
Abstract
Although various types of drugs and therapies are available to treat atherosclerosis, it remains a major cause of mortality throughout the world. Macrophages are the major source of foam cells, which are hallmarks of atherosclerotic lesions. Consequently, the roles of macrophages in the pathophysiology of atherosclerosis are increasingly investigated. Autophagy is a self-protecting cellular catabolic pathway. Since its discovery, autophagy has been found to be associated with a variety of diseases, including cardiovascular diseases, malignant tumors, neurodegenerative diseases, and immune system disorders. Accumulating evidence demonstrates that autophagy plays an important role in inhibiting inflammation and apoptosis, and in promoting efferocytosis and cholesterol efflux. These facts suggest the induction of autophagy may be exploited as a potential strategy for the treatment of atherosclerosis. In this review we mainly discuss the relationship between macrophage autophagy and atherosclerosis and the molecular mechanisms, as well as the recent advances in targeting the process of autophagy to treat atherosclerosis.
Similar content being viewed by others


Introduction
Atherosclerosis (AS) remains a major cause of mortality in developed and many developing countries, and it causes acute cardiovascular events and chronic damage. Furthermore, it is predicted that by the year 2020, cardiovascular diseases, and particularly AS, will become the leading cause of the total burden of disease in the world. Three classes of cells—macrophages, smooth muscle cells (SMCs) and vascular endothelial cells—are important for the initiation and development of AS. Of these three types of cells, macrophages are being increasingly studied by researchers. It is widely recognized that apoptosis in lesional macrophages, along with their defective function in efferocytosis, promotes plaque necrosis1, which leads to plaque instability, thrombosis and, finally, cardiovascular events2. Autophagy (literally meaning “to eat oneself”) is a self-protecting process. It has already been shown that macrophage autophagy plays a protective role in AS and that it functions as a promoter of cholesterol efflux. This provides us with a new therapeutic direction for treating AS3,4. This review will mainly discuss the relationship between macrophage autophagy and atherosclerosis, possible mechanisms, and recent advances in targeting autophagy to treat atherosclerosis.
Macrophages and atherosclerosis
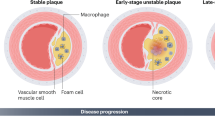
Macrophages play an important role in AS. During the formation of atherosclerotic plaques, circulating monocytes move into the subendothelium of vessel walls and convert into macrophages, which subsequently turn into foam cells after engulfing oxygenized low density lipoproteins (oxLDL) or other modified lipoproteins5. Foam cells are hallmarks of atherosclerotic lesions6. The development of atherosclerotic plaques is classified into four stages: fatty streak, atheromatous plaque, complicated atheromatous plaque and clinical complications in clinical pathology; of these, complicated atheromatous plaque, which features the accumulation of foam cells and a thin fabric cap, has been generally acknowledged as referring to unstable plaques. Ruptures of these plaques are viewed as the main cause of acute cardiovascular events. It has been shown that macrophages aggravate the instability of plaques by secreting many types of inflammatory factors and matrix catabolic enzymes and inducing the death of SMCs5. These inflammatory factors, eg, IL-1, IL-6, IL-12, and tumor necrosis factor, can lead to further inflammatory reactions in atherosclerotic plaques7. The matrix catabolic enzymes delivered by macrophages, which include nonspecific esterase, lysosomal hydrolase and ectoenzyme8, enhance the instability of plaques by damaging the collagen of the fabric cap.
The accumulation of monocyte-derived macrophages is regarded as a crucial step in the formation and development of atherosclerotic plaques, and the extent of monocyte accumulation is enhanced along with the progression of plaques9. This accumulation also contributes to the reduction of the engrafting function of macrophages in plaques10 while worsening the efferocytosis of lesional macrophages as a result of macrophage apoptosis. This, in turn, leads to secondary necrosis and the amplification of plaque inflammation11. In addition to the damaging role of lesional inflammatory reactions, the inhibition of autophagy in macrophages is increasingly being considered to be the main cause of macrophage apoptosis, the mechanisms of which will be discussed in the following sections.
Autophagy and its major signaling pathways
Autophagy is a self-protecting cellular catabolic pathway that relies on lysosomes. Some long-lived proteins, damaged organelles and misfolded proteins are degraded and recycled through the autophagy process12. According to differences in physiological functions and patterns of delivery to the lysosomal lumen, three forms of autophagy has been described: macroautophagy, microautophagy and chaperone-mediated autophagy13. Because macroautophagy has so far been the most extensively studied of the three forms of autophagy, our review will focus on the mechanisms and effects of macroautophagy (hereafter referred to as “autophagy”). As mentioned above, autophagy is a self-guarding function that promotes cellular survival. Some researchers have, however, identified a connection between autophagy and cellular death, which has been called “autophagic cellular death”14. A great deal of evidence supports the idea that autophagy is closely related to the initiation and development of many diseases, including malignant tumors, neurodegenerative diseases, cardiovascular disorders and immunological diseases15,16,17,18.
Autophagy is mainly performed in two steps (Figure 1), as follows. First, following the formation of a cup-shaped phagophore with lipid bilayer membranes, cells are stimulated to form intracellular, spherical double-membraned autophagosomes that enclose misfolded proteins or damaged organelles. Subsequently, autophagosomes dispose of “coat proteins (LC3-II)” on their surface and integrate with lysosomes to form single lipid layer membrane-encased autolysosomes, which have digesting functions and play a role in degradation. Throughout this process, more than 30 types of Atgs (autophagy-related genes) and autophagy-related proteins have a vital impact on the formation of autophagosomes and their fusion with lysosomes. Of these, LC3-II and double-membrane autophagosomes are widely regarded as significant markers that can be used to detect the initiation of autophagy and its extension, and they have been extensively used in autophagy-related fundamental studies19. So far, two major autophagy signaling pathways have been illustrated20,21: an inhibitory pathway, which involves Class I PI3K-mTOR signaling, and an inductive pathway, which involves Class-III PI3K-Beclin1 signaling. In the presence of rich nutrients, growth factors, or insulin interacting with related membrane receptors, Class I PI3K is induced to stimulate the activation of mammalian target of rapamycin (mTOR) and the mTOR complex 1 (mTORC1) via Akt pathway. This, in turn, inhibits Atg1 (ULK1), the first and initiating protein of the autophagy pathway, which prevents autophagosome formation. In a situation characterized by nutrient insufficiency and reactive oxygen species (ROS) stimulation, the Class III PI3K-Beclin1 complex is triggered, which promotes the assembly of the Atg12-Atg5-Atg16L complex and Atg8/LC3. This subsequently stimulates autophagosome formation and induces the autophagy process.

Schematic illustration of the autophagy process in macrophages and recent advances in targeting autophagy for atherosclerotic treatments. Under conditions including nutrient insufficiency, hunger or increased ROS, cup-shaped phagophores with bilayer lipid membranes are formed that are composed of various types of Atgs. Spherical double-membrane autophagosomes subsequently form. On integration with lysosomes, autophagosomes turn into autolysosomes, which function in degradation. Two major signaling pathways regulate this process: an inductive pathway that involves Class-III PI3K-Beclin1 signaling and an inhibitory pathway that involves Class I PI3K-mTOR signaling. Several autophagy inducers are effective at positively intervening in autophagy. Of these, everolimus, resveratrol and berberine inhibit the autophagy suppressor mTOR, and berberine also activates AMPK. The overexpression of β-arrestin-1 and the activation of CB2R are thought to increase the expression of Beclin1 and LC3, contributing to the initiation of autophagy.
Furthermore, there is evidence that IP3R-mediated Ca2+ signaling is an indispensable part of the mTORC1-regulated pathway. Although an autophagy-inhibiting role for intracellular Ca2+ has been reported22, it has been demonstrated that under starvation conditions or exposure to rapamycin, an mTORC1 inhibitor, the endoplasmic reticulum (ER) accumulates large amount of Ca2+ and promotes the release of Ca2+ through IP3Rs, which consequently results in the induction of autophagy23.
Effects of macrophage autophagy on apoptosis, inflam-mation and aggregation
Many researchers have now reached a common consensus that in AS, autophagy, under basal conditions, plays an important role in safeguarding cells against oxidative stress, reducing cell apoptosis and enhancing lesional stability24. Maatinet et al25 showed that 7-ketocholesterol (7-KC), an autophagy inducer, was able to effectively reduce cell death in SMCs, which, as a result, promoted plaque stability. Conversely, excessively induced autophagy may lead to “autophagic cellular death” in SMCs and vascular endothelial cells, driving the low-level synthesis of collagen and the thickening of the fabric cap, which lead to an increased risk of AS-related acute cardiovascular events26. Although in atherosclerotic plaques, autophagy plays important roles in SMCs, vascular endothelial cells and macrophages, autophagy in macrophages is now drawing an increasing amount of attention.
Two brilliant articles published in Cell Metabolism in April, 2012 provided us with a better overall picture of the functions of autophagy. Liao et al1 noted that autophagy prevented lesional macrophage apoptosis and defective efferocytosis and that autophagy deficiency enhanced the total necrotic area in advanced atherosclerotic plaques and the damage caused by oxidative stress in experiments using ldl−/− mice. Razani et al3 provided evidence that autophagy became dysfunctional in AS and that autophagy deficiency, to some extent, led to inflammasome hyperactivation, which further promoted AS progression. The authors of these two articles used LC3/Atg8, p62/SQSTM1 and the double-membrane autophagosomes as autophagy markers. Of these, p62/SQSTM1 is known to be an autophagy degrading substrate, and its accumulation indicates autophagy flux deficiency. Moreover, Liao et al showed that a western diet (WD) increased p62/SQSTM1, contrary to the relative reduction observed in its mRNA expression, further demonstrating the relationship between p62/SQSTM1 accumulation and autophagy flux deficiency. In addition, they noted that the underlying mechanism involving autophagy in atherosclerosis might include the enhancement of apoptosis and NADPH oxidize-mediated oxidative stress in macrophages.
In the last few years, it has been widely acknowledged that the mass aggregation of macrophages and the formation of lipid cores in atherosclerotic plaques are crucial to plaque stability, and they are also regarded as important factors when evaluating the extent and severity of vulnerable plaques in AS27. Plaques featuring a thick fabric cap are abundant in smooth muscle are considered to be relatively stable and to result in fewer plaque ruptures and acute cardiovascular events. Autophagy has far-reaching functions that protect plaque stability. It has already been shown that selectively inhibiting the PI3K/Akt/mTOR pathway in cells can effectively induce autophagy, which successfully reduces macrophage aggregation in atherosclerotic plaques and promotes plaque stability by protecting cells and reducing the secretion of inflammatory factors28. Furthermore, autophagy may function as an inhibitor of apoptosis by decreasing the activation and formation of inflammasomes29.
Effects of macrophage autophagy on cholesterol efflux
Macrophage reverse cholesterol transport (RCT) refers to the efflux of cholesterol from macrophages, using apoA-I or HDL as a carrier, to the liver to be cleared along with bile, ultimately as feces30. Cholesterol efflux from macrophages is therefore the first and primary step of the RCT process31. Because macrophages are the main source of foam cells, it is vital to enhance macrophage cholesterol efflux during AS treatment. Three types of carriers for cholesterol efflux have so far been identified, ABCA1, ABCG1 and SR-B1. ABCA1 mainly presents to apoA-I, while ABCG1 and SR-B1 present to mature HDL32. Recently, Fu et al33 found that ABCA12 deficiency increased ABCA1 degradation, added to the accumulation of cholesterol in macrophages and the formation of foam cells and damaged the process of macrophage RCT, which resulted in the promotion of AS progression. They demonstrated that ABCA12 regulates ABCA1-dependent cholesterol efflux from macrophages. Enhancing the process of cholesterol efflux from macrophages effectively reduces the formation of macrophage-derived foam cells, which contributes to plaque stability.
For quite a long time, lipid droplets (LDs) were widely regarded as simple fat-storing compartments. However, with the increase in our knowledge of LDs, we have found that they tend to be dynamic organelles and we have gained much more respect for the significance of the roles they play in cholesterol efflux6. There is evidence that when cells fail to uptake excess free cholesterol, it is esterified to nontoxic esters (CEs) and then stored in LDs34. It has already been shown that polar lipids in the monolayer, including sterols and phospholipids, constitute the membrane of LDs, which has a core of neutral lipids, such as CEs, sterol esters and triacylglycerols. It has been discovered that several types of proteins are involved in decorating the surface of LDs, including adipophilin, perilipin, TIP47, and DGAT2, which act mainly as markers for detecting LDs in research studies34. As a result, exploring ways to enhance the process of cholesterol efflux from LDs in lesional macrophages has now become the focus of the treatments for AS.
According to a small number of recent studies, autophagy plays an important role in macrophage cholesterol efflux from LDs. This special type of selective autophagy is referred to as “lipophagy”35. Its major mechanism occurs after the fusion of autophagosomes and lysosomes when CEs in LDs are hydrolyzed by specific enzymes into free cholesterols, which can transfer through the cell membrane and then be carried by apoA-I or HDL in back to the liver36,37. Quimet et al38 suggested that autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase (LAL). They demonstrated that LDs were delivered to lysosomes by “lipophagy” in macrophages and that CEs in LDs were hydrolyzed via LAL in lysosomes. Finally, an ABCA1-dependent pathway mediates cholesterol efflux from macrophage foam cells.
Furthermore, Guezennec et al39 demonstrated the Wip1-dependent regulation of autophagy and cholesterol efflux in atherosclerotic lesional macrophages. They indicated that mice deficient in Wip1 phosphatase, a negative regulator of Atm-dependent signaling, inhibited fat accumulation and the conversion of macrophages into foam cells, thus preventing AS through the Atm-mTOR signaling pathway and Wip1-dependent selective autophagy. These data revealed the positive role played by autophagy in macrophage cholesterol efflux.
Pharmacological intervention of autophagy in the treatment of AS
Numerous fundamental and clinical studies have been conducted to target the process of autophagy to treat AS. Based on these studies, several autophagy inducers have been demonstrated to be effective for the alleviation of AS severity in animal models, and some of them have also been reported to be effective and promising therapies for AS treatment in clinical studies (Figure 1).
It has been reported that mTOR inhibitors have therapeutic significance in many types of malignant tumors, including breast cancer40 and renal cell carcinoma41, etc. One of the mTOR inhibitors, everolimus, is among the most studied and best-known autophagy inducers. Researchers have found that in subjects with neurodegenerative diseases, everolimus enhanced the clearance of toxic substances by inhibiting mTOR and inducing autophagy42. Because it has an autophagy-inducing effect, its role in treatments for AS has been widely studied and reported. There is evidence that everolimus promotes plaque stability, and stent-based secretion of everolimus promotes the formation of a stable plaque phenotype43. It was also recently reported by Holdaas and colleagues44 that coronary artery intimal thickening and the incidence of cardiac allograft vasculopathy were significantly reduced by everolimus therapy. In combination with other clinical studies45,46,47, it has been shown that everolimus-eluting stents are the optimal choice for preventing vascular restenosis and the occurrence of cardiac allograft vasculopathy (CAV) after heart transplant or other cardiac surgeries.
Resveratrol is another extensively used autophagy inducer that acts mainly by inhibiting mTOR48. It is a plant-derived polyphenolic substance that has mainly been found to be abundant in grapes, peanuts and red wine49,50. Because of its traditionally recognized anti-oxidative and anti-inflammatory properties, this compound plays a protective role against various types of diseases, including cardiovascular diseases, malignancies, renal diseases, type II diabetes, infections and some neurological disorders51,52,53. Resveratrol has also been shown to play an important role in anti-atherosclerosis and vasorelaxation by inducing autophagy48. For example, Liu and colleagues54 demonstrated that the application of resveratrol facilitated the efferocytosis of oxLDL-induced apoptotic cells in RAW264.7 cells by activating Sirt1-mediated autophagy, which demonstrated a protective role for resveratrol in AS in vitro and indicated that the activation of Sirt1-mediated autophagy by resveratrol might serve as a novel therapy for the treatment of AS. Resveratrol was also reported to have immunomodulatory effects on lesional macrophages by counteracting inflammation in human M1 and M2 macrophages upon challenge with certain types of cholesterol55. In addition, resveratrol supplementation at 3 mg·kg-1·d-1 in a hypercholesterolemic rabbit model led to a reduction of atherosclerotic plaque size and density as well as a decrease in the initial layer thickness, which demonstrated that resveratrol had a potential therapeutic effect on AS56,57. However, researchers have noted that resveratrol does not add to the antiatherogenic effect of atorvastatin when these drugs are used together58. Hence, when resveratrol is applied in combination with other lipid-reducing drugs, their respective pharmacological functions should be fully examined.
Other autophagy inducers have been reported with properties that alleviate or treat AS. For example, berberine, which is extracted from Coptis, has been shown to be a promising and potential therapy for the treatment of various types of diseases because of its diverse pharmacological characteristics, which include anti-microbial, anti-diabetic, anti-hyperlipidemic, anti-inflammatory and anti-oxidant properties59,60,61. Its protective effect against AS was recently discussed by Fan and colleagues59, who demonstrated that berberine treatment inhibited inflammatory reactions in J774A.1 cells by the induction of the autophagy process, which was mediated through the activation of the AMPK/mTOR signaling pathway. These data provide new insight into berberine's molecular mechanisms, its relationship with autophagy and its therapeutic potential for the treatment of AS.
Moreover, our lab has conducted several studies to explore autophagy inducers and their respective roles in diseases18,62,63. For example, β-arrestins, which are traditionally recognized as modulators of the desensitization and inactivation of G protein-coupled receptors (GPCRs), have recently been reported to take part in many signaling pathways, independent of G proteins, as scaffold proteins, and this is increasingly being studied64. A previous study in our lab indicated that β-arrestin-1, but not β-arrestin-2, induced Beclin1-dependent autophagy in neurons, which revealed a neuroprotective role for β-arrestin-1 in cerebral ischemia. In addition, Kim and colleagues65 demonstrated that knockout of β-arrestin-1 in mice significantly enhanced the progression and severity of AS because the mice lacked β-arrestin-1 and therefore its function, which is to reduce neointimal hyperplasia in SMCs. These data indicated that β-arrestin-1 played a protective role in alleviating AS. Based on the consistency of reports on its positive roles, we have reason to believe that macrophage β-arrestin-1 may also significantly contribute to protection against and alleviation of AS. Another type of promising autophagy inducer that is currently being studied in our lab is cannabinoid receptor 2 (CB2R) agonists. We have demonstrated that selectively activating CB2R induces the autophagy process, which in turn inhibits NLRP3 inflammasome initiation and activation in mouse microglia, contributing to the alleviation of experimental autoimmune encephalomyelitis (EAE)18. In combination with the already recognized protective effects of CB2R in AS66,67,68, CB2R agonists are another group of autophagy-related factors that are likely to be useful for the treatment of AS.
Summary
Macrophages play an important role in the initiation and progression of AS. The accumulation of macrophages promotes plaque development and instability69. Lesional macrophage apoptosis and defective efferocytosis also participate in AS progression. In the presence of stimulation by oxidative stress or modified LDL (eg, oxLDL), autophagy is induced in macrophages and acts as a cellular safeguard by clearing damaged organelles and misfolded proteins, protecting cells from apoptosis, preserving macrophage efferocytosis and enhancing cholesterol efflux from macrophage-derived foam cells during different steps of AS (Figure 2). Although the protective mechanisms involved in macrophage autophagy in AS are gradually being revealed, there are still gaps in our knowledge, and many mechanisms are waiting to be explored. Furthermore, despite the development and utility of various autophagy inducers that can be used to treat AS, their best adaptive doses and possible side effects remain to be fully described, and more autophagy inducers that target lesional macrophages require further development and validation.

Schematic illustration of the roles of macrophage autophagy during the different steps of AS. During the initiation of AS, circulating monocytes move into the subendothelium of vessel walls, where they break through the endothelial cells and become macrophages. When macrophages ingest oxLDL or other forms of modified lipoproteins, they turn into foam cells. The accumulation of foam cells results in the formation of atherosclerotic plaques. Macrophage autophagy contributes to the inhibition of foam cell formation by reducing oxLDL ingestion by macrophages and increasing efferocytosis and cholesterol efflux in macrophages. It also inhibits the occurrence of plaque rupture and fall off by inhibiting apoptosis and inflammation in lesional plaques, which alleviates the severity of AS.
References
Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012; 15: 545–53.
Qian YN, Luo YT, Duan HX, Feng LQ, Bi Q, Wang YJ, et al. Adhesion molecule CD146 and its soluble form correlate well with carotid atherosclerosis and plaque instability. CNS Neurosci Ther 2014; 20: 438–45.
Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012; 15: 534–44.
Xu WH, Li ML, Niu JW, Feng F, Jin ZY, Gao S . Intracranial artery atherosclerosis and lumen dilation in cerebral small-vessel diseases: a high-resolution MRI Study. CNS Neurosci Ther 2014; 20: 364–7.
Chen SG, Xiao J, Liu XH, Liu MM, Mo ZC, Yin K, et al. Ibrolipim increases ABCA1/G1 expression by the LXRα signaling pathway in THP-1 macrophage-derived foam cells. Acta Pharmacol Sin 2010; 10: 1343–9.
Farese RV Jr, Walther TC . Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 2009; 139: 855–60.
Apostolakis S, Spandidos D . Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol Sin 2013; 34: 1251–6.
Hume DA . The mononuclear phagocyte system. Curr Opin Immunol 2006; 18: 49–53.
Liu YR, Chen JJ, Dai M . Paeonol protects rat vascular endothelial cells from ox-LDL-induced injury in vitro via downregulating microRNA-21 expression and TNF-α release. Acta Pharmacol Sin 2014; 35: 483–8.
Li Y, Liao F, Yin XJ, Cui LL, Ma GD, Nong XX, et al. An association study on ADAM10 promoter polymorphisms and atherosclerotic cerebral infarction in a Chinese population. CNS Neurosci Ther 2013; 19: 785–94.
Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, et al. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 2014; 56: 219–31.
Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM . BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther 2014; 20: 1045–55.
Yang Z, Klionsky DJ . Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22: 124–31.
Kroemer G, Levine B . Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9: 1004–10.
Qi H, Su FY, Wan S, Chen Y, Cheng YQ, Liu AJ . The antiaging activity and cerebral protection of rapamycin at micro-doses. CNS Neurosci Ther 2014; 20: 991–8.
He PX, Che YS, He QJ, Chen Y, Ding J . G226, a novel epipoly-thiodioxopiperazine derivative, induces autophagy and caspase-dependent apoptosis in human breast cancer cells in vitro. Acta Pharmacol Sin 2014; 35: 1055–64.
Mizumura K, Choi AM, Ryter SW . Emerging role of selec-tive autophagy in human diseases. Front Pharmacol 2014; 5: 244.
Shao BZ, Wei W, Ke P, Xu ZQ, Zhou JX, Liu C . Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP3 inflammasome. CNS Neurosci Ther 2014; 20: 1021–8.
Wei N, Yu SP, Gu XH, Chen DD, Whalin MK, Xu GL, et al. The involvement of autophagy pathway in exaggerated ischemic brain damage in diabetic mice. CNS Neurosci Ther 2013; 19: 753–63.
Salabei JK, Conklin DJ . Cardiovascular autophagy: crossroads of pathology, pharmacology and toxicology. Cardiovasc Toxicol 2013; 13: 220–9.
Ryter SW, Lee SJ, Smith A, Choi AM . Autophagy in vascular disease. Proc Am Thorac Soc 2010; 7: 40–7.
Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142: 270–83.
Decuypere JP, Kindt D, Luyten T, Welkenhuyzen K, Missiaen L, De Smedt H, et al. mTOR-controlled autophagy requires intracellular Ca2+ signaling. PloS One 2013; 8: e61020.
Kiffin R, Bandyopadhyay U, Cuervo AM . Oxidative stress and autophagy. Antioxid Redox Signal 2006; 8: 152–62.
Martinet W, Schrijvers DM, Timmermans JP, Bult H . Interactions between cell death induced by statins and 7-ketocholesterol in rabbit aorta smooth muscle cells. Br J Pharmacol 2008; 154: 1236–46.
Schrijvers DM, De Meyer GR, Martinet W . Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol 2011; 31: 2787–91.
Ling MY, Ma ZY, Wang YY, Qi J, Liu L, Li L, et al. Up-regulated ATP-sensitive potassium channels play a role in increased inflammation and plaque vulnerability in macrophages. Atherosclerosis 2013; 226: 348–55.
Zhai C, Cheng J, Mujahid H, Wang H, Kong J, Yin Y, et al. Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PloS One 2014; 9: e90563.
Li R, Ji Z, Qin H, Kang X, Sun B, Wang M, et al. Interference in autophagosome fusion by rare earth nanoparticles disrupts autophagic flux and regulation of an interleukin-1beta producing inflammasome. ACS Nano 2014; 8: 10280–92.
Khera AV, Rader DJ . Future therapeutic directions in reverse cholesterol transport. Curr Atheroscler Rep 2010; 12: 73–81.
Larkin JC, Sears SB, Sadovsky Y . The influence of ligand-activated LXR on primary human trophoblasts. Placenta 2014; 35: 919–24.
Yu XH, Fu YC, Zhang DW, Yin K, Tang CK . Foam cells in atherosclerosis. Clin Chim Acta 2013; 424: 245–52.
Fu Y, Mukhamedova N, Ip S, D'Souza W, Henley KJ, DiTommaso T, et al. ABCA12 regulates ABCA1-dependent cholesterol efflux from macrophages and the development of atherosclerosis. Cell Metab 2013; 18: 225–38.
Ouimet M, Marcel YL . Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol 2012; 32: 575–81.
Narabayashi K, Ito Y, Eid N, Maemura K, Inoue T, Takeuchi T, et al. Indomethacin suppresses LAMP-2 expression and induces lipophagy and lipoapoptosis in rat enterocytes via the ER stress pathway. Int J Cell Biol 2015; 50: 541–54.
Singh R, Cuervo AM . Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol 2012; 2012: 282041.
Liu K, Czaja MJ . Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 2013; 20: 3–11.
Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL . Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 2011; 13: 655–67.
Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, Bulavin DV . Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab 2012; 16: 68–80.
Martin LA, Andre F, Campone M, Bachelot T, Jerusalem G . mTOR inhibitors in advanced breast cancer: ready for prime time? Cancer Treat Rev 2013; 39: 742–52.
Pal SK, Quinn DI . Differentiating mTOR inhibitors in renal cell carcinoma. Cancer Treat Rev 2013; 39: 709–19.
Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, et al. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol 2007; 3: 331–8.
Watanabe T, Kotani J, Murata Y, Seguchi O, Yanase M, Nakatani T . Tissue characterization of progressive cardiac allograft vasculopathy in patients with everolimus therapy compared with donor-transmitted atherosclerosis assessed using serial intravascular imaging: a case report. Transplant Proc 2014; 46: 2456–61.
Holdaas H, Potena L, Saliba F . mTOR inhibitors and dyslipidemia in transplant recipients: a cause for concern? Transplant Rev 2015; 29: 93–102.
Rosing K, Fobker M, Kannenberg F, Gunia S, Dell'Aquila AM, Kwiecien R, et al. Everolimus therapy is associated with reduced lipoprotein-associated phospholipase A(2) (Lp-Pla(2)) activity and oxidative stress in heart transplant recipients. Atherosclerosis 2013; 230: 164–70.
Hsu S, Koren E, Chan Y, Koscec M, Sheehy A, Kolodgie F, et al. Effects of everolimus on macrophage-derived foam cell behavior. Cardiovasc Revasc Med 2014; 15: 269–77.
Hoymans VY, VAN Dyck CJ, Haine SE, Frederix G, Fransen E, Timmermans JP, et al. Long-term vascular responses to Resolute(R) and Xience V(R) polymer-based drug-eluting stents in a rabbit model of atherosclerosis. J Interv Cardiol 2014; 27: 381–90.
Das M, Das DK . Resveratrol and cardiovascular health. Mol Aspects Med 2010; 31: 503–12.
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006; 444: 337–42.
Calabriso N, Scoditti E, Massaro M, Pellegrino M, Storelli C, Ingrosso I, et al. Multiple anti-inflammatory and anti-atherosclerotic properties of red wine polyphenolic extracts: differential role of hydroxycinnamic acids, flavonols and stilbenes on endothelial inflammatory gene expression. Eur J Nutr 2015. Doi: 10.1007/s00394-015-0865-6.
Ginter E, Simko V, Panakova V . Antioxidants in health and disease. Bratisl Lek Listy 2014; 115: 603–6.
Cobb CA, Cole MP . Oxidative and nitrative stress in neurodege-neration. Neurobiol Dis 2015; 84: 4–21.
Kitada M, Koya D . Renal protective effects of resveratrol. Oxid Med Cell Longev 2013; 2013: 568093.
Liu B, Zhang B, Guo R, Li S, Xu Y . Enhancement in efferocytosis of oxidized low-density lipoprotein-induced apoptotic RAW264.7 cells through Sirt1-mediated autophagy. Int J Mol Med 2014; 33: 523–33.
Buttari B, Profumo E, Segoni L, D'Arcangelo D, Rossi S, Facchiano F, et al. Resveratrol counteracts inflammation in human M1 and M2 macrophages upon challenge with 7-oxo-cholesterol: potential therapeutic implications in atherosclerosis. Oxid Med Cell Longev 2014; 2014: 257543.
Wang Z, Zou J, Cao K, Hsieh TC, Huang Y, Wu JM . Dealcoholized red wine containing known amounts of resveratrol suppresses atherosclerosis in hypercholesterolemic rabbits without affecting plasma lipid levels. Int J Mol Med 2005; 16: 533–40.
Malhotra A, Bath S, Elbarbry F . An organ system approach to explore the antioxidative, anti-inflammatory, and cytoprotective actions of resveratrol. Oxid Med Cell Longev 2015; 2015: 803971.
Berbee JF, Wong MC, Wang Y, van der Hoorn JW, Khedoe PP, van Klinken JB, et al. Resveratrol protects against atherosclerosis, but does not add to the antiatherogenic effect of atorvastatin, in APOE*3-Leiden.CETP mice. J Nutr Biochem 2013; 24: 1423–30.
Fan X, Wang J, Hou J, Lin C, Bensoussan A, Chang D, et al. Berberine alleviates ox-LDL induced inflammatory factors by up-regulation of autophagy via AMPK/mTOR signaling pathway. J Transl Med 2015; 13: 92.
Derosa G, Maffioli P, Cicero AF . Berberine on metabolic and cardiovascular risk factors: an analysis from preclinical evidences to clinical trials. Expert Opin Biol Ther 2012; 12: 1113–24.
Tillhon M, Guamán Ortiz LM, Lombardi P, Scovassi AI . Berberine: new perspectives for old remedies. Biochem Pharmacol 2012; 84: 1260–7.
Wang P, Xu TY, Wei K, Guan YF, Wang X, Xu H, et al. ARRB1/beta-arrestin-1 mediates neuroprotection through coordination of BECN1-dependent autophagy in cerebral ischemia. Autophagy 2014; 10: 1535–48.
Zhang L, Dai F, Sheng PL, Chen ZQ, Xu QP, Guo YQ . Resveratrol analogue 3,4,4′-trihydroxy-trans-stilbene induces apoptosis and autophagy in human non-small-cell lung cancer cells in vitro. Acta Pharmacol Sin 2015; 36: 1256–65.
Shenoy SK, Lefkowitz RJ . Beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 2011; 32: 521–33.
Kim J, Zhang L, Peppel K, Wu JH, Zidar DA, Brian L, et al. β-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res 2008; 103: 70–9.
Jiang LS, Pu J, Han ZH, Hu LH, He B . Role of activated endocan-nabinoid system in regulation of cellular cholesterol metabolism in macrophages. Cardiovasc Res 2009; 81: 805–13.
Delsing DJ, Leijten FP, Arts K, van Eenennaam H, Garritsen A, Gijbels MJ, et al. Cannabinoid receptor 2 deficiency in Haematopoietic cells aggravates early atherosclerosis in LDL receptor deficient mice. Open Cardiovasc Med J 2011; 5: 15–21.
Steffens S, Mach F . Cannabinoid receptors in atherosclerosis. Curr Opin Lipidol 2006; 17: 519–26.
De Meyer I, Martinet W, De Meyer GR . Therapeutic strategies to deplete macrophages in atherosclerotic plaques. Br J Clin Pharmacol 2012; 74: 246–63.
Acknowledgements
This work was supported by a grant from the Shanghai Natural Science Foundation of China (No 13ZR1448400) and and a State Key Program of National Natural Science of China (Grant No 81230083).
Author information
Authors and Affiliations
Corresponding authors
PowerPoint slides
Rights and permissions
About this article

Cite this article
Shao, Bz., Han, Bz., Zeng, Yx. et al. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol Sin 37, 150–156 (2016). https://doi.org/10.1038/aps.2015.87
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.87
Keywords
This article is cited by
-
Macrophage-based therapeutic approaches for cardiovascular diseases
Basic Research in Cardiology (2024)
-
The SGLT2 inhibitor empagliflozin attenuates atherosclerosis progression by inducing autophagy
Journal of Physiology and Biochemistry (2024)
-
Macrophage-specific autophagy-related gene HSPB8 is involved in the macrophage polarization in atherosclerosis
BMC Cardiovascular Disorders (2023)
-
Hypoxia Affects Autophagy in Human Umbilical Vein Endothelial Cells via the IRE1 Unfolded Protein Response
Current Medical Science (2023)
-
CircRNA-PTPRA Knockdown Inhibits Atherosclerosis Progression by Repressing ox-LDL-Induced Endothelial Cell Injury via Sponging of miR-671-5p
Biochemical Genetics (2023)
