
Abstract
A single somatic mutation, V617F, in Janus kinase 2 (JAK2) is one of the causes of myeloproliferative neoplasms (MPNs), including primary myelofibrosis, and the JAK2V617F mutant kinase is a therapeutic target in MPN. However, inhibition of wild-type (WT) JAK2 can decrease the erythrocyte or platelet (PLT) count. Our selective JAK2 inhibitor, NS-018, suppressed the growth of Ba/F3 cells harboring JAK2V617F more strongly than that of cells harboring WT JAK2. The 4.3-fold JAK2V617F selectivity of NS-018 is higher than the 1.0- to 2.9-fold selectivity of seven existing JAK2 inhibitors. NS-018 also inhibited erythroid colony formation in JAK2V617F transgenic mice at significantly lower concentrations than in WT mice. In keeping with the above results, in a JAK2V617F bone marrow transplantation mouse model with a myelofibrosis-like disease, NS-018 reduced leukocytosis and splenomegaly, improved bone marrow fibrosis and prolonged survival without decreasing the erythrocyte or PLT count in the peripheral blood. By exploring the X-ray co-crystal structure of NS-018 bound to JAK2, we identified unique hydrogen-bonding interactions between NS-018 and Gly993 as a plausible explanation for its JAK2V617F selectivity. These results suggest that NS-018 will have therapeutic benefit for MPN patients through both its efficacy and its reduced hematologic adverse effects.
Similar content being viewed by others

Main
Janus kinase 2 (JAK2) is a tyrosine kinase, which has an essential role in the cytokine signaling pathways that regulate hematopoiesis. Germline deletion of JAK2 in mice results in embryonic lethality because of a lack of hematopoiesis,1, 2 and conditional JAK2 deletion in young adult mice severely impairs erythropoiesis and thrombopoiesis.3
A somatic point mutation at codon 617 of JAK2, V617F, occurs in the breakpoint cluster region-abelson-negative myeloproliferative neoplasms (MPNs), including polycythemia vera, essential thrombocythemia and primary myelofibrosis.4, 5, 6, 7, 8 The resulting mutant protein, JAK2V617F, is a constitutively activated kinase that activates multiple downstream signaling pathways, such as the signal transducer and activator of transcription, extracellular signal-regulated kinases and Akt signaling pathways, and it transforms hematopoietic cells to cytokine-independent growth. The expression of JAK2V617F causes MPN-like diseases in mice after bone marrow transplantation (BMT).9, 10, 11, 12 Transgenic mice expressing JAK2V617F also develop MPN-like diseases.13, 14, 15, 16, 17, 18 These findings suggest that the inhibition of aberrant JAK2 activity would have therapeutic benefit, and accordingly several JAK2 inhibitors have been developed for the treatment of MPN.19, 20, 21, 22, 23, 24, 25
JAK2 inhibitors show significant therapeutic benefit by reducing spleen size, relieving debilitating symptoms and improving overall survival in clinical trials of these compounds in the treatment of myelofibrosis.24, 25, 26 Although current JAK2 inhibitors are therapeutically effective, they are reported to have hematologic adverse events, including anemia and thrombocytopenia.24, 25, 26
NS-018, (N-[(1S)-1-(4-fluorophenyl)ethyl]-4-(1-methyl-1H-pyrazol-4-yl)-N′-(pyrazin-2-yl)pyridine-2,6-diamine maleate), is a potent and selective inhibitor of JAK2 and Src-family kinases, which is currently in early-phase clinical trials for MPN. In a previous study, NS-018 prevented the progression of anemia in JAK2V617F transgenic mice as well as improving splenomegaly and survival.27 However, it remains unclear which characteristic feature of NS-018 contributes to this effect, and furthermore, the effect has not yet been confirmed in other animal models. In this paper, we report that NS-018 suppressed the growth of cells harboring JAK2V617F more strongly than that of cells harboring wild-type (WT) JAK2. We also confirmed the effect of NS-018 on red blood cells (RBCs) in another myelofibrosis model mouse.
Materials and methods
Structural analysis
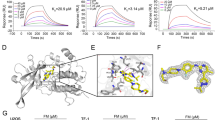
The acquisition of the X-ray co-crystal structure of NS-018 bound to JAK2 was described previously.27 All published X-ray crystal structures were taken from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). Figure 1 and Supplementary Figure 1 were prepared with PyMOL version 1.3 (Schrödinger, New York, NY, USA).

Ribbon-and-stick representation of the X-ray co-crystal structure of NS-018 (blue) bound to JAK2 (gray). Dashed lines represent hydrogen bonds.
Production of retroviruses
Murine JAK2WT (WT) and murine JAK2V617F cDNA15 were cloned into the retroviral vector pMCs-IRES-GFP (Cell Biolabs, San Diego, CA, USA). Transient transfection of Platinum-E retroviral packaging cells (Cell Biolabs) was performed by using the FuGENE6 transfection agent (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Retroviral supernatants were harvested after 48 h and used to transduce the murine interleukin (IL)-3 -dependent pro-B cell line Ba/F3 or bone marrow cells.
Cell culture and growth assay
Ba/F3 cells (Riken BRC Cell Bank, Tsukuba, Ibaraki, Japan) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and 20 ng/ml recombinant murine IL-3 (PeproTech, Rocky Hill, NJ, USA). Ba/F3-JAK2WT and Ba/F3-JAK2V617F cells were generated by retroviral infection with polybrene.
For growth assays, cells were seeded in 96-well plates at 1 × 103 cells/well, treated with serial dilutions of compound, and incubated for 90 h at 37 °C in 5% CO2. Viability was measured by WST-8 assay (Cell Counting Kit-8; Dojindo Laboratories, Kumamoto, Japan). The concentration required to give 50% inhibition (IC50) was estimated with SAS version 9.1.3 (SAS Institute, Cary, NC, USA).
JAK2V617F BMT mice
Murine BMT experiments were performed as previously described.9, 10 Briefly, female BALB/c donor mice (Japan SLC, Hamamatsu, Shizuoka, Japan) were treated with 5-fluorouracil (150 mg/kg, intraperitoneal injection) to increase the number of cycling stem cells for retroviral transduction. Three days after injection, bone marrow cells were harvested by flushing the femurs and tibias of the donor mice with phosphate-buffered saline. Mononuclear cells were isolated by Ficoll-Paque density gradient centrifugation and cultured for 24 h in αMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum and recombinant murine IL-6, stem cell factor, FMS-like tyrosine kinase 3 (flt3) ligand and thrombopoietin (PeproTech; 50 ng/ml each). The cells were treated with retroviral supernatants in RetroNectin-coated dishes (Takara Bio, Otsu, Shiga, Japan) for 72 h. The cells were then injected into the lateral tail vein of BALB/c recipient mice that had been irradiated with 2.5 Gy of gamma rays (2 × 105 cells per mouse).
Ten days after transplantation, NS-018 was orally administered twice daily for 40 days at 50 mg/kg. After treatment, all mice were humanely killed and terminal blood samples and organs were collected. Hematological parameters were determined with an ADVIA 120 hematology system (Siemens Healthcare, Erlangen, Germany). For survival analysis, the statistical significance of differences between vehicle and NS-018 groups was assessed by the log-rank test with SAS version 9.1.3. For blood count and spleen weight analyses, the statistical significance of differences between control and vehicle groups was assessed by the Welch test. If the control and vehicle groups were significantly different, the statistical significance of differences between the vehicle and NS-018 groups was assessed by the Welch test. For histological evaluation, tissues samples from spleens and femurs were fixed in formalin, embedded in paraffin and cut for hematoxylin–eosin staining or Watanabe silver staining according to standard protocols (KAC, Kyoto, Japan). Histological slides were viewed under an Olympus BX50 microscope (Olympus, Tokyo, Japan) and photographed with an Olympus FX380 digital camera (Olympus). Studies were conducted in compliance with the Law for the Humane Treatment and Management of Animals (Law No. 105, 1 October 1973, as revised on 1 June 2006).
Results
X-ray co-crystal structure of NS-018 bound to JAK2
The JAK2 activation loop, which has a DFG (Asp–Phe–Gly) motif at the start, regulates kinase activity by changing its position. The position of Gly993, located immediately N-terminal to the DFG motif, thus depends on the activation state of the kinase.28 The X-ray co-crystal structure of JAK2 in complex with NS-018 (Figure 1) revealed that NS-018 formed hydrogen bonds with the backbone amino and carbonyl groups of Leu932 in the hinge region. In addition, NS-018 interacted with the carbonyl group of Gly993 through two kinds of hydrogen-bonding interactions. First, there is a hydrogen bond between a nitrogen atom of NS-018 and the water molecule depicted by the red sphere in Figure 1. This hydrogen-bonded water molecule forms a second hydrogen bond with the carbonyl group of Gly993. Thus, there are water-mediated hydrogen-bonding interactions between NS-018 and Gly993. Second, there is a CH···O hydrogen bond between an aromatic CH on the fluorobenzene moiety of NS-018 and the carbonyl group of Gly993 (Supplementary Figure 1a). This type of hydrogen-bonding interaction often has a role in the binding of kinase inhibitors to their target kinases.29 Other amino acids shown in Figure 1 form the binding site for NS-018.
Preferential inhibition by NS-018 of the growth of cells harboring JAK2V617F
To compare the inhibitory effect of NS-018 on JAK2WT and JAK2V617F in cells, we assessed the antiproliferative activity of NS-018 against Ba/F3 cells expressing murine JAK2WT or murine JAK2V617F. NS-018 suppressed the growth of Ba/F3-JAK2V617F cells with an IC50 value of 470 nM, whereas it suppressed the growth of Ba/F3-JAK2WT cells stimulated with IL-3 with an IC50 value of 2000 nM (Table 1). Thus, NS-018 showed 4.3-fold selectivity for Ba/F3-JAK2V617F cells over Ba/F3-JAK2WT cells (V617F/WT ratio). Other JAK2 inhibitors also showed selectivity for Ba/F3-JAK2V617F over Ba/F3-JAK2WT cells (Table 1), although the selectivity was lower. For example, INCB018424 (ruxolitinib) and TG101348 showed V617F/WT ratios of 2.0 and 1.5, respectively. Thus, among the eight JAK2 inhibitors tested, NS-018 showed the highest selectivity for JAK2V617F cells.
To further evaluate the functional selectivity of NS-018 for JAK2V617F cells, we performed erythroid colony formation assays with bone marrow cells from JAK2V617F transgenic mice and WT control mice. NS-018 inhibited the formation of erythroid colony-forming units from WT control and JAK2V617F transgenic mice in a dose-dependent manner, but the degree of inhibition was significantly greater in the JAK2V617F transgenic mice (Supplementary Figure 2). Specifically, in JAK2V617F transgenic mice, NS-018 inhibited erythroid colony growth with a mean IC50 value of 360 nM, whereas in WT control mice the corresponding IC50 value was >600 nM. These results show that NS-018 preferentially suppressed the growth of cells harboring JAK2V617F.
Efficacy of NS-018 in JAK2V617F BMT mice
To assess the ability of NS-018 to selectively inhibit JAK2V617F-harboring cells in vivo, we established a JAK2V617F BMT mouse model. BALB/c mice were subjected to BMT with bone marrow donor cells retrovirally transduced to express JAK2WT or JAK2V617F. Only JAK2V617F BMT mice developed myelofibrosis-like disease, including leukocytosis, splenomegaly and bone marrow fibrosis (Supplementary Table 1). They also exhibited higher mortality than control or JAK2WT BMT mice.
We next evaluated the efficacy of NS-018 in this model. When disease was established at 10 days after transplantation, JAK2V617F BMT mice were randomly assigned to either of two groups for treatment with NS-018 or vehicle. NS-018 was administered by oral gavage twice a day for 40 days at a dose of 50 mg/kg, whereas the control group received vehicle only. No signs of gross toxicity were observed during the 40 days of treatment.
During the total of 50 days of the study, four of eight mice in the vehicle-treated group died, whereas no mice in the NS-018-treated group died (Figure 2a). This represents a statistically significant prolongation of survival in the NS-018-treated group (P<0.05).

Effect of NS-018 on survival and peripheral blood counts in JAK2V617F BMT mice. (a) Kaplan–Meier plot of untransplanted control mice treated with vehicle (control) and JAK2V617F BMT mice treated with vehicle (vehicle) or 50 mg/kg NS-018 (NS-018) for 40 days. Statistical significance in survival between the vehicle and NS-018 groups was assessed by the log-rank test (*P<0.05, eight mice per group). (b) White blood cell (WBC), (c) neutrophil (NEUT) and lymphocyte (LYMPH), (d) red blood cell (RBC) and reticulocyte (RETIC) and (e) platelet (PLT) counts. Bars represent the mean±s.e.m. The statistical significance of differences between the control and vehicle groups (#P<0.05) and between the vehicle and NS-018 groups (*P<0.05) were assessed by the Welch test.
Vehicle-treated JAK2V617F BMT mice showed marked leukocytosis (Figure 2b), as evidenced by a 47-fold increase in the mean white blood cell count in the peripheral blood of vehicle-treated mice to 359 × 109/l (compared with 7.6 × 109/l in control mice). NS-018 treatment achieved a 95% suppression of this increase to 17.4 × 109/l. In vehicle-treated JAK2V617F BMT mice, the differential white blood cell count showed that the neutrophils had increased to 80.0%, compared with 7.5% in control mice (Figure 2c). Conversely, the percentage of lymphocytes in vehicle-treated JAK2V617F BMT mice was 10.7%, compared with 88.8% in control mice. NS-018 treatment partially suppressed both the increase in neutrophils and the decrease in lymphocytes, giving 47.9% neutrophils and 42.2% lymphocytes. JAK2V617F BMT mice showed a higher reticulocyte (RETIC) count than control mice but about the same RBC count. NS-018 treatment did not decrease the RBC and only marginally decreased the RETIC count (Figure 2d). JAK2V617F BMT mice showed a 78% decrease in the platelet (PLT) count compared with control mice (Figure 2e), and NS-018 treatment did not further decrease the count.
Vehicle-treated JAK2V617F BMT mice also showed marked splenomegaly, with higher average spleen weights (2.07±0.08 g) than control mice (0.10±0.01 g; Figure 3a). NS-018-treated JAK2V617F BMT mice had a spleen weight of 0.49±0.08 g, or 24% of that of vehicle-treated mice, so that NS-018 largely prevented the development of splenomegaly. The histopathological results also provide evidence that NS-018 treatment suppressed the development of splenomegaly. Spleen sections from vehicle-treated JAK2V617F BMT mice exhibited complete disruption of the normal splenic architecture (Figures 3b and c). The white pulp was partially preserved but blended throughout, and the red pulp was expanded, mainly by myeloid-cell invasion. NS-018 treatment resulted in markedly reduced myeloid-cell invasion and a less disrupted splenic architecture (Figure 3d).

Effect of NS-018 on spleen weight and bone marrow fibrosis in JAK2V617F BMT mice. NS-018 was orally administered for 40 days at 50 mg/kg twice a day. After treatment, spleens and femurs were removed. (a) Spleen weight. Bars represent the mean±s.e.m. The statistical significance of differences between the control and vehicle groups (##P<0.01) and between the vehicle and NS-018 groups (**P<0.01) was assessed by the Welch test. (b–d) Spleen sections stained with hematoxylin and eosin (original magnification × 40). (e–g) Bone marrow tissue sections prepared from femur and stained for reticulin (original magnification × 200).
Finally, NS-018 partially suppressed bone marrow fibrosis in JAK2V617F BMT mice (Figures 3e–g). There was mild-to-moderate reticulin fibrosis in all vehicle-treated mice that survived to the study end point (n=4), whereas in seven of eight mice treated with NS-018 there was only slight-to-little reticulin fibrosis. Only one NS-018-treated mouse showed mild fibrosis. Taken together, these results show that NS-018 reduced leukocytosis and splenomegaly, suppressed bone marrow fibrosis and prolonged survival in JAK2V617F BMT mice without reducing the RBC or PLT counts.
Discussion
In our search for a JAK2 inhibitor with reduced hematologic adverse effects, we evaluated the potential of our novel JAK2 inhibitor NS-018 in preclinical models. We first assessed the selective inhibition of the mutant kinase JAK2V617F by NS-018 in Ba/F3 cells. NS-018 showed higher selectivity for JAK2V617F over JAK2WT (a higher V617F/WT ratio) than existing JAK2 inhibitors such as ruxolitinib and TG101348 (Table 1). The preferential inhibition of the growth of Ba/F3-JAK2V617F cells by JAK2 inhibitors was unrelated to their target specificity among Jak-family kinases (for example, selectivity for JAK1/2 or JAK2).
NS-018 shows potent inhibition of Src-family kinases in addition to JAK2 (ref. 27), and it could be argued that this contributed to its activity against Ba/F3-JAK2V617F cells. However, Src-family kinases expressed in Ba/F3-JAK2V617F cells were not phosphorylated (Supplementary Figure 3) and therefore not activated. Furthermore, the Src-family kinase inhibitor dasatinib did not show potent antiproliferative activity against Ba/F3-JAK2V617F cells (IC50=5000 nM; data not shown). For these reasons, it is unlikely that the Src-inhibitory activity of NS-018 contributed to its antiproliferative activity against Ba/F3-JAK2V617F cells.
The efficacy of several JAK2 inhibitors has been evaluated in JAK2V617F BMT model mice,30, 31 but there are phenotypic differences in disease severity among mouse strains.9 Thus, BMT in BALB/c mice results in more markedly elevated leukocyte counts, a greater degree of splenomegaly and reticulin fibrosis, and higher mortality, than in C57Bl/6 mice. To assess the effect of NS-018 on bone marrow fibrosis, we used the more severely affected strain, BALB/c, to establish a BMT model. Although our model mice showed elevated white blood cell counts, splenomegaly, high mortality and bone marrow fibrosis, as previously reported,9 elevated peripheral RBC and PLT counts were not observed (Supplementary Table 1). Rather, the mice showed thrombopenia 50 days after transplantation (Figure 2e). Although the reason for the differences in pathologic phenotype from the previous report is unclear, possible reasons are the difference in the promoters of the retroviral vectors used and the difference in the progenitor cell types infected with retrovirus.
Although NS-018 treatment markedly reduced leukocytosis in JAK2V617F BMT model mice (Figure 2b), it did not decrease the RBC or PLT counts (Figures 2d and e). Because our BMT model had some unusual features, including the absence of polycythemia and thrombocythemia as described above, this apparent lack of effect of NS-018 on the RBC and PLT counts should be interpreted carefully. However, it is consistent with our previous finding that NS-018 prevents the progression of anemia and the decrease in PLT count in JAK2V617F transgenic mice.27 When we evaluated the another JAK2 inhibitor, R723, in the same model, prevention of progressive anemia was not observed.30 Because JAK2 signaling has important roles in erythropoiesis and thrombopoiesis,1, 2, 3 JAK2 inhibition is thought to have on-target adverse effects such as a decrease in the RBC and PLT counts. In fact, some JAK2 inhibitors do reduce the RBC or PLT count in mice.30, 31, 32, 33 More importantly, the dose-limiting toxicity of ruxolitinib in MPN patients is thrombocytopenia.19, 34 Thus, a JAK2 inhibitor that produces a smaller reduction in the RBC and PLT counts in the therapeutic window would have clinical benefit. As described here, NS-018 preferentially inhibited the growth of Ba/F3-JAK2V617F cells (Table 1) and suppressed erythroid colony formation from bone marrow cells from JAK2V617F transgenic mice (Supplementary Figure 2). Furthermore, NS-018 inhibits erythroid colony formation by peripheral blood mononuclear cells from JAK2V617F+ polycythemia vera patients at concentrations significantly lower than those required for healthy controls.27 These results suggest that NS-018 inhibits the constitutively active JAK2V617F more potently than WT JAK2, which is involved in normal erythropoiesis and thrombopoiesis. This preferential inhibition of JAK2V617F by NS-018 could explain the fact that NS-018 does not reduce the RBC or PLT count at therapeutic doses in mouse models. The concentration range over which NS-018 inhibits JAK2V617-dependent cells but not WT cells in the model mice might be wider than for other JAK2 inhibitors.
In the present study, NS-018 treatment improved bone marrow fibrosis in JAK2V617F BMT mice (Figures 3e–g). This contrasts with our previous study of JAK2V617F transgenic mice in which NS-018 treatment had little effect on the progression of bone marrow fibrosis.27 However, the fibrosis in the transgenic mice was more severe than in the BMT mice used in the present study. In addition, in the transgenic mice NS-018 was administered on weekdays only, whereas in the BMT mice it was administered every day. The lack of dosing of the transgenic mice over the weekend might have reduced the effect of NS-018 on the fibrosis. Some JAK2 inhibitors, including TG101348 and CYT387, improve bone marrow fibrosis in JAK2V617F BMT mice,30, 31 but unfortunately they also produce a decrease in the hematocrit in the therapeutic dose range in mice. Thus, an important and distinctive feature of NS-018 is its apparent lack of a suppressive effect on the RBC and PLT counts in the therapeutic window in mouse models.
To investigate the structural factors determining the preference of NS-018 for JAK2V617F, we explored the X-ray co-crystal structures of the inhibitors listed in Table 1. As shown in Figure 1 and Supplementary Figure 1a, NS-018 interacts with the carbonyl group of Gly993, which is located immediately N-terminal to the DFG motif, through two kinds of hydrogen-bonding interactions that are only operative in activated JAK2. These interactions could explain the high V617F/WT ratio of NS-018. Although similar CH···O hydrogen-bonding interactions with Gly993 are found for AZD1480 (Supplementary Figure 1b), the water-mediated interaction is absent. This could account for the lower V617F/WT ratio of AZD1480 compared with NS-018. CP-690,550 interacts with JAK2 in a unique way (Supplementary Figure 1c); specifically, (1) Gly993 does not interact directly with CP-690,550, and Asp994 is located close to CP-690,550, and (2) a unique interaction is found between the terminal CN group of CP-690,550 and the glycine-rich loop (P-loop).35 Although the reason for CP-690,550’s relatively high V617F/WT ratio is unclear, a different mechanism could contribute to the recognition of the active state of JAK2 by CP-690,550. INCB018424 and CYT387 also have the terminal CN group, so a similar recognition mechanism might be operative for these compounds. A docking model of the interaction of TG101209 with JAK2 has been published,36 but the X-ray structure is still unavailable. It was thus impossible to perform detailed analyses for TG101209 and the structurally similar TG101348. The X-ray structure shows that AT9283 does not interact with Gly993 (Supplementary Figure 1d), and accordingly AT9283 showed no preference for V617F. A recent study of the JAK2 inhibitor LY2784544, which has a high preference for JAK2V617F, also emphasizes the interaction with Gly993 (ref. 37). All these facts point to the importance of considering interactions with Gly993 when the aim is to design inhibitors with higher V617F/WT ratios.
In summary, NS-018 preferentially inhibited the growth of JAK2V617F-harboring cells over JAK2WT-harboring cells. NS-018 was also effective against leukocytosis, splenomegaly and bone marrow fibrosis, and prolonged survival in JAK2V617F BMT mice with no reduction in the RBC or PLT counts. These characteristic features of NS-018 may be explained at least in part by its unique mode of binding to the activated form of JAK2. This may contribute to a therapeutic benefit for MPN patients by allowing the simultaneous satisfaction of the two requirements of efficacy and reduced hematologic adverse effects. The efficacy and safety of NS-018 for the treatment of MPN are expected to be verified by ongoing clinical trials.
References
Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998; 93: 385–395.
Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K . Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998; 93: 397–409.
Park SO, Wamsley HL, Bae K, Hu Z, Li X, Choe SW et al. Conditional deletion of Jak2 reveals an essential role in hematopoiesis throughout mouse ontogeny: implications for Jak2 inhibition in humans. PLoS One 2013; 8: e59675.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054–1061.
James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387–397.
Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem 2005; 280: 22788–22792.
Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG . Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 2006; 107: 4274–4281.
Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL . JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 2006; 108: 1652–1660.
Bumm TG, Elsea C, Corbin AS, Loriaux M, Sherbenou D, Wood L et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res 2006; 66: 11156–11165.
Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One 2006; 1: e18.
Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008; 111: 3931–3940.
Xing S, Wanting TH, Zhao W, Ma J, Wang S, Xu X et al. Transgenic expression of JAK2V617F causes myeloproliferative disorders in mice. Blood 2008; 111: 5109–5117.
Shide K, Shimoda HK, Kumano T, Karube K, Kameda T, Takenaka K et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia 2008; 22: 87–95.
Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG . Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera–like disease. Blood 2010; 115: 3589–3596.
Marty C, Lacout C, Martin A, Hasan S, Jacquot S, Birling MC et al. Myeloproliferative neoplasm induced by constitutive expression of JAK2V617F in knock-in mice. Blood 2010; 116: 783–787.
Li J, Spensberger D, Ahn JS, Anand S, Beer PA, Ghevaert C et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F–positive essential thrombocythemia. Blood 2010; 116: 1528–1538.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 2010; 363: 1117–1127.
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012; 366: 799–807.
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med 2012; 366: 787–798.
Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol 2011; 29: 789–796.
Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or postpolycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010; 115: 1131–1136.
Pardanani A, Vannucchi AM, Passamonti F, Cervantes F, Barbui T, Tefferi A . JAK inhibitor therapy for myelofibrosis: critical assessment of value and limitations. Leukemia 2011; 25: 218–225.
Tefferi A . JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood 2012; 119: 2721–2730.
Tam CS, Verstovsek S . Investigational Janus kinase inhibitors. Expert Opin Investig Drugs 2013; 22: 687–699.
Nakaya Y, Shide K, Niwa T, Homan J, Sugahara S, Horio T et al. Efficacy of NS-018, a potent and selective JAK2/Src inhibitor, in primary cells and mouse models of myeloproliferative neoplasms. Blood Cancer J 2011; 1: e29.
Lindauer K, Loerting T, Liedl KR, Kroemer RT . Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng 2001; 14: 27–37.
Pierce AC, ter Haar E, Binch HM, Kay DP, Patel SR, Li P . CH···O and CH···N hydrogen bonds in ligand design: a novel quinazolin-4-ylthiazol-2-ylamine protein kinase inhibitor. J Med Chem 2005; 48: 1278–1281.
Shide K, Kameda T, Markovtsov V, Shimoda HK, Tonkin E, Fang S et al. R723, a selective JAK2 inhibitor, effectively treats JAK2V617F-induced murine myeloproliferative neoplasm. Blood 2011; 117: 6866–6875.
Wernig G, Kharas MG, Okabe R, Moore SA, Leeman DS, Cullen DE et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell 2008; 13: 311–320.
Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010; 115: 5232–5240.
Wernig G, Kharas MG, Mullally A, Leeman DS, Okabe R, George T et al. EXEL-8232, a small-molecule JAK2 inhibitor, effectively treats thrombocytosis and extramedullary hematopoiesis in a murine model of myeloproliferative neoplasm induced by MPLW515L. Leukemia 2012; 26: 720–727.
Ostojic A, Vrhovac R, Verstovsek S . Ruxolitinib for the treatment of myelofibrosis: its clinical potential. Ther Clin Risk Manag 2012; 8: 95–103.
Williams NK, Bamert RS, Patel O, Wang C, Walden PM, Wilks AF et al. Dissecting specificity in the Janus kinases: the structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J Mol Biol 2009; 387: 219–232.
Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 2007; 21: 1658–1668.
Ma L, Clayton JR, Walgren RA, Zhao B, Evans RJ, Smith MC et al. Discovery and characterization of LY2784544, a small-molecule tyrosine kinase inhibitor of JAK2V617F. Blood Cancer J 2011; 3: e109.
Acknowledgements
We thank Dr T Ego, Nippon Shinyaku Co. Ltd, for technical advice and assistance and Dr Gerald E Smyth, Nippon Shinyaku Co. Ltd, for English language editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Yohei Nakaya, Haruna Naito, Tomoko Niwa and Tatsuya Horio are employees of Nippon Shinyaku Co. Ltd. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Blood Cancer Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Nakaya, Y., Shide, K., Naito, H. et al. Effect of NS-018, a selective JAK2V617F inhibitor, in a murine model of myelofibrosis. Blood Cancer Journal 4, e174 (2014). https://doi.org/10.1038/bcj.2013.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2013.73
Keywords
This article is cited by
-
NS-018 reduces myeloma cell proliferation and suppresses osteolysis through inhibition of the JAK2 and Src signaling pathways
Blood Cancer Journal (2018)
-
Rethinking JAK2 inhibition: towards novel strategies of more specific and versatile janus kinase inhibition
Leukemia (2017)
-
A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis
Leukemia (2017)
