
Abstract
Aim:
A previous report shows that emodin extracted from the Chinese herbs rhubarb and giant knotweed rhizome can ameliorate the anticancer drug cisplatin-induced injury of HEK293 cells. In this study, we investigated whether and how emodin could protect renal tubular epithelial cells against cisplatin-induced nephrotoxicity in vitro.
Methods:
The viability and apoptosis of normal rat renal tubular epithelial cells (NRK-52E) were detected using formazan assay and flow cytometry analysis, respectively. The expression levels of cleaved caspase-3, autophagy maker LC3 I/II, and AMPK/mTOR signaling pathway-related proteins were measured with Western blot analysis. The changes of morphology and RFP-LC3 fluorescence were observed under microscopy.
Results:
Cisplatin (10-50 μmol/L) dose-dependently induced cell damage and apoptosis in NRK-52E cells, whereas emodin (10 and 100 μmol/L) significantly ameliorated cisplatin-induced cell damage, apoptosis and caspase-3 cleavage. Emodin dose-dependently increased LC3-II levels and induced RFP-LC3-containing punctate structures in NRK-52E cells. Furthermore, the protective effects of emodin were abolished by bafilomycin A1 (10 nmol/L), and mimicked by rapamycin (100 nmol/L). Moreover, emodin increased the phosphorylation of AMPK and suppressed the phosphorylation of mTOR. The AMPK inhibitor compound C (10 μmol/L) not only abolished emodin-induced autophagy activation, but also emodin-induced anti-apoptotic effects.
Conclusion:
Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro through modulating the AMPK/mTOR signaling pathways and activating autophagy. Emodin may have therapeutic potential for the prevention of cisplatin-induced nephrotoxicity.
Similar content being viewed by others


Introduction
Cisplatin (cis-diamminedichloroplatinum II) is a chemotherapeutic reagent that is widely used for the treatment of malignant cancers. Despite its effectiveness, its application is restricted by the cytotoxic effect of cisplatin on the kidney1,2. Cisplatin causes nephrotoxicity through the induction of oxidative stress, stress-related cell apoptosis and necrosis, especially in proximal tubules and collection ducts3,4,5.
Autophagy (macroautophagy) is an important cellular process that maintains cellular homeostasis by degrading defective or aged organelles, protein aggregates, and various macromolecules6. Autophagy can be induced under certain pathological circumstances, including starvation, oxidative stress, hypoxia, and ischemia/reperfusion. Both in vivo and in vitro studies have demonstrated that cisplatin induces autophagy in renal tubular cells, which has been considered an adaptive defensive response against cytotoxicity in renal tubular epithelial cells7,8,9. It was recently reported that the activation of autophagy during acute kidney injury (AKI) might exert a protective effect for the survival of renal tubular epithelial cells10.
Emodin (1,3,8-trihydroxy-6-methylanthraquinone) is a natural anthraquinone that is extracted from the Chinese herbs rhubarb and giant knotweed rhizome11. It has been shown that emodin possesses various pharmacological properties, including immunosuppression, anti-inflammation12, anti-proliferation11,13, anticancer14,15 and antioxidant activities16. Previous studies have shown that emodin exerts renal protective effects. It suppresses cell proliferation and fibronectin expression in glomerular mesangial cells cultured under high glucose (HG)11, and Lan et al demonstrated that emodin suppresses connective tissue growth factor (CTGF) overexpression and extracellular matrix (ECM) accumulation through the inhibition of the p38MAPK pathway17.
A recent study found that emodin also protected human kidney (HEK293) cells against cisplatin-induced cell injury via its role as a potent free radical scavenger18. However, the mechanism underlying this effect remains poorly understood. Previous studies have shown that emodin is a potent adenosine mono-phosphate (AMP)-activated protein kinase (AMPK) activator19 and regulates the mammalian target of rapamycin (mTOR) pathway20. Given that the AMPK/mTOR signaling pathway plays an important role in the regulation of autophagy21, we hypothesized that autophagy might be regulated by emodin to mediate its cellular protective effect. In the current study, our data showing that autophagy is induced by emodin through the AMPK/mTOR signaling pathways and plays an important role in the anti-apoptotic effect of emodin against cisplatin-induced cell death.
Materials and methods
Reagents
Emodin, cisplatin, Bafilomycin A1, rapamycin and Dorsomorphin (compound C) were purchased from Sigma-Aldrich Chemical Co (St Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from Biosharp (St Louis, MO, USA). Emodin was dissolved in DMSO to a concentration of 10 mmol/L.
Cell culture
Normal rat renal tubular epithelial cells (NRK-52E) were obtained from the American Type Culture Collection (Manassas, VA, USA) and routinely cultured in Dulbecco's modified Eagle's medium/Ham's F-12 (DMEM/F-12) (Hyclone, ThermoFisher, Beijing, China) supplemented with 5% fetal bovine serum (FBS).
Evaluation of viable cells
The number of viable cells was assessed by trypan blue exclusion. Cells were resuspended in the trypan blue solution (0.4%) after scraping and were then counted under a light microscope with a hemacytometer. A water-soluble tetrazolium salt (WST) assay (formazan assay) was also performed using a Cell Counting Kit-8 (Dojindo Laboratory, Shanghai, China). At least three independent experiments were conducted.
Western blot analysis
Western blot analysis was performed as previously described22. Anti- microtubule-associated protein 1 light chain 3 (LC3) A/B (D3U4C), anti-caspase-3, anti-cleaved caspase-3 (Asp175), anti-phospho-mTOR (Ser2481), anti-mTOR, anti-phospho-AMPKα (Thr172), anti-AMPKα, anti-phospho-p70S6 kinase (Ser371), and anti-β-actin antibodies, as well as horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulins, were purchased from Cell Signaling Technology (Beverly, MA, USA). The results were quantified using Image-Pro Plus 6.0 software (Media Cybernetic, Washington, USA) and were normalized to the densitometric signal of β-actin.
Flow cytometric (FACS) analysis of apoptosis
The apoptosis of NRK-52E cells was assessed by using flow cytometry (Becton Dickinson) to analyze Annexin V-FITC and PI-stained cells labeled using the Annexin V-FITC Apoptosis Detection Kit (KeyGEN BioTECH, Nanjing, China) according to the manufacturer's protocol. Briefly, at the end of the experimental period, cells were harvested in EDTA-free trypsin, centrifuged at 1000 r/min for 5 min, and washed with PBS. Cells were re-suspended with binding buffer, incubated for 15 min with Annexin V-FITC and PI, and subjected to FACS analysis. For the experiment, Annexin V-FITC green fluorescence was detected by the channel FITC (FL1), and PI red fluorescence was detected by the channel PI (FL3). The percentage of Annexin V-FITC and PI stained cells was assessed using Accuri C6 software (Becton Dickinson).
Transient transfection
NRK-52E cells were transfected with the pmRFP-LC3 plasmid using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The pmRFP-LC3 (#21075) plasmid was obtained from Addgene. NRK-52E cells transfected with pmRFP-LC3 were then incubated with or without 100 μmol/L emodin for 1 h and were observed for the formation of autophagosomes under a fluorescent microscope.
Statistical analysis
The data are expressed as the mean±standard error(SEM). Statistical analysis was performed using the non-parametric Mann-Whitney U-test to compare data in different groups. A P-value < 0.05 was considered to be statistically significant.
Results
Cisplatin induces apoptosis in renal tubular cells
Previous studies have suggested that cisplatin might induce apoptosis in renal tubular epithelial cells8. In our study, NRK-52E cells were exposed to different concentrations of cisplatin (from 10 to 50 μmol/L), and cell viability was evaluated. As expected, cisplatin resulted in concentration-dependent cell death. At a low concentration, cisplatin exhibited minor toxicity by inducing the death of several cells, as revealed by the appearance of round cells and reduced formazan formation. At high concentrations (20 and 50 μmol/L), however, cisplatin caused severe cellular damage (Figure 1A and 1B). These data indicate that cisplatin induced NRK-52E cell death in a dose-dependent manner.

Cisplatin induces apoptosis in renal tubular cells. (A and B) NRK-52E cells treated with or without (Ctrl) different concentrations of cisplatin (10–50 μmol/L) for 24 h were subjected to phase-contrast microscopy (A) and an assessment of cell viability by formazan assay (B). (C) NRK-52E cells were treated with 50 μmol/L cisplatin at different time points and subjected to Western blot analysis of caspase-3 and cleaved caspase-3 expression. The β-actin level is shown as a loading control. (D) Quantification of individual signals normalized to the level of β-actin is presented. (E and F) Cells treated with or without 50 μmol/L cisplatin for 24 h were subjected to Annexin V and PI assay (E) and quantification of the percentage of apoptotic cells (F). Mean±SEM. n=3. bP<0.05.
We also examined the expression of apoptosis-related markers after exposing cells to 50 μmol/L cisplatin for different time intervals. As shown in Figure 1C, the expression of cleaved caspase-3 increased when cells were treated with cisplatin for more than 12 h (Figure 1C and 1D). Taken together, these data confirmed the presence of a time- and concentration-dependent pro-apoptotic effect of cisplatin on NRK-52E cells. Additionally, after 24 h of treatment, FACS analysis revealed that an increased fraction of apoptotic cisplatin-treated cells (8.9% early apoptotic cells and 17.4% late apoptotic cells) was detected (Figure 1E and 1F).
Emodin ameliorates cisplatin-triggered apoptosis
Some studies have shown that emodin exerts a renoprotective effect in cisplatin-induced renal injury18,23. To examine whether cisplatin-triggered apoptosis can be inhibited by emodin in renal tubular cells, NRK-52E cells were treated with cisplatin with or without emodin for up to 24 h, and microscopic analysis was performed. The cisplatin-treated cells exhibited abundant cellular death that was markedly attenuated by treatment with emodin, especially at 100 μmol/L (Figure 2A). Quantitative analysis showed that the number of apoptotic cells was significantly decreased by emodin (Figure 2B).

Emodin ameliorates cisplatin-triggered apoptosis. (A and B) NRK-52E cells were treated with 50 μmol/L cisplatin together with the indicated concentrations of emodin for 24 h and were subjected to phase-contrast microscopy (A) and quantification of the number of apoptotic cells (B). (C) Cells were treated with cisplatin in the absence or presence of emodin for 24 h, then were washed repeatedly and reseeded in fresh culture medium without cisplatin or emodin and cultured for another 24 h. Cells were harvested and counted by trypan blue exclusion. (D) Cells were treated with 50 μmol/L cisplatin with or without 100 μmol/L emodin for 12 h or 24 h and were subjected to Western blot analysis of caspase-3 and cleaved caspase-3 levels. The level of β-actin is shown as a loading control. (E) Quantification of individual signals normalized to the level of β-actin is presented. Mean±SEM. n=4. bP<0.05.
To further test the effect of emodin on cell viability, NRK-52E cells were treated with cisplatin in the absence or presence of emodin for 24 h, then the cells were washed repeatedly and reseeded in fresh culture medium without cisplatin or emodin and cultured for another 24 h. We found that the number of emodin-treated NRK-52E cells was much higher than cisplatin-treated cells (Figure 2C). Notably, treatment with emodin alone did not induce apoptosis within 24 h (data not shown).
The anti-apoptotic effect of emodin was further confirmed by examining the level of cleaved caspase-3. Western blot analysis showed that compared with the cisplatin-treated group, intervention with emodin restored the level of caspase-3 and significantly suppressed the expression of cleaved caspase-3 (Figure 2D and 2E). Taken together, these results indicate that emodin could effectively protect against cisplatin-induced apoptosis in NRK-52E cells.
Induction of autophagic activity by emodin in NRK-52E cells
We observed that cells treated with 100 μmol/L emodin exhibited a prominent vacuolization of the cytoplasm, which is a typical morphological change resulting from autophagy. Autophagy plays a critical role in maintaining cell homeostasis and might serve as an anti-apoptotic mechanism. We therefore examined whether autophagy could be induced in NRK-52E cells exposed to emodin at different concentrations and different time points. To this end, we examined changes in the expression of autophagy biomarkers, including microtubule-associated protein 1 light chain 3 (LC3) and its lipidated form (LC3-II)24. The results showed that treatment with emodin increased the level of LC3-II in a dose- (Figure 3A and 3B) and time-dependent (Figure 3C and 3D) manner.

Induction of autophagic activity by emodin in NRK-52E cells. (A) NRK-52E cells were treated with the indicated concentrations of emodin for 1 h and were subjected to Western blot analysis of LC3-I and LC3-II protein. Hanks' balanced salt solution was used as a positive control. The level of β-actin is shown as a loading control. (B) Quantification of individual signals normalized to the level of β-actin is presented. (C) Cells were treated with 50 μmol/L emodin for 0–6 h and subjected to Western blot analysis of LC3-I and LC3-II protein levels. The level of β-actin is shown as a loading control. (D) Quantification of individual signals normalized to the level of β-actin is presented. (E) Fluorescent microscopic analysis of NRK-52E cells transfected with pmRFP fluorescent-tagged LC3 plasmid and exposed to 100 μmol/L emodin for 1 h. The red color indicates autophagosomes (arrows). (F) Cells were treated with 50 μmol/L cisplatin for 0.5, 1, 2, 6 or 12 h and were subjected to Western blot analysis of LC3-I and LC3-II protein levels. The level of β-actin is shown as a loading control. (G) Quantification of individual signals normalized by the level of β-actin is presented. (H) Cells were treated with 50 μmol/L cisplatin with or without 100 μmol/L emodin for 1 h and were subjected to Western blot analysis of LC3-I and LC3-II protein levels. The level of β-actin is shown as a loading control. (I) Quantification of individual signals normalized to the level of β-actin is presented. Mean±SEM. n=4. bP<0.05.
There are certain limitations of only testing LC3 conversion for the activity of autophagy25, and the GFP-LC3 or RFP-LC3 labeling method is often recommended as an additional approach. Thus, NRK-52E cells were transiently transfected with a plasmid expressing pmRFP-tagged LC3 and were then exposed to emodin. As shown in Figure 3E, following emodin exposure, cells contained an increased number of punctate structures at 1 h, whereas cells without emodin treatment showed a diffuse distribution of red fluorescence, which indicated an increase in the formation of autophagosomes. Together, these results suggest that emodin effectively induced autophagic activation in NRK-52E cells.
Cisplatin could induce autophagy7,8,9, which may exert protective effects against cisplatin-induced apoptosis. Consistent with previous reports, when NRK-52E cells were exposed to 50 μmol/L cisplatin, LC3-II markedly increased and reached a maximum level at 1 h and 6 h (Figure 3F and 3G). We further tested whether emodin affects cisplatin-induced autophagy. As shown in Figure 3H and 3I, cisplatin induced an increase in LC3-II that was further enhanced by 100 μmol/L emodin treatment. Together, our findings indicated that emodin induces autophagy and enhances the activation of autophagy induced by cisplatin.
Autophagy mediates the cytoprotective effect of emodin against Cisplatin-triggered apoptosis
We next asked whether autophagy was involved in the protective effect against cisplatin-induced apoptosis. To this end, rapamycin, which has been reported to activate autophagy by inhibiting the mTOR signaling pathway26, was utilized. As expected, Western blot analysis showed that autophagic activity was significantly induced by rapamycin treatment at different time points (Figure 4A and 4B).

Emodin-induced autophagy mediates the cytoprotective effect against cisplatin-triggered apoptosis. (A) NRK-52E cells were treated with 100 nmol/L rapamycin (Rap) at different times and were subjected to Western blot analysis of the LC3-I and LC3-II protein levels. The level of β-actin is shown as a loading control. (B) Quantification of individual signals normalized to the level of β-actin is presented. (C) Cells were treated with 50 μmol/L cisplatin with or without 100 nmol/L Rap for 24 h and were subjected to phase-contrast microscopy. (D and E) Cells were treated with 50 μmol/L cisplatin with or without 100 nmol/L Rap for 24 h and were subjected to Annexin V and PI assays (D), and the percentage of apoptotic cells was quantified (E). (F) Cells were treated with 50 μmol/L cisplatin with or without 100 μmol/L emodin and 10 nmol/L bafilomycin A1 (BA) for 24 h and were subjected to phase-contrast microscopy. Mean±SEM. n=4. bP<0.05.
To examine whether the activation of autophagy by rapamycin reproduced the anti-apoptotic effect of emodin, cells were exposed to cisplatin in the absence or presence of rapamycin and were subjected to morphological observation (Figure 4C) and FACS analysis (Figure 4D and 4E). As shown in Figure 4C, microscopic analyses showed that rapamycin attenuated cisplatin-induced cell death. FACS analysis revealed that the percentage of apoptotic cells did not differ significantly among the three treatment combinations during the early stage (2-6 h) of cisplatin treatment (data not shown). After 24 h, approximately 12.7% early apoptotic and 11.3% late apoptotic cells were detected in cisplatin-treated NRK-52E cells (Figure 4D and 4E). However, in the rapamycin-treated group, the percentages of early and late apoptotic cells dropped to approximately 8.8% and 5.5%, respectively (Figure 4D and 4E).
To further test whether the anti-apoptotic effects of emodin were mediated by the activation of autophagy, bafilomycin A1, an autophagic inhibitor that blocks autophagosome-lysosome fusion, was applied25. As shown in Figure 4F, cisplatin-induced apoptosis was substantially attenuated in the emodin-treated group. However, when autophagy flux was interrupted by bafilomycin A1, the protective effects of emodin were abolished. Overall, these data demonstrate that autophagy induced by emodin mediates its cytoprotective effects against cisplatin-triggered apoptosis.
Emodin induces autophagy by regulating the AMPK/mTOR signaling pathways
To identify the mechanism by which emodin induces autophagy, we examined the effects of emodin on AMPK activity and mTOR signaling, both of which are well-known upstream regulators of autophagy. When NRK-52E cells were exposed to different concentrations of emodin (10 and 50 μmol/L), as shown in Figure 5A and 5B, emodin time-dependently decreased the phosphorylation of p70 ribosomal S6 kinase (p70S6K) at both concentrations. Consistently, treatment with emodin markedly suppressed the phosphorylation of mTOR and induced the phosphorylation of AMPK (Figure 5C and 5D).

Emodin induces autophagy by regulating the AMPK/mTOR signaling pathways. (A) NRK-52E cells were treated with different concentrations of emodin (10 and 50 μmol/L) for 0–6 h and were subjected to Western blot analysis of p-p70S6K protein level. The level of β-actin is shown as a loading control. (B) Quantification of individual signals normalized to the level of β-actin is presented. (C) Cells were treated with 50 μmol/L emodin for 0–6 h and were subjected to Western blot analysis of p-mTOR, mTOR, p-AMPK and AMPK protein levels. The level of β-actin is shown as a loading control. (D) Quantification of individual signals normalized to the level of β-actin is presented. (E) Cells were treated with 50 μmol/L emodin with or without 10 μmol/L compound C for 1 h and were subjected to Western blot analysis of p-AMPK, LC3-I and LC3-II protein levels. The level of β-actin is shown as a loading control. (F) Quantification of individual signals normalized to the level of β-actin is presented. (G) Fluorescent microscopic analysis of NRK-52E cells transfected with pmRFP fluorescent-tagged LC3 plasmid and exposed to 50 μmol/L emodin with or without 10 μmol/L compound C for 1 h. The number of punctate structures was quantified (H). Mean±SEM. n=3. bP<0.05.
To further investigate the role of AMPK activation in emodin-induced autophagy and other anti-apoptotic effects, compound C, a well-known AMPK inhibitor, was used. As expected, compound C potently suppressed emodin-induced AMPK phosphorylation. In parallel, emodin-induced LC3 conversion was abolished by compound C (Figure 5E and 5F). Additionally, as shown in Figure 5G and 5H, following emodin exposure, pmRFP-tagged LC3-transfected cells exhibited increased punctate structures. When compound C was added, the number of punctate structures was significantly decreased.
We next examined the influence of compound C on the anti-apoptotic effect of emodin, as indicated by cellular morphological changes and the level of cleaved caspase-3. As expected, the further induction of LC3 conversion caused by emodin in addition to cisplatin was significantly inhibited by treatment with compound C (Figure 6A and 6B). As shown in Figure 6C, the protective effect of emodin on cisplatin-induced apoptosis was largely abolished by the inhibition of AMPK activation. Additionally, Western blot analysis showed that intervention with compound C significantly increased the level of cleaved caspase-3 (Figure 6D and 6E). Overall, these data demonstrated that the emodin-induced activation of AMPK was important both in the induction of autophagy and for the cytoprotective effect of emodin against cisplatin-triggered apoptosis.

Inhibition of AMPK activation abolished the anti-apoptotic effect of emodin. (A) Cells were treated with 50 μmol/L cisplatin with or without 50 μmol/L emodin and 10 μmol/L compound C for 1 h and were subjected to Western blot analysis of LC3-I and LC3-II protein levels. The level of β-actin is shown as a loading control. (B) Quantification of individual signals normalized to the level of β-actin is presented. (C) Cells were treated with 50 μmol/L cisplatin with or without 50 μmol/L emodin and 10 μmol/L compound C for 24 h and subjected to phase-contrast microscopy. (D) Cells were treated with 50 μmol/L cisplatin with or without 50 μmol/L emodin and 10 μmol/L compound C for 24 h and were subjected to Western blot analysis of caspase-3 and cleaved caspase-3 protein levels. The level of β-actin is shown as a loading control. (E) Quantification of individual signals normalized by the level of β-actin is presented. Mean±SEM. n=3. bP<0.05.
Discussion
In the present investigation, we demonstrated that emodin, a bioactive substance found in rhubarb, suppressed cisplatin-induced renal tubular epithelial cell apoptosis. We found that this protective effect was mediated by the activation of autophagy. Previous reports have shown that rhubarb and emodin possess renal protective potential23,27. Our current findings present a new mechanism for the renal-protective effect of emodin.
Tubular epithelial cell apoptosis is an important pathogenic alteration that leads to Cisplatin-related renal injury. Several reports have suggested that emodin has anti-apoptotic effects. For example, Dai et al reported that emodin could protect WI-38 cells from Cisplatin-induced apoptosis by affecting the cell cycle28. Consistently, we observed that emodin protected cells against Cisplatin cytotoxicity via the induction of autophagy. However, there have been reports that emodin promotes apoptosis, especially in tumor cells29. This discrepancy could be due to different experimental conditions, such as the use of different cell types or intervention approaches. Interestingly, autophagy is considered an alternative cell death mechanism. Autophagic and apoptotic cell death are, in fact, the two main manifestations of programmed cell death30,31. The over-induction of autophagy may also cause cell damage. Although the detailed mechanisms are unknown, it is possible that the degree of autophagy induced by emodin might be dependent on cell type and may therefore differentially affect cells.
Autophagic and apoptotic cell death are the two primary manifestations of programmed cell death. Autophagy (type II programmed cell death) has been considered an alternative cell death mechanism30,31 and is an intracellular metabolic process that results in the digestion of dysfunctional organelles and proteins32. Both autophagy and apoptosis are basic physiologic processes that play vital roles in maintaining cellular homeostasis. An increasing number of in vivo and in vitro studies have shown that apoptosis is one of the consequences of cisplatin treatment in renal cells33. There are also studies indicating that cisplatin is able to induce autophagy in some renal cell lines, such as renal tubular epithelial cells7,8,9. However, the relationship between cisplatin-induced apoptosis and autophagy remains controversial.
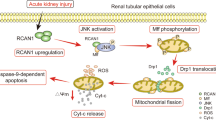
In our study, autophagy might serve as the central protective mechanism mediating the anti-apoptotic effect of emodin. Emodin pretreatment further strengthened the autophagic activity induced by cisplatin, especially at an early phase, whereas cisplatin-induced apoptosis was significantly alleviated. Similarly, emodin has been reported to trigger autophagy in glioma and human vascular smooth muscle cells34,35. However, the mechanisms by which emodin activates autophagy are unknown. Previous studies have demonstrated that emodin is a potent AMPK activator via the inhibition of mitochondrial respiratory complex I activity and thus leads to increased Ca2+/calmodulin-dependent protein kinase kinase activity19. Other studies have shown that aloe-emodin can directly bind mTORC2 and inhibit its kinase activity to suppress tumor growth20. Our data indicated that emodin-induced autophagy was also mediated by the AMPK/mTOR pathway. We hypothesized that emodin induces autophagy by activating AMPK and inhibiting the mTOR signaling pathway, and it thus exerts a cytoprotective effect against cisplatin-triggered apoptosis (Figure 7).

The hypothesized role of emodin in cisplatin-induced renal tubular cell apoptosis. Emodin induces autophagy by activating the AMPK pathway and inhibiting the mTOR pathway, thereby exerting a cytoprotective effect against cisplatin-triggered apoptosis.
Oxidative stress plays a critical role in the pathogenesis of cisplatin nephrotoxicity. It was recently reported that cisplatin treatment generates significant oxidant loading, which results in accelerated oxidation reactions in kidney tissues36. Previous studies also showed that emodin exerts antioxidant effects. Ali et al reported that treatment with emodin resulted in a remarkable and significant restoration of anti-oxidative capability in cisplatin-induced nephrotoxicity23. Mostafa also demonstrated that emodin suppressed cisplatin-induced oxidative stress and, hence, protected HEK 293 cells from cellular damage18. Mitochondrial damage results in increased ROS production, which leads to the release of pro-apoptotic proteins and results in cellular apoptosis37. Impaired mitochondria can also be degraded by autophagy in a process referred to as mitochondrial autophagy38. Impaired mitochondria are specifically engulfed by autophagosomes and are selectively degraded via fusion with lysosomes, thereby maintaining a stable intracellular environment. From this viewpoint, the autophagic activity induced by emodin may help rebalance mitochondrial metabolism, thereby reducing the generation of ROS and attenuating cisplatin-induced oxidative stress.
In summary, we found that autophagy is activated in cisplatin-treated NRK-52E cells and that the strengthening of the activation of autophagy by emodin plays a cytoprotective role against cisplatin-triggered apoptosis. These effects are mediated by the induction of AMPK and the suppression of mTOR/p70S6K signaling. This study confirmed that emodin ameliorates cisplatin-induced renal tubular cell apoptosis and provided additional evidence in support of the clinical usage of emodin in the treatment of cisplatin-related kidney diseases.
Author contribution
Hong LIU, Liu-bao GU, and Wei SUN designed the research; Hong LIU and Liu-bao GU performed the research; Yue TU, Hao HU, and Yan-ru HUANG contributed helpful discussions and technical assistance; Hong LIU, Liu-bao GU, and Wei SUN analyzed the data; Hong LIU and Liu-bao GU wrote the manuscript.
References
Sahni V, Choudhury D, Ahmed Z . Chemotherapy-associated renal dysfunction. Nat Rev Nephrol 2009; 5: 450–62.
Yao X, Panichpisal K, Kurtzman N, Nugent K . Cisplatin nephrotoxicity: a review. Am J Med Sci 2007; 334: 115–24.
Ozkok A, Edelstein CL . Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res Int 2014; 2014: 967826.
Tsuruya K, Ninomiya T, Tokumoto M, Hirakawa M, Masutani K, Taniguchi M, et al. Direct involvement of the receptor-mediated apoptotic pathways in cisplatin-induced renal tubular cell death. Kidney Int 2003; 63: 72–82.
Mitazaki S, Hashimoto M, Matsuhashi Y, Honma S, Suto M, Kato N, et al. Interleukin-6 modulates oxidative stress produced during the development of cisplatin nephrotoxicity. Life Sci 2013; 92: 694–700
Mizushima N . Autophagy: process and function. Genes Dev 2007; 21: 2861–73.
Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z . Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 2008; 74: 631–40.
Yang C, Kaushal V, Shah SV, Kaushal GP . Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 2008; 294: F777–87.
Kaushal GP, Kaushal V, Herzog C, Yang C . Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 2008; 4: 710–2.
Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, et al. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 2008; 4: 783–91.
Li X, Liu W, Wang Q, Liu P, Deng Y, Lan T, et al. Emodin suppresses cell proliferation and fibronectin expression via p38MAPK pathway in rat mesangial cells cultured under high glucose. Mol Cell Endocrinol 2009; 307: 157–62.
Chang CH, Lin CC, Yang JJ, Namba T, Hattori M . Anti-inflammatory effects of emodin from ventilago leiocarpa. Am J Chin Med 1996; 24: 139–42.
Liu Y, Jia L, Liu ZC, Zhang H, Zhang PJ, Wan Q, et al. Emodin ameliorates high-glucose induced mesangial p38 over-activation and hypocontractility via activation of PPARgamma. Exp Mol Med 2009; 41: 648–55.
Chang YC, Lai TY, Yu CS, Chen HY, Yang JS, Chueh FS, et al. Emodin induces apoptotic death in murine myelomonocytic leukemia WEHI-3 cells in vitro and enhances phagocytosis in leukemia mice in vivo. Evid Based Complement Alternat Med 2011; 2011: 523–96.
Liu A, Chen H, Wei W, Ye S, Liao W, Gong J, et al. Antiproliferative and antimetastatic effects of emodin on human pancreatic cancer. Oncol Rep 2011; 26: 81–9.
Yon JM, Baek IJ, Lee BJ, Yun YW, Nam SY . Emodin and [6]-gingerol lessen hypoxia-induced embryotoxicities in cultured mouse whole embryos via upregulation of hypoxia-inducible factor 1alpha and intracellular superoxide dismutases. Reprod Toxicol 2011; 31: 513–8.
Lan T, Liu W, Xie X, Xu S, Huang K, Peng J, et al. Sphingosine kinase-1 pathway mediates high glucose-induced fibronectin expression in glomerular mesangial cells. Mol Endocrinol 2011; 25: 2094–105.
Waly MI, Ali BH, Al-Lawati I, Nemmar A . Protective effects of emodin against cisplatin-induced oxidative stress in cultured human kidney (HEK 293) cells. J Appl Toxicol 2013; 33: 626–30.
Song P, Kim JH, Ghim J, Yoon JH, Lee A, Kwon Y, et al. Emodin regulates glucose utilization by activating AMP-activated protein kinase. J Biol Chem 2013; 288: 5732–42.
Liu K, Park C, Li S, Lee KW, Liu H, He L, et al. Aloe-emodin suppresses prostate cancer by targeting the mTOR complex 2. Carcinogenesis 2012; 33: 1406–11.
Shaw RJ . LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol 2009; 196: 65–80.
Gu L, Johno H, Nakajima S, Yoshitomi T, Takahashi S, Kitamura M . Intervention in genotoxic stress-induced senescence by cordycepin through activation of eIF2alpha and suppression of Sp1. Toxicol Sci 2013; 134: 345–54.
Ali BH, Al-Salam S, Al Husseini IS, Al-Lawati I, Waly M, Yasin J, et al. Abrogation of cisplatin-induced nephrotoxicity by emodin in rats. Fundam Clin Pharmacol 2013; 27: 192–200.
Tanida I, Ueno T, Kominami E . LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36: 2503–18.
Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research. Cell 2010; 140: 313–26.
He C, Klionsky DJ . Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43: 67–93.
Wei J, Ni L, Yao J . Experimental treatment of rhubarb on mesangio-proliferative glomerulonephritis in rats. Zhonghua Nei Ke Za Zhi 1997; 36: 87–9.
Dai Z, Zhong LF . Inhibitory effect of emodin on WI-38 cell apoptosis induced by cisplatin. Chin J Pharmacol Toxicol 2003; 17: 276–80.
Wang W, Sun Y, Li X, Li H, Chen Y, Tian Y, et al. Emodin potentiates the anticancer effect of cisplatin on gallbladder cancer cells through the generation of reactive oxygen species and the inhibition of survivin expression. Oncol Rep 2011; 26: 1143–8.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451: 1069–75.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–52.
Boya P, Reggiori F, Codogno P . Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15: 713–20.
Rovetta F, Stacchiotti A, Consiglio A, Cadei M, Grigolato PG, Lavazza A, et al. ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Exp Cell Res 2012; 318: 238–50.
Mijatovic S, Maksimovic-Ivanic D, Radovic J, Miljkovic D, Harhaji L, Vuckovic O, et al. Anti-glioma action of aloe emodin: the role of ERK inhibition. Cell Mol Life Sci 2005; 62: 589–98.
Wang X, Zou Y, Sun A, Xu D, Niu Y, Wang S, et al. Emodin induces growth arrest and death of human vascular smooth muscle cells through reactive oxygen species and p53. J Cardiovasc Pharmacol 2007; 49: 253–60.
Cetin R, Devrim E, Kilicoglu B, Avci A, Candir O, Durak I . Cisplatin impairs antioxidant system and causes oxidation in rat kidney tissues: possible protective roles of natural antioxidant foods. J Appl Toxicol 2006; 26: 42–6.
Ghelli A, Zanna C, Porcelli AM, Schapira AH, Martinuzzi A, Carelli V, et al. Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J Biol Chem 2003; 278: 4145–50.
Lemasters JJ . Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005; 8: 3–5.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No 81373607 to Wei SUN); the Jiangsu Scientific Research Innovation Fund for post-graduate students (SJLX15_0445 to Hong LIU); the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD; to Wei SUN); and Science and Technology Funding for Life and Health Care of Jiangsu Province (BL2012032 to Wei SUN).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, H., Gu, Lb., Tu, Y. et al. Emodin ameliorates cisplatin-induced apoptosis of rat renal tubular cells in vitro by activating autophagy. Acta Pharmacol Sin 37, 235–245 (2016). https://doi.org/10.1038/aps.2015.114
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.114
Keywords
This article is cited by
-
The underlying mechanisms of cisplatin-induced nephrotoxicity and its therapeutic intervention using natural compounds
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
Combining use of phillyrin and autophagy blocker exerts suppressive effect on nasopharyngeal carcinoma cell malignancy and autophagy via AMPK/mTOR/p70s6k signaling pathway
Molecular & Cellular Toxicology (2023)
-
Synergistic protective effects of lycopene and N-acetylcysteine against cisplatin-induced hepatorenal toxicity in rats
Scientific Reports (2021)
-
Microcalorimetric evaluation of the effects of three anthraquinone derivatives from Chinese Rhubarb and the synergistic effect of the mixture on Staphylococcus aureus
Journal of Thermal Analysis and Calorimetry (2020)
-
Emodin alleviates alternatively activated macrophage and asthmatic airway inflammation in a murine asthma model
Acta Pharmacologica Sinica (2018)
