
Abstract
Background:
The epidermal growth factor receptor (EGFR) is overexpressed in colorectal cancer (CRC), and is correlated with poor prognosis, making it an attractive target for monoclonal antibody (mAb) therapy. A component of the therapeutic efficacy of IgG1 mAbs is their stimulation of antibody-dependent cellular cytotoxicity (ADCC) by natural killer (NK) cells bearing the CD16 receptor. As NK cells are functionally impaired in cancer patients and may be further compromised upon chemotherapy, it is crucial to assess whether immunotherapeutic strategies aimed at further enhancing ADCC are viable.
Methods:
CRC patients before, during and after chemotherapy were immunophenotyped by flow cytometry for major white blood cell populations. ADCC-independent NK cell functionality was assessed in cytotoxicity assays against K562 cells. ADCC-dependent killing of EGFR+ A431 cancer cells by NK cells was measured with a degranulation assay where ADCC was induced by GA201, an anti-EGFR mAb glyco-engineered to enhance ADCC.
Results:
Here, we confirm the observation that NK cells in cancer patients are dysfunctional. However, GA201 was able to induce robust NK cell-dependent cytotoxicity in CRC patient NK cells, effectively overcoming their impairment.
Conclusions:
These findings support the evaluation of the therapeutic potential of GA201 in combination with chemotherapy in CRC patients.
Similar content being viewed by others


Main
Colorectal cancer (CRC) is the second most frequent cause of cancer death in the western world. In the United States alone, the American Cancer Society estimates more than 140 000 new cases and 50 000 deaths for 2013. Overall, the lifetime risk of developing CRC is 5.1% (American Cancer Society, 2013). A modest increase in average survival can be achieved with current therapeutic strategies, yielding 10–12 months with single agent 5-fluorouracil and increasing to 16–20 months upon addition of oxaliplatin and irinotecan, compared with 6 months with palliative care alone. When administered as a first-line treatment, combination chemotherapy can substantially improve response rates (31%–56%), median progression-free survival (6.5–9 months) and median overall survival (14.5–21.4 months) (Giacchetti et al, 1999; de Gramont et al, 2000; Douillard, 2000; Saltz, 2000; Goldberg et al, 2004; Grothey and Goetz, 2004; Tournigand et al, 2004; Kohne et al, 2005; Laurent-Puig et al, 2009; Maughan et al, 2011). Although these results are encouraging, much remains to be achieved in CRC therapy.
Small-molecule drugs have been joined by innovative biologics in the treatment of CRC. This is the case, for instance, of the anti-epidermal growth factor receptor (EGFR) monoclonal antibodies (mAbs) cetuximab and panitumumab. EGFR is a growth factor receptor affecting cell adhesion, survival and proliferation that is overexpressed in 75% of CRC and correlated with poor prognosis (Nicholson et al, 2001; Giacomelli et al, 2003; Azria et al, 2005; Spano et al, 2005; Markman et al, 2010). By blocking ligand binding to EGFR, cetuximab and panitumumab prevent mitogenic signalling, ultimately hampering cell division. Intimately connected to their mechanism of action, the benefit of anti-EGFR mAbs is bypassed by gain-of-function mutations in KRAS, a signalling molecule downstream of EGFR (Amado et al, 2008; Karapetis et al, 2008; Bokemeyer et al, 2009; Van Cutsem et al, 2009). Indeed, in subjects with KRAS-mutant tumours, the proliferative signal is constitutively active and therefore unaffected by receptor blockade (Ciardiello and Tortora, 2008). However, mAbs can be further optimised for greater therapeutic potential. In addition to being highly specific inhibitors, antibodies are naturally endowed with several immunomodulatory properties. Such properties may be exploited to achieve immunotherapeutic effects, in synergy with receptor blockade and possibly in combination with chemotherapy.
The immune system naturally monitors and continuously eliminates nascent transformed cells, a realisation that dates back to at least 1957 (Burnet, 1957). Integral to this process of immune surveillance is the natural killer (NK) cell, a type of leukocyte that has a major role in the rejection of tumours. NK cells kill target cells by releasing small granules of apoptosis-inducing and lytic proteins, such as granzymes and perforin (Vivier et al, 2008). NK cells are lymphocytes identified by the surface marker CD56 (in humans) and by the lack of T- and B-cell antigen receptors or lineage markers, such as CD3. Most NK cells express CD16 (FcγRIIIa), an activating low-affinity receptor for the Fc domain of IgG-isotype antibodies. Through CD16, NK cells recognise IgG-coated cells and promptly kill them, a process called antibody-dependent cell-mediated cytotoxicity (ADCC). Not all IgG subclasses are equally able to elicit ADCC. For instance, IgG1 antibodies are more ADCC-efficient than IgG2 (Nimmerjahn and Ravetch, 2005; Schneider-Merck et al, 2010).
Therefore, it comes as no surprise that one component of the therapeutic efficacy of IgG1 mAbs is immunological. Indeed, a common single-nucleotide polymorphism in CD16 (V158F) is correlated with clinical responses to cetuximab (Zhang et al, 2007) and the anti-CD20 mAb rituximab (Cartron et al, 2002). Mechanistically, this polymorphism was found to modulate the affinity of CD16 for antibodies, in turn affecting the potency of ADCC (Taylor et al, 2009). These observations, connecting antibody/Fc receptor affinity, ADCC efficiency and clinical outcomes, are crucial, as they lead to the prediction that enhancements of mAb affinity for Fc receptors may result in significant clinical benefits. One way to increase the binding between Fc receptors and mAbs is to manipulate protein glycosylation, which is known to affect protein–protein interactions. Pioneering work demonstrated that antibodies containing engineered bisected, afucosylated oligosaccharides in their Fc domain show greatly increased affinity for CD16, ultimately resulting in ADCC amplification (Umana et al, 1999; Ferrara et al, 2006, 2011). Importantly, glyco-engineering improves the therapeutic potential of mAbs in both low (158F) and high (158 V) affinity variants of CD16 (Wu et al, 1997; Mossner et al, 2010; Gerdes et al, 2013). Moreover, this therapeutic mechanism is likely to be important when simple interference with receptor/ligand interactions fails as a therapeutic strategy, as in the case of EGFR/EGF inhibition in tumours with activating KRAS mutations (Gerdes et al, 2013).
For ADCC-based immunotherapy of cancer patients to be effective, it is essential that pivotal immune effector cells, such as NK cells, are present and have sufficient functional capacity. However, treatments inducing leukopenia, such as chemotherapy, may negatively impact on the immunotherapeutic contribution of mAbs. Understanding whether ADCC-eliciting antibodies retain effectiveness even in combination with chemotherapy is critical to pave the way to novel therapeutic strategies against cancer. This is even more compelling in the light of recent findings calling into question the clinical efficacy of the IgG1 mAb cetuximab in synergy with oxaliplatin-containing chemotherapy (Maughan et al, 2011), in contrast to earlier positive reports with both FOLFIRI and FOLFOX-4 (Bokemeyer et al, 2009; Van Cutsem et al, 2009).
Therefore, we studied NK cell cytotoxic responses (ADCC and also CD16-independent responses) in metastatic CRC patients before oxaliplatin/irinotecan-containing chemotherapy, on active therapy or following two lines of standard treatment. ADCC was induced using a novel glyco-engineered anti-EGFR mAb, GA201 (RG7160), which showed superior potency compared with cetuximab (Gerdes et al, 2013), and is currently in phase I/II clinical trials for colorectal and head and neck cancers. We found that, despite defects in CD16-independent cytotoxic functions, NK cell numbers and ADCC were largely preserved in patients, regardless of their therapeutic stage, giving full scope to combining chemotherapy and ADCC-triggering immunotherapy.
Materials and methods
Patient samples
Blood samples were drawn following informed consent for study from age-matched healthy controls at disease progression or colorectal carcinoma patients at presentation with metastatic disease (met CRC), before therapy; on active chemotherapy, 2–3 weeks post recent cycle of oxaliplatin-based (FOLFOX/CAPOX) or irinotecan-based (FOLFIRI/single-agent/+Capecitabine) regimen before cycles 2–11 of first or second line; or at disease progression, after at least 4 weeks since failure of two lines of standard treatment. An aliquot of blood was used for direct immunophenotyping. Peripheral blood mononuclear cells (PBMCs) were isolated using the standard Ficoll density gradient and cryopreserved in freezing medium (10% DMSO, 40% FBS, 50% Cellgro SCGM).
Immunophenotyping and cell counts
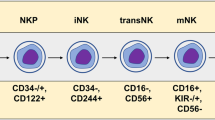
The Multitest IMK kit with Trucount beads (BD Biosciences, San Diego, CA, USA) was used to enumerate mature human T cells (CD3+), helper/inducer T cells (CD3+ CD4+), suppressor/cytotoxic T cells (CD3+ CD8+), B cells (CD19+) and NK cells (CD3− CD56+/CD16+) directly in unprocessed whole blood. Deeper phenotyping of NK cells requires accurate measurement of CD16. In our pilot experiments, we found that CD16 is not readily detectable in whole blood, whereas it is after removing plasma, probably due to the binding of CD16 by the Fc domain of antibodies present in the serum. Consequently, we immunophenotyped NK cells for CD16 in freshly isolated PBMCs.
Functional assays of NK cell cytotoxicity
Cryopreserved PBMCs were thawed and rested for 16 h in Cellgro SCGM serum-free medium (Cellgenix, Freiburg, Germany) supplemented with 50 IU ml−1 recombinant human lL-2 (Peprotech, Rocky Hill, NJ, USA) before use in functional assays.
Direct NK cell cytotoxicity was measured in a standard assay against the erythroleukaemia cell line K562 (NKTEST, Glycotope, Berlin, Germany). Briefly, CFSE-prelabelled K562 cells (provided by the manufacturer) were incubated with donor PBMCs at different effector:target ratios (6 : 1, 12 : 1 and 25 : 1). After 4 h, killed target cells were identified by uptake of the fluorescent DNA stain 7-aminoactinomycin D, which can only penetrate dead cells through compromised plasma membranes. Results were adjusted for the rate of spontaneous cell death (in the absence of effector cells) and for the NK cell frequency in thawed PBMCs as determined by flow cytometry. Note that it is well known that NK cells are the only population within PBMCs mediating K562 cell killing.
To induce ADCC in NK cells, we co-incubated donor PBMCs with the EGFR+ epidermoid carcinoma cell line A431 (obtained from ATCC, Manassas, VA, USA) at 1 : 1 ratio in the presence of 10 μg ml−1 GA201 anti-EGFR antibody. Because EGFR-positive cells grow as adherent cultures, a degranulation assay is more suited to test the NK cell cytotoxic function than the NKTEST. To detect degranulation, an APC-conjugated anti-CD107a antibody was added to the co-culture at 1 μg ml−1. After 1 h, the protein transport-inhibitor monensin (0.1 mM) was added to prevent CD107a endosomal recycling and improve the CD107a staining, as per the standard protocols (Alter et al, 2004). After an additional 2 h, cells were harvested, stained for CD56 and CD3 and acquired with a FACSCanto II flow cytometer (BD Biosciences). Degranulating NK cells were identified as CD3− CD56+ CD107a+.
Statistical analysis
Data were analysed with Prism 6 (Graphpad, San Diego, CA, USA) and R 3.0.1 (http://cran.r-project.org/bin/windows/base/old/3.0.1/) using the packages lme4, ez, gmodels and multcomp.
When few patient cohorts were compared with a single control group (Figures 1 and 2), a Fisher’s LSD test was performed. When only a meaningful subset of pairwise comparisons was selected for testing (Figure 3), a Sidak test was performed. P-values were coded with asterisks.

Counts of white blood cells, lymphocytes and subsets in peripheral blood. (A) Blood cell counts. Total white cell (top) or lymphocyte (bottom) counts per microlitre. (B) Lymphocyte subset counts per microlitre of blood. Cohorts are: age-matched healthy controls; and CRC patients at presentation with metastatic disease, on active chemotherapy or second-line chemotherapy failures. Mean and s.d. are shown in red. Patient cohorts were contrasted to the healthy control group using a Fisher’s LSD test. *P<0.05. All other comparisons did not reveal significant differences.

NK cell frequency and counts in peripheral blood. (A) Frequency in lymphocytes and (B) absolute counts of CD56/CD16+ CD3− NK cells in age-matched healthy controls, CRC patients at presentation with metastatic disease, on active chemotherapy or second-line chemotherapy failures. (C) Percentage of CD16+ in CD56+ CD3− NK cells. Mean and s.d. are shown in red. Patient cohorts were contrasted to the healthy control group using a Fisher’s LSD test. No comparisons revealed significant differences.

ADCC by NK cells triggered by anti-EGFR monoclonal antibodies. (A) Representative plots of degranulating (CD107a+) NK cells (CD3− CD56+) from a healthy donor. ADCC was triggered by anti-EGFR antibodies in the presence of EGFR+ A431 target cells. (B) Percentage of CD107a+ NK cells in cohorts (age-matched healthy controls, CRC patients at presentation with metastatic disease, on active chemotherapy or second-line chemotherapy failures). Mean and s.d. are shown in red. Comparisons were tested for statistical significance using a Sidak test. ****P<0.0001; ***P<0.001; NS, not significant.
CD16-independent NK cell cytotoxic responses (Figure 4) were fitted to a linear mixed effect model. The subject ID was modelled as a random effect, while log-transformed NK:Target ratio and patient cohort were modelled as fixed effects. The conditional coefficient of determination was calculated according to Nakagawa and Schielzeth (2013). Pairwise contrasts of intercepts or slopes between healthy controls and each patient cohort were tested for significance, with P-values adjusted for multiple comparisons using the single-step method.

CD16-independent NK cell cytotoxicity. (A) Representative plots of NK cell cytotoxic assays in healthy donors, measuring 7-AAD uptake by CFSE+ K562 target cells in the absence (background) or in the presence of increasing numbers of NK cells. (B) Top, specific killing as a function of effector:target ratio normalised for NK cell frequency within PBMCs for each subject. Cohorts are: age-matched healthy controls; and CRC patients at presentation with metastatic disease, on active chemotherapy or second-line chemotherapy failures. Bottom, linear mixed effect model (R2=0.90), split in panels by cohort. The predicted activity is shown with 95% confidence intervals. Patient cohorts were contrasted to healthy donors. *Significant difference of slope at α=0.05.
Results
CRC patients show a largely intact immune system regardless of chemotherapy stage
We immunophenotyped age-matched healthy controls and CRC patients (pre-chemotherapy, undergoing chemotherapy and second-line failure post chemotherapy) by flow cytometry. The numbers of white blood cells (WBCs) and lymphocytes were comparable in healthy donors and patients (Figure 1A), with the exception of patients on active chemotherapy, who showed reduced WBC counts, as expected. When lymphocytes were segregated into subsets, we found comparable counts for B cells (CD19+), NKT cells (CD56+ CD3+) and T cells, either unsegregated or split into subsets (CD4+, CD8+ or double negative), across all cohorts of healthy donors and patients (Figure 1B).
Then, we focused on NK cells. The percentage of NK cells in lymphocytes was comparable in healthy donors and patients (Figure 2A). Similarly, even if actual NK cell counts appeared slightly reduced in all the patient cohorts, no statistically significant differences were found (Figure 2B). In addition, NK cell expression of CD16, the major receptor mediating ADCC, was preserved in patients (Figure 2C).
Patients on before and post chemotherapy, but not on active chemotherapy, show impaired CD16-independent NK cell-mediated cytotoxicity
To assess the functional potential of NK cells in patients, we first investigated whether antibody-independent cytotoxicity was intact by measuring the ability of NK cells to kill the classical target cells K562 (Figure 4A). NK cell cytotoxicity against the erythroleukaemic K562 cells is not driven by CD16 but by a variety of activating receptors, such as NKG2D (Chen et al, 2007). As shown in Figure 4B, pre- and post-chemotherapy patients exhibited impaired killing capacity when compared with controls. Conversely, patients actively undergoing chemotherapy retained their killing capacity.
ADCC responses to GA201 were robust in healthy donors as well as in all the patient cohorts
CD16-dependent cytotoxic function (ADCC) of NK cells was assessed by measuring the accumulation of CD107a (LAMP-1) from cytolytic granules at the cell surface, as a result of degranulation. The EGFR+ epidermoid carcinoma cell line A431 targeted by the anti-EGFR mAb GA201 was used to trigger ADCC (Figure 3A).
In both healthy donors and all patient cohorts, GA201 was able to induce robust NK cell degranulation, whereas the IgG2a mAb panitumumab was ineffective (Figure 3B). Importantly, ADCC in pre-chemotherapy patients was as efficient as in patients actively undergoing chemotherapy (Figure 3B). This suggests that the impairment of patients’ NK cells noted above (Figure 4B) affects only CD16-independent functions, sparing ADCC capabilities, which are central to immunotherapeutic approaches based on glyco-engineered mAbs. Moreover, while the magnitude of responses of failures to two lines of chemotherapy was lower than those of patients on pre-chemotherapy or on chemotherapy, it was still substantially high in absolute terms.
Altogether, these findings indicate that immunotherapeutic approaches exploiting ADCC in CRC patients may be viable even in synergy with chemotherapy.
Discussion
A large body of literature has demonstrated an accumulation of inflammatory cells in the direct vicinity of solid tumours. Several studies found that the infiltration of NK cells in malignant tumours was associated with a favourable outcome (Coca et al, 1997; Ishigami et al, 2000; Villegas et al, 2002; Menon et al, 2004). Others highlighted that colorectal tumours were only sparsely infiltrated by NK cells compared with cytotoxic T cells, despite loss of MHC class I expression by the tumour cells (Sandel et al, 2005). This was unusual in that NK cells, but not cytotoxic T cells, are in charge of clearing MHC class I-negative targets (Raulet, 2006), leading the authors to speculate that NK cell-escaping tumor variants had been selected. As such, they suggest that an NK cell-centric immune therapy approach may restore the contribution of the immune system to tumour surveillance and control (Sandel et al, 2005).
ADCC-inducing mAbs precisely leverage NK cell cytotoxic abilities. Most NK cells express high levels of the activating Fcγ receptor CD16, and we found this not to be impaired in patients with metastatic CRC, either on or after chemotherapy. NK cell levels themselves were, at most, only marginally affected (if at all) by disease and/or therapy. This is of particular importance because if NK cells were compromised by chemotherapy, then the benefit provided by mAbs given in combination therapies would be limited to the blockade of receptor signalling. Not only were NK cell levels normal but we also found that CD16-dependent cytotoxicity was largely intact in patients’ NK cells, as demonstrated by the ability of the ADCC-capable GA201 antibody to elicit degranulation in a substantial fraction of NK cells. This contrasts with the observation that CD16-independent natural cytotoxicity was impaired in pre- and post-chemotherapy patients (but not in patients actively undergoing chemotherapy, possibly because chemotherapy-induced inflammation may have had a stimulatory adjuvant effect on NK cells). It is entirely conceivable that CD16-dependent and -independent functions are differentially impacted upon by disease and/or therapy (Levy et al, 2011). However, it may also be the case that the strong stimulation provided by glyco-engineered antibodies is able to overcome, at least to some degree, the impaired NK cell function, whereas the lower magnitude of physiological activation triggered by K562 cells could be inadequate in rescuing the functionally compromised cells. Whatever may be the case, ultimately the data presented show that NK cells can be targeted by immunotherapeutic strategies.
The present study builds upon and extends previous important observations. First, it is established that GA201 elicits amplified NK cell-mediated ADCC when compared with non-engineered antibodies, such as cetuximab (Gerdes et al, 2013). Second, such ADCC enhancement has the potential to benefit those patients in whom cetuximab is either partially or totally ineffective, such as carriers of low-affinity CD16 polymorphism or KRAS gain-of-function mutations, respectively (Wu et al, 1997; Mossner et al, 2010; Gerdes et al, 2013). The failure of cetuximab to benefit patients with KRAS-mutated tumours (Lievre et al, 2006) suggests that natural unmanipulated ADCC cannot act as a failsafe mechanism when receptor blocking is not a viable therapeutic option, providing further grounds to artificially enhancing ADCC via bioengineering. Indeed, GA201 activity is robust irrespective of the genetics of CD16 and KRAS (Gerdes et al, 2013). Furthermore, we now show that GA201 is able to elicit NK cell activation in patients, regardless of therapeutic stage (pre-treatment, on active chemotherapy and following second-line failure post chemotherapy). Our findings strongly support the need for evaluation of enhanced-ADCC therapies in clinical trials of CRC patients following two lines of chemotherapy and possibly even patients on active chemotherapy.
Although we focused on anti-EGFR mAbs against CRC, in principle our approach may be generalised. Indeed, a number of malignancies are currently being targeted by mAb therapies, from breast cancer (trastuzumab, anti-HER2/neu) to B-cell lymphomas (rituximab, anti-CD20). Endowing ligand-blocking mAbs with enhanced ADCC-triggering capabilities in such diseases may prove beneficial. For instance, in the first stage of a phase 3 study, obinutuzumab/GA101, a type 2 anti-CD20 antibody glyco-engineered using the same strategy behind GA201, showed improvement of progression-free survival in people with chronic lymphocytic leukaemia (Roche, 2013). However, as mAb manipulation becomes increasingly common, it brings the need of pre-emptively assessing NK cell functionality in the particular disease under scrutiny, similarly to our present investigation in the context of anti-EGFR therapy of CRC. This is even more important because it is current clinical practice to combine chemotherapy with mAbs. As biotechnology redefines our current notion of drugs, classical clinical studies of effectiveness will increasingly need to be accompanied by research focusing on the underlying biological mechanism to provide informative feedback supporting clinical decisions and/or technological refinement.
References
Alter G, Malenfant JM, Altfeld M (2004) CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods 294 (1-2): 15–22.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26 (10): 1626–1634.
American Cancer Society (2013) What are the key statistics about colorectal cancer? Available at: http://www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-key-statistics American Cancer Society: Atlanta, GA.
Azria D, Bibeau F, Barbier N, Zouhair A, Lemanski C, Rouanet P, Ychou M, Senesse P, Ozsahin M, Pelegrin A, Dubois JB, Thezenas S (2005) Prognostic impact of epidermal growth factor receptor (EGFR) expression on loco-regional recurrence after preoperative radiotherapy in rectal cancer. BMC Cancer 5: 62.
Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P (2009) Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 27 (5): 663–671.
Burnet M (1957) Cancer; a biological approach. I. The processes of control. Br Med J 1 (5022): 779–786.
Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H (2002) Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99 (3): 754–758.
Chen X, Trivedi PP, Ge B, Krzewski K, Strominger JL (2007) Many NK cell receptors activate ERK2 and JNK1 to trigger microtubule organizing center and granule polarization and cytotoxicity. Proc Natl Acad Sci USA 104 (15): 6329–6334.
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358 (11): 1160–1174.
Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez M, Vallejo C, Martos J, Moreno M (1997) The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 79: 2320–2328.
de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer G, Papamichael D, Le Bail N, Louvet C, Hendler D, de Braud F, Wilson C, Morvan F, Bonetti A (2000) Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 18 (16): 2938–2947.
Douillard JY (2000) Irinotecan and high-dose fluorouracil/leucovorin for metastatic colorectal cancer. Oncology (Williston Park) 14 (12 Suppl 14): 51–55.
Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, Umana P, Benz J (2011) Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA 108 (31): 12669–12674.
Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P (2006) The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem 281 (8): 5032–5036.
Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, Moessner E, Freytag O, Friess T, Ries CH, Bossenmaier B, Mueller HJ, Umana P (2013) GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res 19 (5): 1126–1138.
Giacchetti S, Itzhaki M, Gruia G, Adam R, Zidani R, Kunstlinger F, Brienza S, Alafaci E, Bertheault-Cvitkovic F, Jasmin C, Reynes M, Bismuth H, Misset JL, Levi F (1999) Long-term survival of patients with unresectable colorectal cancer liver metastases following infusional chemotherapy with 5-fluorouracil, leucovorin, oxaliplatin and surgery. Ann Oncol 10 (6): 663–669.
Giacomelli L, Gianni W, Belfiore C, Gandini O, Repetto L, Filippini A, Frati L, Agliano AM, Gazzaniga P (2003) Persistence of epidermal growth factor receptor and interleukin 10 in blood of colorectal cancer patients after surgery identifies patients with high risk to relapse. Clin Cancer Res 9 (7): 2678–2682.
Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, Alberts SR (2004) A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 22 (1): 23–30.
Grothey A, Goetz MP (2004) Oxaliplatin plus oral fluoropyrimidines in colorectal cancer. Clin Colorectal Cancer 4 (Suppl 1): S37–S42.
Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, Aridome K, Hokita S, Aikou T (2000) Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 88: 577–583.
Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359 (17): 1757–1765.
Kohne CH, van Cutsem E, Wils J, Bokemeyer C, El-Serafi M, Lutz MP, Lorenz M, Reichardt P, Ruckle-Lanz H, Frickhofen N, Fuchs R, Mergenthaler HG, Langenbuch T, Vanhoefer U, Rougier P, Voigtmann R, Muller L, Genicot B, Anak O, Nordlinger B (2005) Phase III study of weekly high-dose infusional fluorouracil plus folinic acid with or without irinotecan in patients with metastatic colorectal cancer: European Organisation for Research and Treatment of Cancer Gastrointestinal Group Study 40986. J Clin Oncol 23 (22): 4856–4865.
Laurent-Puig P, Lievre A, Blons H (2009) Mutations and response to epidermal growth factor receptor inhibitors. Clin Cancer Res 15 (4): 1133–1139.
Levy EM, Roberti MP, Mordoh J (2011) Natural killer cells in human cancer: from biological functions to clinical applications. J Biomed Biotechnol 2011: 676198.
Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66 (8): 3992–3995.
Markman B, Javier Ramos F, Capdevila J, Tabernero J (2010) EGFR and KRAS in colorectal cancer. Adv Clin Chem 51: 71–119.
Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, Idziaszczyk S, Harris R, Fisher D, Kenny SL, Kay E, Mitchell JK, Madi A, Jasani B, James MD, Bridgewater J, Kennedy MJ, Claes B, Lambrechts D, Kaplan R, Cheadle JP (2011) Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet 377 (9783): 2103–2114.
Menon A, Janssen-van Rhijn C, Morreau H, Putter H, Tollenaar R, van de Velde C, Leuren G, Kuppen P (2004) Immune system and prognosis in colorectal cancer: a detailed immunohistochemical analysis. Lab Invest 84: 493–501.
Mossner E, Brunker P, Moser S, Puntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, van Puijenbroek E, Ferrara C, Sondermann P, Jager C, Strein P, Fertig G, Friess T, Schull C, Bauer S, Dal Porto J, Del Nagro C, Dabbagh K, Dyer MJ, Poppema S, Klein C, Umana P (2010) Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 115 (22): 4393–4402.
Nakagawa S, Schielzeth H (2013) A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol Evol 4 (2): 133–142.
Nicholson RI, Gee JM, Harper ME (2001) EGFR and cancer prognosis. Eur J Cancer 37 (Suppl 4): S9–15.
Nimmerjahn F, Ravetch JV (2005) Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 310 (5753): 1510–1512.
Raulet DH (2006) Missing self recognition and self tolerance of natural killer (NK) cells. Semin Immunol 18 (3): 145–150.
Roche (2013) Obinutuzumab (GA101) significantly improved progression-free survival in people with chronic lymphocytic leukemia (CLL). Available at: http://www.roche.com/media/media_releases/med-cor-2013-01-31.htm Roche: Basel, Switzerland.
Saltz L (2000) Irinotecan-based combinations for the adjuvant treatment of stage III colon cancer. Oncology (Williston Park) 14 (12 Suppl 14): 47–50.
Sandel MH, Speetjens FM, Menon AG, Albertsson PA, Basse PH, Hokland M, Nagelkerke JF, Tollenaar RA, van de Velde CJ, Kuppen PJ (2005) Natural killer cells infiltrating colorectal cancer and MHC class I expression. Mol Immunol 42 (4): 541–546.
Schneider-Merck T, Lammerts van Bueren JJ, Berger S, Rossen K, van Berkel PH, Derer S, Beyer T, Lohse S, Bleeker WK, Peipp M, Parren PW, van de Winkel JG, Valerius T, Dechant M (2010) Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol 184 (1): 512–520.
Spano JP, Lagorce C, Atlan D, Milano G, Domont J, Benamouzig R, Attar A, Benichou J, Martin A, Morere JF, Raphael M, Penault-Llorca F, Breau JL, Fagard R, Khayat D, Wind P (2005) Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol 16 (1): 102–108.
Taylor RJ, Chan SL, Wood A, Voskens CJ, Wolf JS, Lin W, Chapoval A, Schulze DH, Tian G, Strome SE (2009) FcgammaRIIIa polymorphisms and cetuximab induced cytotoxicity in squamous cell carcinoma of the head and neck. Cancer Immunol Immunother 58 (7): 997–1006.
Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, Landi B, Colin P, Louvet C, de Gramont A (2004) FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 22 (2): 229–237.
Umana P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE (1999) Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 17 (2): 176–180.
Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360 (14): 1408–1417.
Villegas F, Coca S, Villarrubia V, Jiminez R, Chillon M, Jareno J, Zuil M, Callol L (2002) Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 35: 23–28.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S (2008) Functions of natural killer cells. Nat Immunol 9 (5): 503–510.
Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, Salmon JE, Kimberly RP (1997) A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest 100 (5): 1059–1070.
Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, Chang HM, Borucka E, Lurje G, Sherrod AE, Iqbal S, Groshen S, Lenz HJ (2007) FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol 25 (24): 3712–3718.
Acknowledgements
We are grateful to Simon Hollingsworth, current affiliation AstraZeneca, UK for contributions to the study design and biomarker program; Luigi Manenti and the Roche Glycart pre-clinical team for providing GA201 and very helpful discussions. Special thanks to Ria Boot, Samantha Barrett, Juel Tuazon, Ileana Samidis, Kylie Gyertson and Oncology Research Nurses at Guy’s and St Thomas’ Hospital NHS Trust, London. We are indebted to Linda Barber for her help in the setting up of flow cytometry-based assays. Research support: We acknowledge financial support from Roche and from the Department of Health, including a post-doctoral fellowship for DEO, via the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) award to Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. These studies were also supported by infrastructure support from NIHR through a BRC award to the Institute of Psychiatry in partnership with King’s College Hospital NHS Trust and King’s College London. Work in the FF laboratory is also supported by Rosetrees Trust, Elimination of Leukaemia Fund, the Experimental Cancer Medicine Centre at King’s College London and Leukaemia Lymphoma Research in UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Oppenheim, D., Spreafico, R., Etuk, A. et al. Glyco-engineered anti-EGFR mAb elicits ADCC by NK cells from colorectal cancer patients irrespective of chemotherapy. Br J Cancer 110, 1221–1227 (2014). https://doi.org/10.1038/bjc.2014.35
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.35
Keywords
This article is cited by
-
The role of natural killer cell in gastrointestinal cancer: killer or helper
Oncogene (2021)
-
Detecting intratumoral heterogeneity of EGFR activity by liposome-based in vivo transfection of a fluorescent biosensor
Oncogene (2017)
-
Current controversies in the management of metastatic colorectal cancer
Cancer Chemotherapy and Pharmacology (2015)
