
Abstract
Background:
Classification of endometrial carcinomas (ECs) by morphologic features is inconsistent, and yields limited prognostic and predictive information. A new system for classification based on the molecular categories identified in The Cancer Genome Atlas is proposed.
Methods:
Genomic data from the Cancer Genome Atlas (TCGA) support classification of endometrial carcinomas into four prognostically significant subgroups; we used the TCGA data set to develop surrogate assays that could replicate the TCGA classification, but without the need for the labor-intensive and cost-prohibitive genomic methodology. Combinations of the most relevant assays were carried forward and tested on a new independent cohort of 152 endometrial carcinoma cases, and molecular vs clinical risk group stratification was compared.
Results:
Replication of TCGA survival curves was achieved with statistical significance using multiple different molecular classification models (16 total tested). Internal validation supported carrying forward a classifier based on the following components: mismatch repair protein immunohistochemistry, POLE mutational analysis and p53 immunohistochemistry as a surrogate for ‘copy-number’ status. The proposed molecular classifier was associated with clinical outcomes, as was stage, grade, lymph-vascular space invasion, nodal involvement and adjuvant treatment. In multivariable analysis both molecular classification and clinical risk groups were associated with outcomes, but differed greatly in composition of cases within each category, with half of POLE and mismatch repair loss subgroups residing within the clinically defined ‘high-risk’ group. Combining the molecular classifier with clinicopathologic features or risk groups provided the highest C-index for discrimination of outcome survival curves.
Conclusions:
Molecular classification of ECs can be achieved using clinically applicable methods on formalin-fixed paraffin-embedded samples, and provides independent prognostic information beyond established risk factors. This pragmatic molecular classification tool has potential to be used routinely in guiding treatment for individuals with endometrial carcinoma and in stratifying cases in future clinical trials.
Similar content being viewed by others

Main
Endometrial cancers (EC) are the most prevalent gynaecologic malignancies in the developed world and are the fourth most common cancer in women overall (Siegel et al, 2015). Incidence rates have increased markedly over the last decades attributable at least in part to the global epidemic of obesity (Sheikh et al, 2014). Although the majority of women with EC have good outcomes, women with advanced disease or more aggressive subtypes may not be curable with adjuvant therapy. In Canada over the last decade, the annual percentage increase in age-standardised mortality rate for EC is greater than any other cancer in women (Society CC Canadian Cancer Statistics, 2014) and we are in desperate need of new approaches, including diagnostic tools, to manage this cancer.
There are many unanswered questions in EC pertaining to diagnosis and optimal management. Management considerations include which surgery to perform by either a generalist or subspecialist, which if any adjuvant therapies to administer, surveillance strategies and fertility-sparing options in young women. Currently, there are multiple systems of risk-group stratification based on post-surgical staging pathologic examination (principally histotype, tumour grade and stage) that may help guide treatment(s) (Creutzberg et al, 2000; Fanning, 2001; Keys et al, 2004; Mariani et al, 2008; Kwon et al, 2009; Colombo et al, 2013; AlHilli et al, 2014; Bendifallah et al, 2015; Kong et al, 2015). However, pathologists are unable to reproducibly diagnose histotype and grade of EC; lack of consensus between expert pathologists has been demonstrated, even with the addition of immunohistochemistry (Guan et al, 2011; Gilks et al, 2013; Han et al, 2013; Hoang et al, 2013). This lack of reproducibility is a major barrier to improving care for women with this disease, and treatments vary within and between cancer centres globally. Assessment of treatment efficacy when there is variable histotype and grade assignment hinders our ability to determine optimal management.
Molecular classification of EC has shown great promise, proving to be reproducible, and demonstrating associations with clinical outcomes (Salvesen et al, 2009; Le Gallo et al, 2012; McConechy et al, 2012; Cancer Genome Atlas Research N et al, 2013). The Cancer Genome Atlas (TCGA) identified four genomic subgroups. The group with POLE mutations and its corresponding ‘ultramutated’ phenotype was a novel finding, and particularly interesting given the very favourable outcomes even with high-grade tumours. This has been validated in other series of cases with POLE mutations (Meng et al, 2014; Billingsley et al, 2015; Church et al, 2015). The other distinct subgroups identified from the TCGA data included microsatellite instability (MSI), copy-number low (CN low) and copy-number high (CN high), the latter consisting mostly of cases diagnosed by referring centre pathologists as high-grade serous cancers. For new cases of EC, categorisation into one of these four subgroups could potentially provide prognostic and predictive information for individuals. Therefore, an ability to classify cases in this manner might offer an improvement on the current clinical/pathology-based risk group system (Murali et al, 2014). Unfortunately, methodologies used for the TCGA study to identify these four genomic subgroups, including genome sequencing, were costly, complex and unsuitable for wider clinical application. Our goal was to determine whether the same molecular subgroups could be identified and the survival curves reproduced with assays that could be used in routine clinical practice.
Our first aim was to design simple, lower cost, molecular-based classification methodologies that can recover the TCGA subtypes described earlier. These proposed classifiers were tested and compared both on the TCGA data and on a separate cohort of patients from our centre (n=152). In our second aim, the pragmatic molecular classification model selected based on results of the first aim was compared with contemporary clinical risk group stratification. Improvement in outcome prediction resulting from the addition of a molecular classifier should lead to better management for women with EC.
Materials and Methods
Samples and clinical data
Patient cohort-vancouver
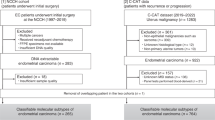
A retrospective cohort of 152 patients with primary endometrial carcinoma was identified from the Vancouver General Hospital cases banked in the OVCARE Tissue Bank Repository, Vancouver, BC, Canada (McConechy et al, 2012). These patients were diagnosed with EC between 2002 and 2009. Patients were excluded if they had a diagnosis of a concurrent cancer that was being treated at the same time as their endometrial cancer or any previous treatment, which may have influenced her outcome (e.g., prior radiotherapy). Exclusion criteria also included uterine pre-cancers, cancers metastatic to the uterus or no definitive surgery performed (no hysterectomy). Patients had comprehensive data collected including details of pathology, surgery, chemotherapy, radiation and outcomes with a minimum of 2 years potential follow-up. Patient management was according to BC Cancer Agency Guidelines (http://www.bccancer.bc.ca/HPI/CancerManagementGuidelines). Research ethics approval for the Tissue/Biospecimen Bank and this project was granted from the University of British Columbia Institutional Review Board and all patients underwent informed written consent for the use of their biospecimens for research purposes.
Patient cohort details TCGA, outcomes definitions, management of missing data and assay methods
Development of a molecular classification model
In the TCGA cohort of fully evaluable cases (n=232), roughly 7% of cases were grouped as POLE ultramutated phenotype, 28% were designated with microsatellite instability (MSI), 26% copy-number high (CN high) and 39% copy-number low (CN low) (Cancer Genome Atlas Research Network et al, 2013). When applying the new classifier tool to the TCGA cohort, our primary objective was to classify all patients into the TCGA clusters, but also to minimise the number of false negatives in the CN high poor prognosis group, to avoid under-treating women who may have aggressive disease.
In reproducing the TCGA clusters considerations at each step included:
-
1
POLE: In TCGA, the POLE ultramutated cluster was identified based on a POLE mutation, a high percent of C to A transversions and low percent of C to G transversions, as well as more than 500 SNVs. For our classifier, PTEN was initially included in the assessment of the models along with POLE mutations because, in the TCGA mutational analysis, although PTEN mutations were seen to some degree across MSI and CN low subgroups, PTEN and POLE mutations were noted to co-occur in almost all of the ‘ultramutated’ subgroup and seemed to better define this category. Hence, we initially proposed two methods to identify the ultramutated subgroup; one using the POLE mutation status alone, and one that uses both the POLE and PTEN mutation status.
-
2
MSI: The MSI group in the TCGA analysis was based on results from the MSI assay using seven markers (Cancer Genome Atlas Research N et al, 2013). In our models, we identified the MSI phenotype subsequent to POLE ultramutated, as done in TCGA and also considered switching the order to identify the MSI cluster first. In practice this is practical as it would be useful to have this information as early as possible to enable referral to hereditary cancer programs for Lynch syndrome testing. We moved from using the MSI assay to using MMR IHC testing (MLH1, MSH2, MSH6 and PMS2), which we have shown to be highly concordant with MSI assay (McConechy et al, 2015) and more cost effective and practical.
-
3
CN: In TCGA, copy number was assessed with Affymetrix SNP 6.0 microarrays using DNA originating from frozen tissue. Moving forward, we wished to have a more cost-effective method that could be achieved on FFPE material; thus, we mined the TCGA data and found that copy-number status at three specific loci (FGFR (4p16.3), SOX17 (8q11.23) and MYC (8q24.12) were most predictive of overall copy-number status, for example, using just these three loci we were able to identify all cases within the CN high cluster. We looked to assess these three loci by FISH (Supplementary Methods). In addition, TP53 was noted to be mutated in most of the copy-number high cases in the TCGA cohort and in silico analysis demonstrated that p53 status was able to reproduce the CN high/low survival curves. TP53 mutation status was not equivalent to CN high subgroup in TCGA but identified a subgroup of EC cases with distinctly worse outcomes. Therefore, CN status was determined as follows: (i) FISH determination of copy-number status at three loci most associated with CN high subgroup in TCGA (FGFR (4p16.3), SOX17 (8q11.23) and MYC (8q24.12), scored on two thresholds, and (ii) p53 status determined by IHC or TP53 sequencing, yielding four possible ways to classify CN high following determination of MSI and POLE groups.
Varying the combinations of the features described above resulted in eight different ways to classify patients in the TCGA cohort, and a total of 16 in the Vancouver cohort. We were able to directly compare the performance of these different scenarios within the TCGA cohort, because the genomic-based data labels and outcome details were available to us (with the exception of immunohistochemistry and FISH). As the TCGA equivalent genomics data were not available for the Vancouver cohort, performance measures of the more selective molecular components were based on survival outcomes. We were able to reproduce the four genomic subgroups and survival curves in both the TCGA and Vancouver cohorts, as shown in Supplementary Figure 1 and Figure 1, respectively.


Kaplan–Meier survival analyses and log-rank statistics of eight possible models for pragmatic molecular classification of endometrial cancers applied to the Vancouver cohort ( n =143). Overall survival (OS), disease-specific survival (DSS) and recurrence-free survival (RFS) are shown for each model and molecular subgroups are distinguished by colour (POLE (blue), MMR IHC abn (yellow), p53 wt (green) and p53 abn (red)). Model 8 is outlined in red and is the model that was used for subsequent univariate and multivariate analysis, was combined with either European Society of Medical Oncologists clinical risk groups or pathological parameters.
Statistical methods
Univariable analyses of molecular classifier categories against overall survival (OS), disease-specific survival (DSS) and recurrence-free survival (RFS) were examined both using Kaplan–Meier plots with log-rank significance testing and Cox proportional hazard regression models. Multivariable cox proportional hazard regression model analysis was performed to assess any additional prognostic information that would be added by the molecular classifier beyond the clinical risk group classification and the standard prognostic factors (age, BMI, grade, stage, histology, LVSI and treatment). To assess the additional prognostic information added by the classifier model to clinicopathological parameters, two sets of multivariable analyses were performed: (i) multivariable analyses with ESMO clincial risk groups, (ii) multivariable analyses with individual clinicopathological parameters: age, BMI, grade, stage, histology and LVSI.
Where the percent censoring exceeded 80%, a Firth bias reducing correction was applied to obtain estimates. P-values from omnibus likelihood ratio test in all Cox models were reported. Smoothed plots of weighted Schoenfeld residuals were used to assess proportional hazard assumptions (Grambsch et al, 1995). Only complete observations were used for model fitting. A missing value analysis was done to explore the distribution of missing values and to ensure they are missing at random.
The performance of the models was first assessed visually based on their ability to reproduce a similar pattern as the TCGA-identified (integrated genomic data-based) groups. Furthermore, the performance was quantified by computing accuracy measures (in the TCGA cohort where true labels are available) and Harrell’s C-index in the TCGA and in the Vancouver cohort when considering survival outcome. The C-index is a measure of the discriminative ability of the model. A C-index of 0.5 indicates that the model has no discriminative ability and a C-index of 1 indicates that a model perfectly distinguishes between those who have an event and those who do not.
Bootstrapping techniques (Steyerberg et al, 2001) were used for internal validation in both the TCGA data and our own cohort. Validation using bootstrap re-sampling would estimate the likely performance of the model on a new sample of patients from a same clinical setting. One thousand bootstrap samples are used; in each bootstrap iteration, a sample of size equal to the original cohort is drawn with replacement from the original cohort. Models assessed with the C-index were developed in the bootstrap samples and tested in those subjects not included in the bootstrap sample (Efron and Tibshirani, 1997). In comparing the TCGA-predicted subtypes with the actual labels, the sensitivity and the specificity were obtained from the bootstrap samples alone.
To address the second aim, clinical risk groups were assigned according to the European Society of Medical Oncologists (ESMO) criteria (Colombo et al, 2013) and compared with molecular subgroups in both the TCGA (Supplementary Figure 2) and new endometrial carcinoma cohorts.
The association of TCGA-inspired endometrial subtypes (POLE/MMR IHC abn/p53 wt/p53 abn) with other variables such as demographic (age), clinical (treatment), pathological (stage (FIGO 2009), grade, histology and LVSI) was tested with non-parametric tests. Kruskal–Wallis rank sum test was used for continuous variables (age and BMI), and Fisher’s exact test was used for all other variables/categorical (stage, grade, histology, LVSI, any positive nodes and initial adjuvant treatment and clinical risk groups).
Statistical significance level was set to 0.05. P-values reported were not corrected for multiple comparisons. All statistical analyses were performed using the statistical software R v3.1.0 (R Core Team, 2014).
Results
Application of the molecular classification tool to a new cohort of endometrial carcinomas
For the Vancouver cohort, beginning with 152 patients, one patient was excluded for having undergone neoadjuvant chemotherapy, seven cases were excluded who failed sequencing or had no DNA available for POLE or TP53 sequencing, and one case had insufficient tumour tissue remaining to enable MMR IHC status to be determined, leaving 143 fully evaluable cases. ‘MSI’ status in the Vancouver cohort was determined by MMR IHC, as we have demonstrated the high concordance with MSI assay in ECs (McConechy et al, 2015). In total, 41 of 143 fully evaluable cases had abnormal MMR IHC (29%) (‘MMR IHC abn’), consistent with the TCGA data (Table 1).
POLE exonuclease domain (EDM) mutations were found in 13 cases in the total cohort. In one case, (VOA 843, Supplementary Table 1) a low level (5%) validated POLE mutation was found in exon 12 that is not a known hot spot mutation, and this tumour also demonstrated isolated MSH6 loss with IHC. As the first step in our classifier model was to assess MMR IHC this case was classified as MMR IHC abnormal (not grouped with ‘POLE’). Of the remaining cases classified as ‘POLE’ mutant (12 of 143 (8.4%) cases), they were exclusively stage I, with 5 of 12 (42%) grade 3, and all but one case (92%) showing endometrioid histology (Table 1). In this small cohort all tumours with POLE EDM mutations had normal MMR IHC. TP53 mutations were identified in 3 of 12 POLE mutated cases by sequencing and 1 of 12 cases by abnormal p53 IHC (score 0 or 2+). Full details of the subset of cases in the Vancouver cohort with POLE mutations, including chromosome, genomic position and amino-acid change, as well as TP53 and PTEN mutation details, and the status of MMR IHC (MLH1, MSH2, MSH6, PMS2) and p53 IHC for these cases are given in Supplementary Table 1. There were no recurrences or deaths in the cases with POLE EDM mutations, with an observation time of over 5 years for this subgroup.
Using p53 IHC status as a surrogate for ‘copy-number high’ to identify p53 abnormal (‘p53 abn’) subgroup, 25 cases had aberrant p53 equalling 17.5% of the total cohort of 143, or 28% of cases following the exclusion of those classified as ‘MMR IHC abn’ and ‘POLE’ positive (Table 1). Using TP53 sequencing for determination of ‘p53 abn’ revealed mutations in 27 cases in the total cohort or 19 of the 88 (22%) cases remaining after the exclusion of those classified as ‘MMR IHC abn’ and ‘POLE’ EDM mutation positive. Supplementary Table 2 includes the specifics on POLE, TP53, PTEN mutations and MMR and p53 IHC for the full cohort (n=153).
FISH testing was interpretable in 121 cases. Results for threshold 1 (T1) suggest copy-number high status in 12 cases, 11 of which also had TP53 mutations; however, an additional 15 cases had TP53 mutations and were not designated ‘CN high’ by FISH. Similarly, for Threshold 2 (T2) only 9 cases met criteria of ‘CN high’ designation, 8 of which also had TP53 mutations but 18 other cases had TP53 mutations and did not qualify for ‘CN high’ status based on FISH T2. Within the non-MMR IHC abn, non-POLE cohort, and using TP53 mutation status for comparison the kappa statistic for level of agreement between testing methods for T1 was 0.66 (95% CI 0.42–0.84) with a sensitivity, specificity, positive (PPV) and negative predictive value (NPV) of 1, 0.56, 0.88 and 1, respectively. For T2 the kappa statistic was 0.49 (95% CI 0.24–0.72) with corresponding accuracy of 1, 0.39, 0.85 and 1. We also compared p53 IHC to TP53 mutation status in the whole cohort and in the cohort remaining after removal of ‘MMR IHC abn’ and ‘POLE’ mutated cases (n=88) and found a kappa statistic of 0.77 (95% CI 0.59–0.92) with sensitivity, specificity, PPV and NPV of 0.9, 0.94, 0.98 and 0.74, respectively.
The Vancouver cohort included the major histological subtypes (83% endometrioid histotype, the remainder being of serous or mixed histotypes, with the exception of a single undifferentiated carcinoma), all stages and grades (Table 1) similar in distribution to both TCGA (Supplementary Table 3) and the general population. The estimated median follow-up time in the Vancouver cohort, as calculated by the reverse Kaplan–Meir method (Schemper and Smith, 1996), is 5 years. The median observation time is 4.67 years. A total of 27 recurrences and 28 deaths were observed. A comparison of patient demographics and clinicopathologic details for the full cohort and within ‘MMR IHC abn’, ‘POLE’, ‘p53 wt and ‘p53 abn’ categories based on a pragmatic classification is given in Table 1. The distribution of multiple parameters differed across the molecular subgroups, notably an increased presence of LVSI in the ‘MMR IHC abn’ and ‘p53 abn’ subgroups, and one-third of both ‘MMR IHC abn’ and ‘p53 abn’ cases having node positive disease. Stage was also more advanced in the ‘MMR IHC abn’ and ‘p53 abn’, with 71 and 79% of cases with disease beyond the uterus, respectively. Not surprisingly, average age was highest in the ‘p53 abn’ group with the highest proportion of serous/non endometrioid cases. Women with MMR IHC abn cases were also older, likely secondary to the higher proportion of MMR IHC loss at MLH1 in this cohort, with a known increased frequency of methylation in older individuals.
We had 16 different possible combinations with which we could analyse outcomes for a molecular classifier, based on defining MMR IHC as normal or abnormal first or after classification of POLE cases (one decision), POLE mutations or POLE and PTEN mutations together (two ways to categorise this step) and copy number that could be determined by four different options in surrogate testing (four ways to categorise: p53 IHC, TP53 mutations, FISH for three loci T1 and FISH T2). As the cases with POLE EDM mutations all had normal MMR IHC, changing the order of these two tests in the model for this cohort (e.g. stepwise analysis of MMR IHC first then POLE status vs POLE first then MMR IHC) made no difference, thus Kaplan–Meier analyses and the log-rank statistic for 8 models not 16 as shown in Figure 1. Statistical significance of the log-rank test is noted in the majority of these models, with the exception of FISH T2 (Models 3 and 6). Although FISH testing of three loci (MYC, SOX17 and FGFR3) segregated by the first threshold (T1) may act as a surrogate test for copy number, it is suboptimal for clinical use because (1) we achieved results in only a subset of cases, (2) there was a high level of subjectivity in scoring and (3) the log-rank test for T1 did not reach statistical significance for all outcome parameters (Models 2 and 5). Addition of PTEN mutation status to POLE mutation categorization, however, seemingly helpful in our initial discovery phase with the TCGA data (Supplementary Figure 1), did not add apparent benefit to the models in our Vancouver data set (Models 4, 5, 6, 7). Of note, 11 of 12 cases with POLE mutations also harbour PTEN mutations in this data set.
Assessment of the molecular classification tool compared with traditional clinical/pathological risk groups
ESMO clinical risk group stratification assigned based on complete clinicopathologic data from staging was also demonstrated to be associated with OS, DSS and PFS in our Vancouver data set (P<0.005 for all) (data not shown).
Harrell’s C-Index measuring the discriminative ability of a model to predict an event (e.g., outcomes; OS, DSS and RFS) is shown for each of eight models in Figure 2. We have also shown the C-index for ESMO clinical risk group stratification, clinical risk group stratification combined with molecular classification or pathologic parameters (each component of grade, stage, LVSI and so on added to the model) combined with molecular classification demonstrating the improved ability to discriminate EC outcomes when both traditional and molecular tools are used, with confidence intervals no longer crossing the threshold of 0.5 (Figure 2). Kaplan–Meier analyses for the eight models, C-indices, sensitivity and specificity for the molecular classifier models with and without ESMO clinical risk group stratification were also applied to the TCGA cohort but appear less able to discern outcomes (Supplementary Figure 1).

Harrell’s C-Index for Models 1 to 8, ESMO clinical risk group, and combined molecular and risk groups or pathologic parameters as applied to the Vancouver cohort ( n =143). A C-index of 0.5 (dotted line) indicates that the model has no discriminative ability and a C-index of 1 indicates that a model perfectly distinguishes between those who have an event and those who do not. The pragmatic model chosen to move forward with is outlined in red. Also outlined are the indices for the molecular classifier combined with clinical risk groups or pathological parameters, suggesting an improved ability to discriminate outcomes when taken together.
Figure 3 shows a model option (Model 8 in Figure 1, column 8 in Figure 2), based on pragmatic surrogate molecular assays inclusive of: (1) MMR IHC abnormalities (‘MMR IHC abn’), (2) ‘POLE’ EDM mutations and (3) p53 status determined by IHC, as a surrogate to delineate ’p53 wt’ and ‘p53 abn’ groups. Subsequent tables and comparisons of the molecular classifier in univariate and multivariable analysis used this model specifically for classification.

Univariable analysis was performed to test for associations of known prognostic impact with outcomes (OS, DSS and RFS). These include the molecular subtypes resulting from the chosen model (Model 8), demographic and clinicopathologic parameters as well as the clinical risk groups (ESMO) (Table 2). Increased hazard ratios (HR) were demonstrated for p53 abn molecular subgroup, stage indicative of disease beyond the uterine corpus (e.g., >stage I), grade, presence of lymphatic or vascular space invasion, positive lymph nodes or receiving adjuvant treatment (Table 2).
Multivariable analysis was performed to determine whether the molecular classifier adds any additional prognostic information to the ‘traditional’ clinicopathologic risk group categorisation, defined here by ESMO criteria (encompassing stage, grade and histology), and suggests that the molecular tool remains prognostic independently of the ESMO risk groups (Table 3) for example, both predictors are significant in the model, and the hazard ratio for p53 abn group (1.94) is on the same scale as for ESMO risk group (1.98). We also determined whether the molecular classifier was of additional prognostic benefit to demographic or pathology risk factors (as were tested in univariable analysis). Comparing molecular vs clinical models (summative of age, BMI, stage, grade, histology, LVSI and nodal status), the molecular classifier appears to be prognostic for OS, as well as DSS and RFS (Supplementary Table 4) after accounting for the additional demographic and pathology parameters. However, given the low number of events and high number of parameters assessed these results must be interpreted with caution. Visual examination of the Schoenfeld residual plots indicate no evidence of classifier model (MMR/POLE mut/p53 IHC) violating the proportional hazard assumption (data not shown).
Cross tabulation of the four molecular subgroups generated from MMR IHC/POLE mut/p53 IHC with ESMO risk groups in the Vancouver data set is shown in Figure 4 (and in Supplementary Figure 2 for the TCGA cohort). It is apparent these classification systems are identifying different subgroups of women in both cohorts but more profoundly in our new Vancouver series where more precise ESMO classification was achievable. Focusing on the Vancouver cohort, the ‘low-risk’ clinical risk group can be seen across all four molecular subgroups, including over 30% of the POLE mutated cases but also almost 10% of p53 abn tumours. Greater diversity in outcomes in the ‘low-risk’ group is also noted as three recurrences and four deaths were observed in this assigned cohort, exceeding the POLE molecular group (0 events). Not surprisingly most of the p53 abn cases were ‘high risk’, however, approximately half of the cases with POLE mutations and MMR IHC abn phenotype are also ‘high-risk’ patients who under standard clinical care would go on to receive chemotherapy and radiation (Figure 4).

Cross-tabulation of clinicopathologic risk groups (ESMO) with molecular classification by proposed model: MMR IHC/ POLE mut/p53 IHC. Approximately half of the POLE and MMR IHC abn molecular subgroups are noted to include cases that would be designated as ‘high risk’ by traditional clinical risk group stratification. The p53 abn molecular subgroup includes ∼25% ‘low’ and ‘intermediate’ risk cases who would usually be designated to receive minimal (e.g., vaginal brachytherapy) or no therapy. Although both molecular subgroups and clinical risk groups were associated with outcomes, they may identify different women with EC.
Discussion
Endometrial carcinomas, in particular high-grade cancers, cannot be reliably classified by histomorphologic criteria, even by expert pathologists and with the addition of immunohistochemistry (Gilks et al, 2013; Han et al, 2013; Hoang et al, 2013). Interobserver agreement among pathologists for morphologic risk factors such as grade and LVSI is poor (K=0.35 and 0.23, respectively) (Guan et al, 2011), and histotype shows only a moderate degree of interobserver agreement (K=0.58) (Han et al, 2013). If we are to move towards precision medicine, more reliable systems of categorising EC are needed to determine efficacy and appropriateness of treatments (Murali et al, 2014; Bendifallah et al, 2015). Mutational profiling of endometrial cancers has shown promise (Ferguson et al, 2005; Salvesen et al, 2009; Le Gallo et al, 2012; McConechy et al, 2012; Cancer Genome Atlas Research N et al, 2013; Stelloo et al, 2015) but methodologies to assign genomic subgroups can be expensive and complex, and consequently may not be achievable at all centres. Herein, we present a molecular classifier for endometrial cancers that is based on the discoveries of the TCGA, but pared down to key components evaluable by relatively simple molecular methods. These methods were applied to a new training set of cases in which we have detailed clinicopathologic data and outcomes. We are able to reproduce the four subgroups with distinct survival curves as identified in TCGA, with significant P-values achieved in survival analyses. Although our data suggest that p53 IHC and TP53 mutation status results are not completely equivalent, both methods of assessment were successful in identifying the ‘p53 abn’ molecular subgroup. Lower cost and wide availability of p53 IHC in all pathology departments support IHC as the preferred tool. Moving forward, the findings from this cohort as they relate to the assessment of p53 status will need to be confirmed in a larger data set. Removing the FISH assessment, both for practical reasons (work intensive, subjective, results achievable in lower number of cases, and higher cost vs p53 IHC) and due to lower performance compared with other models, seems prudent. At present, we have no surrogate for the critically important POLE mutation detection and we will carry forward with next-generation sequencing to achieve this in the confirmation cohort. We are working to better characterise POLE-mutated cases in terms of immunophenotype that may influence our approach in the future.
Thus, three major indispensable components in the model are maintained: MMR IHC for MSI phenotype, POLE mutation status and p53 status. The order of the determination of these three major components is also worth consideration. Initially we proposed pulling out MMR IHC abnormal cases first, prompting hereditary cancer referral and yielding information that could be important for both patients and physicians to learn of early. A young woman diagnosed with endometrial cancer may be considering conservative management (e.g., oral or local progesterone therapy), but if she carries a germline MMR gene mutation with increased associated lifetime risk of colon, uterine and ovarian carcinoma this would likely change her course with a recommendation made to pursue definitive surgical management, or may alter her decision to preserve her ovaries (as well as prompting colonoscopy screening) (Lu et al, 2005). Surgery with a specialist (gynaecologic oncologist) for comprehensive staging rather than general gynaecologist might be favoured secondary to a higher likelihood of advanced stage, higher grade and LVSI in these patients. Finally, identification of MMR IHC abn tumours may prove to have predictive implications in EC, as observed in colorectal cancers (Bertagnolli et al, 2009; Sargent et al, 2010; Sinicrope et al, 2011), that would influence choice of treatment. At present, MMR IHC results can be available at the same time as initial pathologic diagnosis of malignancy; POLE sequencing is not widely available and takes weeks; however, we anticipate access to, and turnaround time for POLE testing will improve greatly in the next several years. POLE mutation status identifies women with the most favourable outcomes (Cancer Genome Atlas Research N et al, 2013; Meng et al, 2014; Church et al, 2015), seeming to supersede other prognostic factors such as high-grade disease. In a cohort of this size, and given that all our cases classified as ‘POLE’ had normal MMR IHC, we were unable to determine whether changing the order of molecular assessments, such that POLE EDM mutations were detected first, would be more informative.
Ultimately, the classifier model we have chosen to carry forward (Figure 3) is based on performance (survival analyses, Harrell’s C-index), practicality of methods and clinical utility. This classifier will be assessed, according to the Institute of Medicine guidelines (2012) for the development of ‘omics based tests, for confirmation in an independent sample set, then ultimately locked down for validation testing.
In addition to testing the classifier model in hysterectomy specimens we have commenced assessment in cases of matched endometrial biopsy or dilatation and curettage (D&C). Data from other series suggest that endometrial samplings (pipelle or D&C) are highly accurate (>97% sensitivity) at detecting cancer (Stovall et al, 1991), but grade and histotype may be discrepant with the final diagnoses based on examination of the hysterectomy specimens in up to one-half of cases (Francis et al, 2009; Karateke et al, 2011); in contrast, molecular parameters are highly concordant between biopsy and hysterectomy (Stelloo et al, 2014). If we can demonstrate equivalence of a molecular classification system in diagnostic endometrial samples and prognostic significance of a classifier, then women and their physicians could have valuable information that would help them guide decision making at the earliest time point in their cancer journey (e.g., at diagnosis). Decisions could be made before surgical staging regarding the urgency and extent of surgery, anticipated adjuvant therapy and follow-up plans. This information would be particularly helpful in guiding young women, with 14% of endometrial cancers arising in women <50 years of age and 5% in women younger than 40 (Burleigh et al, 2015) Consideration of fertility-sparing options or conservation of ovaries/hormonal function can be weighed against the risk of metastatic, or concurrent ovarian disease or worsened prognosis with deferred surgery.
Our goal is to improve upon the current system of clinicopathologic risk group stratification that is based on stage and the irreproducible variables of grade and histotype assignment (Fanning, 2001; Keys et al, 2004; Mariani et al, 2008; Kwon et al, 2009; Colombo et al, 2013; Kong et al, 2015), and is not highly predictive of outcomes (Bendifallah et al, 2015). Using the ESMO criteria (Colombo et al, 2013) we have demonstrated both in the TCGA data set and in the training set of cases from our centre that the clinicopathologic risk groups are not equivalent to the molecular subgroups identified. What is evident is the number of cases that would be considered ‘undertreated ‘or ‘overtreated’ depending on categorisation. For example, over one quarter of the CN high molecular subgroups were designated as low or intermediate risk and may have been undertreated, with subsequent recurrence and death. Half of the POLE molecular subgroup and MSI subgroups were identified as ‘high risk’ based on grade, stage and/or histotype. These women would have received chemotherapy and radiation based on our centres’ and consensus guidelines. The prognosis for women with POLE mutations is excellent, as observed across several series including our own (Cancer Genome Atlas Research N et al, 2013; Meng et al, 2014; Church et al, 2015). Whether that is because they received this aggressive treatment or is independent of this remains to be determined. It may be that the POLE ultramutated phenotype is exquisitely sensitive to therapy or, as has been suggested previously, has a higher immune infiltrate (Hussein et al, 2014) that may be further stimulated by the introduction of treatment(s). However, it may be that these women had an excellent prognosis, independent of treatment and received toxic therapies with long-term treatment side effects with no survival benefit.
Clinical risk groups were associated with outcomes in both TCGA and our own cohorts and should not be abandoned, but in terms of managing an individual the inconsistency of histotype and grade classification means that the same women may receive vastly different treatments depending on where her pathology is read. For example, a woman with a pathology report, indicating an endometrial high-grade serous cancer invading less than half her myometrial wall, with all other sites negative for disease (stage IA) receives systemic chemotherapy and pelvic radiation based on being high risk. That same woman whose pathology is interpreted at another centre as high-grade endometrioid endometrial cancer would receive vaginal vault radiation only (intermediate risk). Molecular classification adds prognostic information for these women and can directly impact care (e.g., referral for hereditary testing). It will likely prove to be more reproducible than histopathological assessment, but this needs to be formally evaluated. The combination of both clinical/pathologic parameters (either summarised as ESMO risk groups or taken separately, e.g., LVSI and grade) and molecular parameters appears to be an improvement upon either system alone (yields a higher C-index).
Limitations to this study include a relatively small sample size that did not allow us to definitively determine the optimal order of molecular testing. In addition, the distribution of mismatch repair deficient cases that also harbour POLE mutations varies in the literature (Billingsley et al, 2015; Church et al, 2015) and were rare in our small series; therefore, it remains uncertain how best to classify cases with both POLE EDM mutations and MMR IHC abn. Finally, our ‘training set’ of∼150 cases reported herein was a retrospective cohort with potential selection bias related to being drawn from a tertiary cancer treatment centre. We need to validate the utility of this molecular classification tool in a larger independent cohort of endometrial carcinomas. We are working towards the confirmation and validation of this pragmatic molecular classification, abiding by REMARK criteria and following the IOM guidelines (2012).
In summary, we have demonstrated that a set of simple assays, applicable to formalin-fixed paraffin-embedded samples, can reproduce the four TCGA genomically defined prognostic subgroups. These subgroups are associated with clinical outcomes, and identify women who may have a risk of recurrence of their EC that is very different than what is designated by traditional clinical risk group assessment. We see an opportunity to test this classifier across cancer centres and on preoperative endometrial samplings, thus influencing management from time of diagnosis. Independent of any prognostic ability, molecular classification has the ability to direct clinical care, such as referral to hereditary cancer programs for Lynch syndrome testing for abnormal MMR IHC. Molecular classification in ECs would also allow stratification of cases for clinical trials and assessment of treatment efficacy within specific molecular subgroups. This has been a game-changing approach in ovarian cancers (Kobel et al, 2008; Kurman and Shih Ie, 2011; Despierre et al, 2014) and has the potential to greatly advance progress in endometrial cancer research.
Change history
14 July 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Micheel CM, Nass SJ, Omenn, GS (eds) (2012) Evolution of Translational Omics: Lessons Learned and the Path Forward. Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials; Board on Health Care Services; Board on Health Sciences Policy; Institute of Medicine. National Academies Press: Washington (DC).
AlHilli MM, Mariani A, Bakkum-Gamez JN, Dowdy SC, Weaver AL, Peethambaram PP, Keeney GL, Cliby WA, Podratz KC (2014) Risk-scoring models for individualized prediction of overall survival in low-grade and high-grade endometrial cancer. Gynecol Oncol 133 (3): 485–493.
Efron B, Tibshirani R (1997) Improvement on cross-validation: the .632+ bootstrap method. J Am Stat Assoc 92 (438): 548–560.
Bendifallah S, Canlorbe G, Collinet P, Arsene E, Huguet F, Coutant C, Hudry D, Graesslin O, Raimond E, Touboul C, Darai E, Ballester M (2015) Just how accurate are the major risk stratification systems for early-stage endometrial cancer? Br J Cancer 112 (5): 793–801.
Bertagnolli MM, Niedzwiecki D, Compton CC, Hahn HP, Hall M, Damas B, Jewell SD, Mayer RJ, Goldberg RM, Saltz LB, Warren RS, Redston M (2009) Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 27 (11): 1814–1821.
Billingsley CC, Cohn DE, Mutch DG, Stephens JA, Suarez AA, Goodfellow PJ (2015) Polymerase varepsilon (POLE) mutations in endometrial cancer: Clinical outcomes and implications for Lynch syndrome testing. Cancer 121 (3): 386–394.
Burleigh A, Talhouk A, Gilks CB, McAlpine JN (2015) Clinical and pathological characterization of endometrial cancer in young women: identification of a cohort without classical risk factors. Gynecol Oncol in press.
Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, Zhang W, Mills GB, Kucherlapati R, Mardis ER, Levine DA (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497 (7447): 67–73.
Church DN, Stelloo E, Nout RA, Valtcheva N, Depreeuw J, ter Haar N, Noske A, Amant F, Tomlinson IP, Wild PJ, Lambrechts D, Jurgenliemk-Schulz IM, Jobsen JJ, Smit VT, Creutzberg CL, Bosse T (2015) Prognostic significance of POLE proofreading mutations in endometrial cancer. J Natl Cancer Inst 107 (1): 402.
Colombo N, Preti E, Landoni F, Carinelli S, Colombo A, Marini C, Sessa C Group EGW (2013) Endometrial cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 24 (Suppl 6): vi33–vi38.
Creutzberg CL, van Putten WL, Koper PC, Lybeert ML, Jobsen JJ, Warlam-Rodenhuis CC, De Winter KA, Lutgens LC, van den Bergh AC, van de Steen-Banasik E, Beerman H, van Lent M (2000) Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: multicentre randomised trial. PORTEC Study Group. Post Operative Radiation Therapy in Endometrial Carcinoma. Lancet 355 (9213): 1404–1411.
Despierre E, Yesilyurt BT, Lambrechts S, Johnson N, Verheijen R, van der Burg M, Casado A, Rustin G, Berns E, Leunen K, Amant F, Moerman P, Lambrechts D, Vergote I, Eortc GCG Group EGTR (2014) Epithelial ovarian cancer: rationale for changing the one-fits-all standard treatment regimen to subtype-specific treatment. Int J Gynecol Cancer 24 (3): 468–477.
Fanning J (2001) Long-term survival of intermediate risk endometrial cancer (stage IG3, IC, II) treated with full lymphadenectomy and brachytherapy without teletherapy. Gynecol Oncol 82 (2): 371–374.
Ferguson SE, Olshen AB, Viale A, Barakat RR, Boyd J (2005) Stratification of intermediate-risk endometrial cancer patients into groups at high risk or low risk for recurrence based on tumor gene expression profiles. Clin Cancer Res 11 (6): 2252–2257.
Francis JA, Weir MM, Ettler HC, Qiu F, Kwon JS (2009) Should preoperative pathology be used to select patients for surgical staging in endometrial cancer? Int J Gynecol Cancer 19 (3): 380–384.
Gilks CB, Oliva E, Soslow RA (2013) Poor interobserver reproducibility in the diagnosis of high-grade endometrial carcinoma. Am J Surg Pathol 37 (6): 874–881.
Grambsch PM, Therneau TM, Fleming TR (1995) Diagnostic plots to reveal functional form for covariates in multiplicative intensity models. Biometrics 51 (4): 1469–1482.
Guan H, Semaan A, Bandyopadhyay S, Arabi H, Feng J, Fathallah L, Pansare V, Qazi A, Abdul-Karim F, Morris RT, Munkarah AR, Ali-Fehmi R (2011) Prognosis and reproducibility of new and existing binary grading systems for endometrial carcinoma compared to FIGO grading in hysterectomy specimens. Int J Gynecol Cancer 21 (4): 654–660.
Han G, Sidhu D, Duggan MA, Arseneau J, Cesari M, Clement PB, Ewanowich CA, Kalloger SE, Kobel M (2013) Reproducibility of histological cell type in high-grade endometrial carcinoma. Modern Pathol 26 (12): 1594–1604.
Hoang LN, McConechy MK, Kobel M, Han G, Rouzbahman M, Davidson B, Irving J, Ali RH, Leung S, McAlpine JN, Oliva E, Nucci MR, Soslow RA, Huntsman DG, Gilks CB, Lee CH (2013) Histotype-genotype correlation in 36 high-grade endometrial carcinomas. Am J Surg Pathol 37 (9): 1421–1432.
Hussein YR, Weigelt B, Levine DA, Schoolmeester JK, Dao LN, Balzer BL, Liles G, Karlan B, Kobel M, Lee CH, Soslow RA (2014) Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Modern Pathol 28 (4): 505–514.
Karateke A, Tug N, Cam C, Selcuk S, Asoglu MR, Cakir S (2011) Discrepancy of pre- and postoperative grades of patients with endometrial carcinoma. Eur J Gynaecol Oncol 32 (3): 283–285.
Keys HM, Roberts JA, Brunetto VL, Zaino RJ, Spirtos NM, Bloss JD, Pearlman A, Maiman MA, Bell JG Gynecologic Oncology G (2004) A phase III trial of surgery with or without adjunctive external pelvic radiation therapy in intermediate risk endometrial adenocarcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 92 (3): 744–751.
Kobel M, Kalloger SE, Boyd N, McKinney S, Mehl E, Palmer C, Leung S, Bowen NJ, Ionescu DN, Rajput A, Prentice LM, Miller D, Santos J, Swenerton K, Gilks CB, Huntsman D (2008) Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med 5 (12): e232.
Kong TW, Chang SJ, Paek J, Lee Y, Chun M, Ryu HS (2015) Risk group criteria for tailoring adjuvant treatment in patients with endometrial cancer: a validation study of the Gynecologic Oncology Group criteria. J Gynecol Oncol 26 (1): 32–39.
Kurman RJ, Shih Ie M (2011) Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm. Hum Pathol 42 (7): 918–931.
Kwon JS, Qiu F, Saskin R, Carey MS (2009) Are uterine risk factors more important than nodal status in predicting survival in endometrial cancer? Obstet Gynecol 114 (4): 736–743.
Le Gallo M, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, Price JC, Zhang S, England BM, Godwin AK, Sgroi DC . Program NIHISCCS Hieter P, Mullikin JC, Merino MJ, Bell DW (2012) Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet 44 (12): 1310–1315.
Lu KH, Dinh M, Kohlmann W, Watson P, Green J, Syngal S, Bandipalliam P, Chen LM, Allen B, Conrad P, Terdiman J, Sun C, Daniels M, Burke T, Gershenson DM, Lynch H, Lynch P, Broaddus RR (2005) Gynecologic cancer as a "sentinel cancer" for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol 105 (3): 569–574.
Mariani A, Dowdy SC, Cliby WA, Gostout BS, Jones MB, Wilson TO, Podratz KC (2008) Prospective assessment of lymphatic dissemination in endometrial cancer: a paradigm shift in surgical staging. Gynecol Oncol 109 (1): 11–18.
McConechy MK, Ding J, Cheang MC, Wiegand KC, Senz J, Tone AA, Yang W, Prentice LM, Tse K, Zeng T, McDonald H, Schmidt AP, Mutch DG, McAlpine JN, Hirst M, Shah SP, Lee CH, Goodfellow PJ, Gilks CB, Huntsman DG (2012) Use of mutation profiles to refine the classification of endometrial carcinomas. J Pathol 228 (1): 20–30.
McConechy MK, Talhouk A, Li-Chang HH, Leung S, Huntsman DG, Gilks CB, McAlpine JN (2015) Detection of DNA mismatch repair (MMR) deficiencies by immunohistochemistry can effectively diagnose the microsatellite instability (MSI) phenotype in endometrial carcinomas. Gynecol Oncol 137 (2): 306–310.
Meng B, Hoang LN, McIntyre JB, Duggan MA, Nelson GS, Lee CH, Kobel M (2014) POLE exonuclease domain mutation predicts long progression-free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol Oncol 134 (1): 15–19.
Murali R, Soslow RA, Weigelt B (2014) Classification of endometrial carcinoma: more than two types. Lancet Oncol 15 (7): e268–e278.
Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM, Raeder MB, Sos ML, Engelsen IB, Trovik J, Wik E, Greulich H, Bo TH, Jonassen I, Thomas RK, Zander T, Garraway LA, Oyan AM, Sellers WR, Kalland KH, Meyerson M, Akslen LA, Beroukhim R (2009) Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci USA 106 (12): 4834–4839.
Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, Zaniboni A, Seitz JF, Sinicrope F, Gallinger S (2010) Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 28 (20): 3219–3226.
Schemper M, Smith TL (1996) A note on quantifying follow-up in studies of failure time. Control Clin Trial 17 (4): 343–346.
Sheikh MA, Althouse AD, Freese KE, Soisson S, Edwards RP, Welburn S, Sukumvanich P, Comerci J, Kelley J, LaPorte RE, Linkov F (2014) USA Endometrial Cancer Projections to 2030: should we be concerned? Future Oncol 10 (16): 2561–2568.
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA 65 (1): 5–29.
Sinicrope FA, Foster NR, Thibodeau SN, Marsoni S, Monges G, Labianca R, Kim GP, Yothers G, Allegra C, Moore MJ, Gallinger S, Sargent DJ (2011) DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst 103 (11): 863–875.
Society CC Canadian Cancer Statistics (2014) http://www.cancer.ca/~/media/cancer.ca/CW/cancer%20information/cancer%20101/Canadian%20cancer%20statistics/Canadian-Cancer-Statistics-2014-EN.pdf.
Stelloo E, Bosse T, Nout RA, MacKay HJ, Church DN, Nijman HW, Leary A, Edmondson RJ, Powell ME, Crosbie EJ, Kitchener HC, Mileshkin L, Pollock PM, Smit VT, Creutzberg CL (2015) Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Modern Pathol e-pub ahead of print 27 February 2015; doi:10.1038/modpathol.2015.43.
Stelloo E, Nout RA, Naves LC, Ter Haar NT, Creutzberg CL, Smit VT, Bosse T (2014) High concordance of molecular tumor alterations between pre-operative curettage and hysterectomy specimens in patients with endometrial carcinoma. Gynecol Oncol 133 (2): 197–204.
Steyerberg EW, Harrell FE Jr, Borsboom GJ, Eijkemans MJ, Vergouwe Y, Habbema JD (2001) Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. J Clin Epidemiol 54 (8): 774–781.
Stovall TG, Photopulos GJ, Poston WM, Ling FW, Sandles LG (1991) Pipelle endometrial sampling in patients with known endometrial carcinoma. Obstet Gynecol 77 (6): 954–956.
R Core Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria http://www.R-project.org/.
Acknowledgements
We are grateful for the work of Christine Chow, Margaret Luk and Ying Ng as well as support received from the Sarabjit Gill Fund, BC Cancer Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Talhouk, A., McConechy, M., Leung, S. et al. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer 113, 299–310 (2015). https://doi.org/10.1038/bjc.2015.190
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.190
Keywords
This article is cited by
-
Molecular-based classification of endometrial carcinoma in Northern Thailand: impact on prognosis and potential for implementation in resource-limited settings
BMC Women's Health (2023)
-
Retrospective analysis of the 18F-FDG PET/CT cutoff value for metabolic parameters was performed as a prediction model to evaluate risk factors for endometrial cancer
Radiation Oncology (2023)
-
CXCR3 predicts the prognosis of endometrial adenocarcinoma
BMC Medical Genomics (2023)
-
The impact of lymphovascular space invasion on survival in early stage low-grade endometrioid endometrial cancer
European Journal of Medical Research (2023)
-
Alteration in molecular properties during establishment and passaging of endometrial carcinoma patient-derived xenografts
Scientific Reports (2023)
