation patterns was early KRAS mutation followed by CDKN2A then loss of TP53 and SMAD447,48. Our data were consistent with previous reports that TP53 (67%) was the most common co-mutation with KRAS followed by CDKN2A (17%), SMAD4 (11%), and ARID1A (6%) (Fig. 4b)12. We tested the KRAS/CDKN2A/TP53 disease progression model by classified four distinct molecular subtypes of metastatic patients in patients who had been tested for KRAS, TP53, and CDKN2A mutations (n = 232). We found patients with triple negative (KRAS−/TP53−/CDKN2A−) tumors demonstrated the best OS (median 28 months) while CDKN2A predominant tumors had the worst OS (median OS12 months, p = 0.014) (Fig. 5c). In our study, CDKN2A mutation included any mutation (either missense or deletion of CDKN2A). Germline CDKN2A mutation is associated with an increased risk of melanoma and pancreatic cancer, and somatic CDKN2A loss is common in pancreatic cancer49,50,51. Patients with resected PDAC and somatic CDKN2A loss had worse survival (median DFS 11.5 and OS 19.7) compared to patients with wildtype CDKN2A (median DFS 14.8 and median OS 24.6)39. In another study of 100 patients with PDAC (both metastatic and nonmetastatic included), CDKN2A mutations were also associated with shorter OS (22 months vs 35 months; P = 0.018)52. In KRAS-mutated lung cancer, CDKN2A mutation was associated with worse survival on imunotherapy53. In a mouse model, CDKN2A loss accelerated KRASG12D-driven tumor growth54. A therapeutic approach targeting CDKN2A in KRAS-mutated PDAC is under investigation; however, clinical activity of CDK4/6 inhibitors was not seen in early-phase trials55,56. The location of the methylthioadenosine phosphorylase gene (MTAP) is adjacent to CDKN2A and the majority of PDAC tumors with CDKN2A loss also had MTAP loss57,58,59. The surrogate role of CDKN2A is not clear, and the reported rate of MTAP loss in our cohort was low; the detection method for MTAP loss has not yet validated by comparative genomic hybridization for pancreatic cancer in our NGS testing panel.
Univariate OS analysis in our study did not show statistically significant association of either TP53 or CDKN2A co-mutation with OS, but we did find that ARID1A mutation was associated with poor OS (median 18 vs 31 months, HR: 1.6, 95% CI 0.99–2.6, p = 0.05), and SMAD4 mutation was associated with better OS (median 35 vs 27 months, HR: 0.67, 95% CI 0.46–0.99, p = 0.046) (Fig. 5b). SMAD4 is a tumor suppressor gene, and reported results about the prognostic value of SMAD4 have been inconsistent39,60,61,62. While an association between SMAD4 inactivation in resected PDAC and poor prognosis has been reported, a separately reported meta-analysis did not show association between SMAD4 mutation and OS61,62. Our data showed a 13% SMAD4 mutation rate, and it was associated with better OS. Further studies with larger sample sizes and different populations are needed to reconcile these varying results. ARID1A was found to be significantly co-mutated with CDKN2A (OR: 2.7, 95% CI 1.18–6.02, FDR-corrected p = 0.095), and with SMARCA4 (OR: 5.17, 95% CI 1.15–18.44, FDR-corrected p = 0.1). KRASG12R mutated patients had lower rates of ARID1A co-mutation compared with KRASG12D (0% vs 8% in p = 0.02) (Fig. 4b). Similar findings were also observed in the validation cohort from the PanCAN KYT® dataset. Both ARID1A and SMARCA4 are Switch/Sucrose Nonfermentable (SWI/SNF) chromatin remodeling complex genes that are important for epigenetic reprogramming in PDAC63. Context-specific tumor suppressive or oncogenic functions of SWI/SNF chromatin regulation was noticed in PDAC64,65. In mouse models, disrupted ARID1A promoted the carcinogenesis from KRAS-mutated premalignant intraductal papillary mucinous neoplasms (IPMN) to PDAC44. In KRAS-mutated colon cancer, a similar tumor-supporting role of ARID1A was required for MEK/ERK signaling66. Our results of worse OS with ARID1A mutation support the oncogenic role of ARID1A and the potential benefit of targeting ARID1A in PDAC. ARID1A regulates DNA damage checkpoints and sensitizes cells to DNA damage response (DDR) targeting agents67,68,69. The ATM-TP53 signaling pathway is critical in DDR targeting in pancreatic cancer70. Interestingly, in both of our cohorts, TP53 mutation was mutually exclusive with ATM mutation. Our findings of worse OS with ARID1A mutation and mutual exclusivity of TP53 and ATM mutation in PDAC provided insights on PDAC therapeutic vulnerabilities.
The limitations of this study are heterogeneities in both patient populations and tumor mutation testing methods and gene panels. Only patients who had tissue molecular testing done at MD Anderson were included in this study; patients who had tests performed on other panels were not included. This is a retrospective study in a single tertiary cancer institution with ascertainment bias. The external validation cohort had limited clinical information, and treatment history was not available. Tumor genomic factors may not be the main contributor for KRAS mutation allele specificities. Correlative tissue and blood samples from patients were not available to evaluate other non-genomic factors that may account for the differences in clinical outcome observed.
In summary, we reported the KRAS mutation allele-specific clinical outcomes in patients with PDAC using a single institution retrospective study and an external validation cohort. Our findings suggested that KRAS targeting and combination strategies may warrant mutant allele-specific approaches with consideration of the mutations co-occurring with KRAS. In our analysis of 803 patients with PDAC, we found that KRAS mutation status and mutation allele subtypes were associated with OS. Patients with KRAS wildtype and KRASG12R-mutated tumors survived longer than patients with KRASG12D or KRASQ61-mutated tumors, and this observation was confirmed in an external validation cohort. We also found enrichment of KRASG12D mutations in patients with metastatic disease and KRASG12R mutations in patients with well to moderately differentiated tumors. Moreover, we found co-mutations could contribute to KRAS allele-specific clinical outcomes. We found worse OS in ARID1A-mutated patients and a lower co-mutation rate of ARID1A in KRASG12R. Our findings of different clinical outcomes by KRAS mutation subtypes and co-mutation status suggest an allele- and co-mutation-specific impact of KRAS mutations on pancreatic cancer outcomes and provide guidance in improving approaches to target KRAS in pancreatic cancer.
Methods
The MD Anderson Cancer Center Institutional Review Board (IRB) approved the collection of demographic, clinical, and pathological information under IRB protocols 09-0373 and 2023-0091. This study using human data complied with all relevant ethical regulations including the Declaration of Helsinki. Informed consent was waived, as per the IRB guidelines for retrospective studies of previously collected clinical and molecular information. The Palantir Foundry software system (Palantir, Denver, CO) was used to query the MD Anderson EHR to identify patients with a confirmed diagnosis of PDAC who underwent somatic tumor tissue mutation testing at MD Anderson from 3/14/1997 to 4/27/2023 for inclusion in the study.
Patient demographic, histopathology, tumor grade, surgical history, and mutational profile data were collected from the MD Anderson EHR and tumor registry data using the Foundry system. Histologic classification and grade were collected from the patients’ pathology reports. Molecular testing was performed at MD Anderson’s molecular diagnostics laboratory, which is College of American Pathologists (CAP) accredited and Clinical Laboratory Improvement Amendments (CLIA) certified. The gene panels used evolved during the study inclusion period, with expanding lists of genes over time. The information on tumor genomic alterations (GAs) was extracted from the available clinical and molecular data. Deidentified information was used for analysis.
For the co-mutation analysis, only patients who were tested with multigene panels were included (n = 513). The Oncoplot function within MAFtools was used to visualize the somatic mutation distribution. The function performs pair-wise Fisher’s exact test to uncover mutually exclusive or co-occurring gene sets and an FDR-corrected p < 0.1 was considered significant. To better understand the co-mutation patterns with KRAS and the rest of the genes, a heatmap was constructed to demonstrate the co-mutation landscape of KRAS mutation status, as well as the status of the different KRAS alleles, and the rest of the genes analyzed (Fig. 4b). The percentage of co-occurrence between KRAS alleles and pathogenic mutations in the genes listed in the heatmap were determined using in-house R scripts. Fisher’s exact test was used to test for significance in co-occurrence between KRAS alleles and pathogenic mutations. Based on the co-mutation patterns observed, we divided patients into 4 molecularly distinct PDAC co-mutation subtypes to visualize and test the relationship between co-mutation pattern and OS.
Statistical analysis
Differences in disease stage and tumor grade between patients with different KRAS mutations were assessed using Chi-square and Fischer’s exact tests. Overall survival (OS) was calculated from the date of initial diagnosis until death or last known contact. OS curves were estimated using the Kaplan–Meier method, and the difference in survival curves was tested using the log-rank test. Univariate Cox proportional hazards models were used to estimate hazard ratios (HRs) and test the associations of KRAS mutation status, KRAS mutation allele subtypes, and other driver mutations with OS.
In the co-mutation analysis, the somatic interactions function within MAFtools was used to detect mutually exclusive or co-occurring mutation events. Pair-wise Fisher’s exact test was used to uncover mutually exclusive or co-occurring gene sets with Benjamini–Hochberg multiplicity correction, and a false discovery rate (FDR)-corrected p < 0.1 was considered significant. The OS curves for the 4 co-mutation subtypes were estimated with the Kaplan–Meier method and compared using the log-rank test.
GraphPad Prism version 9 (GraphPad Software, San Diego, California USA) and Rstudio 2020 (RStudio, PBC. Boston, MA) were used for the statistical analyses and data visualization71. All tests were two-sided, and statistical significance was identified by a p-value < 0.05.
PanCAN’s know your Tumor® program and dataset
PanCAN, in partnership with Tempus (Tempus Labs Inc., Chicago, IL), offers the Know Your Tumor® (KYT) precision medicine service to patients with pancreatic cancer. KYT data is available through the PanCAN SPARK platform (www.pancan.org/spark). Tempus processes, sequences and conducts group-level bioinformatics analyses on tumor biopsy samples. Data is derived from the Tempus xT NGS panel that covers 648 genes with actionable oncologic mutations. Variants are called from the resulting alignment files using an analysis pipeline that detects SNPs and indels using Freebayes and Pindel72,73. A filtered variant file that contains biologically relevant DNA variants, as determined by the Tempus pipeline, were used for all KYT-related analyses. Patients with PDAC who had their tumor sequenced by Tempus were included in the analysis. Pathogenic or likely pathogenic mutations were determined by Tempus’ proprietary Knowledge Database which is based on the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) guidelines for variant classification. All mutation data was converted to Mutation Annotation Format (MAF) to enable use of the functions in the Bioconductor R package, MAFtools74. The Oncoplot function within MAFtools was used to visualize the somatic mutation distribution across the KYT cohort. The somatic interactions function within MAFtools was used to detect mutually exclusive or co-occurring mutation events. The function performs pair-wise Fisher’s exact test to uncover mutually exclusive or co-occurring gene sets with Benjamini-Hochberg multiplicity correction, and an FDR-corrected p < 0.1 was considered significant. The percentage of co-occurrence between KRAS alleles and pathogenic mutations in the genes listed in the heatmap in Fig. 6 were determined using in-house R scripts. Fisher’s exact test was used to test for significance in co-occurrence between KRAS alleles and other pathogenic mutations. Overall survival (OS) was calculated from the date of initial diagnosis until death or last known contact. OS curves by KRAS mutation and subtype status were estimated using the Kaplan–Meier method, and the difference in survival curves was tested using the log-rank test.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data generated in this study are available upon request from the corresponding author. The MD Anderson institutional IRB does not allow for these data to be deposited into an external repository. PanCAN KYT data is available for access upon completion of a data-sharing agreement on the following domain: pancan.org/spark.
Code availability
No custom code was used in this study. Details of bioinformatics analytics are available upon request.
References
Mizrahi, J. D., Surana, R., Valle, J. W. & Shroff, R. T. Pancreatic cancer. Lancet 395, 2008–2020 (2020).
Rahib, L., Wehner, M. R., Matrisian, L. M. & Nead, K. T. Estimated projection of US cancer incidence and death to 2040. JAMA Network Open 4, e214708–e214708 (2021).
Conroy, T. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825 (2011).
Tempero, M. A. et al. Pancreatic adenocarcinoma, version 2.2021, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 19, 439–457 (2021).
Von Hoff, D. D. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 (2013).
Wang-Gillam, A. et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 387, 545–557 (2016).
Biankin, A. V. et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012).
Jones, S. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. science 321, 1801–1806 (2008).
Waters, A. M. & Der, C. J. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med. 8, a031435 (2018).
Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203.e113 (2017).
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016).
Lee, J. K. et al. Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. NPJ Precis. Oncol. 6, 91 (2022).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Govindan, R. et al. Phase 1 study of AMG 510, a novel molecule targeting KRAS G12C mutant solid. Ann. Oncol. 860, v159–v193 (2019).
Hong, D. S. et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 383, 1207–1217 (2020).
Hallin, J. et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 (2020).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
Strickler, J. H. et al. Sotorasib in KRAS p.G12C–mutated advanced pancreatic cancer. N. Engl. J. Med. 388, 33–43 (2022).
Bekaii-Saab T. S. et al. Adagrasib in advanced solid tumors harboring a KRASG12C mutation. J. Clin. Oncol. 41, 4097–4106.
Hallin, J. et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 28, 2171–2182 (2022).
Tokatlian, T. et al. Chimeric antigen receptors directed at mutant KRAS exhibit an inverse relationship between functional potency and neoantigen selectivity. Cancer Res. Commun. 2, 58–65 (2022).
Leidner, R. et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N. Engl. J. Med. 386, 2112–2119 (2022).
Jänne, P. A. et al. Adagrasib in non–small-cell lung cancer harboring a KRASG12C mutation. N. Engl. J. Med. 387, 120–131 (2022).
Dy, G. K. et al. Long-term outcomes and molecular correlates of sotorasib efficacy in patients with pretreated KRAS G12C-mutated non–small-cell lung cancer: 2-year analysis of CodeBreaK 100. J. Clin. Oncol. 41, 3311–3317 (2023).
Johnson, C., Burkhart, D. L. & Haigis, K. M. Classification of KRAS-activating mutations and the implications for therapeutic intervention. Cancer Discov. 12, 913–923 (2022).
Hobbs, G. A. & Der, C. J. RAS mutations are not created equal. Cancer Discov. 9, 696–698 (2019).
Ricciuti, B. et al. Comparative analysis and isoform-specific therapeutic vulnerabilities of KRAS mutations in non-small cell lung cancer. Clinical Cancer Res. 28, 1640–1650 (2022).
Hunter, J. C. et al. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 13, 1325–1335 (2015).
Kobayashi, Y. et al. Silent mutations reveal therapeutic vulnerability in RAS Q61 cancers. Nature 603, 335–342 (2022).
Ricciuti, B. et al. Dissecting the clinicopathologic, genomic, and immunophenotypic correlates of KRAS(G12D)-mutated non-small-cell lung cancer. Ann. Oncol. 33, 1029–1040 (2022).
Rachakonda, P. S. et al. Somatic mutations in exocrine pancreatic tumors: association with patient survival. PLoS One 8, e60870 (2013).
Skoulidis, F. & Heymach, J. V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 19, 495–509 (2019).
Diehl, A. C. et al. KRAS mutation variants and co-occurring PI3K pathway alterations impact survival for patients with pancreatic ductal adenocarcinomas. Oncologist 27, 1025–1033 (2022).
Alfaro-Munoz, K. et al. Building a data foundation: how MD anderson and palantir are partnering to accelerate research and improve patient care. J. Clin. Oncol. 37, e18077–e18077 (2019).
Zeineddine, F. A. et al. Survival improvement for patients with metastatic colorectal cancer over twenty years. NPJ Precis. Oncol. 7, 16 (2023).
Goldstein, J. B. et al. Tackling “big data” for accelerating cancer research. J. Clin. Oncol. 34, e23160–e23160 (2016).
Pishvaian, M. J. et al. Molecular profiling of patients with pancreatic cancer: initial results from the know your tumor initiative. Clin. Cancer Res. 24, 5018–5027 (2018).
Qian et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 4, e173420–e173420 (2018).
Hou, P. et al. Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov. 10, 1058–1077 (2020).
Dey, P. et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov. 10, 608–625 (2020).
Kemp, S. B. et al. Efficacy of a small-molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov. 13, 298–311 (2023).
Hobbs, G. A. et al. Atypical KRAS(G12R) mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov. 10, 104–123 (2020).
Kimura, Y. et al. ARID1A maintains differentiation of pancreatic ductal cells and inhibits development of pancreatic ductal adenocarcinoma in mice. Gastroenterology 155, 194–209.e192 (2018).
Huynh, M. V. et al. Functional and biological heterogeneity of KRAS(Q61) mutations. Sci. Signal. 15, eabn2694 (2022).
Zhang, Z., Morstein, J., Ecker, A. K., Guiley, K. Z. & Shokat, K. M. Chemoselective covalent modification of K-Ras(G12R) with a small molecule electrophile. J. Am. Chem. Soc. 144, 15916–15921 (2022).
Hu, H.-f et al. Mutations in key driver genes of pancreatic cancer: molecularly targeted therapies and other clinical implications. Acta Pharmacologica Sinica 42, 1725–1741 (2021).
Notta, F. et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538, 378–382 (2016).
Vasen, H. F. et al. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int. J. Cancer 87, 809–811 (2000).
Maitra, A. & Hruban, R. H. Pancreatic cancer. Annu. Rev. Pathol. 3, 157–188 (2008).
Klatte, D. C. F. et al. Pancreatic cancer surveillance in carriers of a germline CDKN2A pathogenic variant: yield and outcomes of a 20-year prospective follow-up. J. Clin. Oncol. 40, 3267–3277 (2022).
Doyle, A. et al. The impact of CDKN2A mutations on overall survival in pancreatic adenocarcinoma. J. Clin. Oncol. 37, 278–278 (2019).
Zhao, D. et al. Clinical and molecular features of KRAS-mutated lung cancer patients treated with immune checkpoint inhibitors. Cancers 14, 4933 (2022).
Aguirre, A. J. et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 17, 3112–3126 (2003).
Goodwin, C. M. et al. Combination therapies with CDK4/6 inhibitors to treat KRAS-mutant pancreatic cancer. Cancer Res. 83, 141–157 (2023).
Baghdadi, T. A. et al. Palbociclib in patients with pancreatic and biliary cancer with CDKN2A alterations: results from the targeted agent and profiling utilization registry study. JCO Precis. Oncol. 3, 1–8 (2019).
Hustinx, S. R. et al. Concordant loss of MTAP and p16/CDKN2A expression in pancreatic intraepithelial neoplasia: evidence of homozygous deletion in a noninvasive precursor lesion. Mod. Pathol. 18, 959–963 (2005).
Ngoi, N. et al. Genomic classification of clinically advanced pancreatic ductal adenocarcinoma (PDAC) based on methylthioadenosine phosphorylase (MTAP) genomic loss (MTAP loss). J. Clin. Oncol. 40, 604–604 (2022).
Hustinx, S. R. et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: a potential new target for therapy. Cancer Biol. Ther. 4, 83–86 (2005).
Hahn, S. A. et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271, 350–353 (1996).
Shugang, X. et al. Prognostic value of SMAD4 in pancreatic cancer: a meta-analysis. Transl. Oncol. 9, 1–7 (2016).
Su, H. & Wang, C. Prognostic value of SMAD4 in resectable pancreatic cancer. Postępy Higieny i Medycyny Doświadczalnej 76, 324–332 (2022).
Shain, A. H. et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc. Natl. Acad. Sci. USA 109, E252–E259 (2012).
Halbrook, C. J., Lyssiotis, C. A., Pasca di Magliano, M. & Maitra, A. Pancreatic cancer: advances and challenges. Cell 186, 1729–1754 (2023).
Ferri-Borgogno, S. et al. Paradoxical role of AT-rich interactive domain 1A in restraining pancreatic carcinogenesis. Cancers 12, 2695 (2020).
Sen, M. et al. ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells. Clin. Epigenetics 11, 92 (2019).
Mandal, J., Mandal, P., Wang, T.-L. & Shih, I.-M. Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response. J. Biomed. Sci. 29, 71 (2022).
Shen, J. et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 5, 752–767 (2015).
Mullen, J., Kato, S., Sicklick, J. K. & Kurzrock, R. Targeting ARID1A mutations in cancer. Cancer Treat. Rev. 100, 102287 (2021).
Crowley, F., Park, W. & O’Reilly, E. M. Targeting DNA damage repair pathways in pancreas cancer. Cancer Metastasis Rev. 40, 891–908 (2021).
RStudio: integrated development for R RStudio, PBC; 2020. http://www.rstudio.com/.
Garrison EM, G. Haplotype-based variant detection from short-read sequencing. https://arxiv.org/pdf/1207.3907.pdf (2012).
Ye, K., Schulz, M. H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28, 1747–1756 (2018).
Acknowledgements
This work was supported by the Col. Daniel Connelly Memorial Fund, the National Cancer Institute (Cancer Center Support Grant P30 CA016672), the Cancer Prevention & Research Institute of Texas (CPRIT, RR180035 to J.P.S.), and a Conquer Cancer Career Development Award (to J.P.S.). The Pancreatic Cancer Action Network (PanCAN) provided access to the validation dataset from PanCAN’s Know Your Tumor® program. Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the American Society of Clinical Oncology® or Conquer Cancer.
Author information
Authors and Affiliations
Contributions
D.Z. and J.P.S. conceptualized the paper, provided oversight, and contributed to patient enrollment and treatment, patient assessment, data interpretation, and writing the manuscript. D.Z., A.Y., and M.Y. conceived and designed the study and contributed to literature search, data acquisition, data analysis, data interpretation, and manuscript writing. J.W., M.O., and S.K. contributed to patient enrollment, treatment, assessment, and data interpretation. K.A., S.D., and L.M. contributed to data acquisition, data analysis, and data interpretation. M.K., P.E., S.C., J.P., and R.S. contributed to the statistical analysis and writing of the manuscript. All authors read, reviewed, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
D.Z. has clinical trial contracts with Mirati and CARsgen and served as a member of the advisory board for Affini-T.K.A., S.D., and L.M. are employees of the Pancreatic Cancer Action Network, a nonprofit cancer advocacy organization that receives donations from private and commercial entities and may benefit indirectly financially and non-financially from this publication. S.P. reports research funding from Mirati Therapeutics, Lilly, Xencor, Novartis, Rgenix, Bristol-Myers Squibb, Astellas Pharma, Framewave, 4D Pharma, Boehringer Ingelheim, NGM Biopharmaceuticals, Janssen, Arcus Biosciences, Elicio Therapeutics, Bionote, Ipsen, Zymeworks, Pfizer, ImmunoMET, Immuneering, Amal Therapeutics. S.P. is a member of the advisory board for Zymeworks, Ipsen, Novartis, Janssen, Boehringer Ingelheim, AskGene Pharma, BPGbio, Jazz Pharmaceuticals, AstraZeneca, US WorldMeds, Nihon Medi-Physics Co., Ltd., Alligator Bioscience. M.L. consults for Pfizer, Delcath, Janssen, BioNTech, G1 Therapeutics, Imvax, and Bayer and received research funding from institutions from EpimAb BioTherapeutics, Merck, Erasca, Boehringer Ingelheim, Arcus Biosciences, Repare Therapeutics, Trisalus Life Sciences, and Xilis. A.M. is a consultant for Tezcat Biotechnologies and is a co-inventor on a patent that has been licensed from Johns Hopkins University by Thrive Earlier Detection (an Exact Sciences company). A.M. serves on the scientific and medical advisory board for Pancreatic Cancer Action Network and Pancreatic Cancer UK. J.P.S. receives grants, research support, or collaborates with Celsius Therapeutics, BostonGene, Caris Life Sciences, Natera, Xilis, Palantir, and Genentech. J.P.S. reports consulting and stock ownership with Engine Biosciences, NaDeNo Nanoscience.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yousef, A., Yousef, M., Chowdhury, S. et al. Impact of KRAS mutations and co-mutations on clinical outcomes in pancreatic ductal adenocarcinoma. npj Precis. Onc. 8, 27 (2024). https://doi.org/10.1038/s41698-024-00505-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-024-00505-0
