
Abstract
Aim:
To elucidate how krüppel-like factor (KLF5) activates cyclin D1 expression in Ang II-induced vascular smooth muscle cells (VSMC) proliferation.
Methods:
An adenoviral vector containing the full-length cDNA of KLF5 and a recombinant plasmid expressing c-Jun were constructed. MTT assay and flow cytometric analysis were used to determine the effect of Ang II on cell growth. The luciferase assay and chromatin immunoprecipitation were used to detect the relationship between KLF5 and c-Jun in transactivation of cyclin D1 gene expression.
Results:
Ang II upregulated the expression of KLF5 with concurrent acceleration of the cell cycle progression in VSMCs. Ang II induced KLF5 activation via the ERK and p38 MAPK pathways triggered by AT-1 receptor. High DNA binding activity and functional interaction of KLF5 and c-Jun were found in Ang II-induced VSMCs. Cotransfection of KLF5 and c-Jun expression vectors significantly increased cyclin D1 promoter activity.
Conclusion:
KLF5 is a downstream signal of the ERK 1/2 and p38 MAPK pathways, and activates the transcription of cyclin D1 gene via functional interaction with c-Jun in Ang II-induced VSMC proliferation.
Similar content being viewed by others


Introduction
Multiple lines of experimental and clinical evidence indicate that angiotensin II (Ang II) not only induces hypertension, but also directly contributes to pathophysiological vascular thickening and atherosclerosis in humans1. A large number of studies of the functions of Ang II in vitro and in vivo have indicated that Ang II activates multiple intracellular signaling cascades, such as those mediated by mitogen-activated protein kinase (MAPK), and regulates various transcription factors, and causes cell growth in vascular smooth muscle cells (VSMCs)1, 2, 3. A growing body of evidence shows that Ang II type 1 (AT-1) receptor but not AT-2 is involved in the signal transduction underlying Ang II-induced VSMC growth1, such as activation of JNK4 and p385. However, little is known about the role of these signal cascades in Ang II-induced proliferative response.
Krüppel-like factor 5 (KLF5), a member of the Sp/KLF family of zinc finger factors, is a key regulator of cardiovascular remodeling6. One mechanism by which KLF5 accomplishes its proliferative effect is to transcriptionally activate several cell cycle-promoting genes, including cyclin D1, cyclin B1, and Cdc27, 8, 9. Our recent study has demonstrated the interaction of KLF5 and c-Jun in Ang II-induced suppression of p21 expression10. In view of involvement of KLF5 and cyclin D1 in VSMC proliferation, we hypothesize that KLF5 might be required for Ang II-mediated expression of cyclin D1 and cell proliferation in VSMC.
In this study, we report that KLF5 plays a crucial role in Ang II-induced cyclin D1 expression and proliferation of VSMCs. We show that Ang II induces KLF5 expression and activation via ERK and p38 MAPK pathways. A functional interaction was found between KLF5 and c-Jun, which enhanced the transactivating activity of the both proteins, and consequently led to a synergistic increase in the expression of cyclin D1 gene. These results indicate that KLF5 is a downstream target of the ERK 1/2 and p38 MAPK pathways and enhances the transcriptional ability of cyclin D1 gene to promote proliferation in Ang II-induced VSMCs.
Materials and methods
Construction of recombinant plasmids and adenoviral vectors
The pGL3-CD1-Luc luciferase reporter plasmid containing the cyclin D1 promoter (from -1745 to +134) was a generous gift from Dr Richard G PESTELL (Northwestern University Medical School, Chicago, IL). Full-length mouse KLF5 cDNA was cloned into pEGFP-N1 and pAd/CMV/V5-DEST Gateway Vector (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol to obtain the pEGFP-KLF5 and Ad-KLF5 constructs, respectively. To construct the c-Jun expression plasmid, c-Jun cDNA was obtained from the pBIISK(−)-Jun plasmid via EcoR I digestion and then inserted into the pcDNA3.1 vector and sequenced. All clones were verified by sequencing (data not shown).
Cell culture and treatment
VSMCs were isolated from the thoracic aorta of 90–110 g male Sprague-Dawley rats as described previously11. VSMCs and the COS-7 cell line were grown in low glucose DMEM (Dulbecco's modified Eagle's medium) (Invitrogen), supplemented with 10% FBS (fetal bovine serum) under 5% CO2 at 37 °C. VSMCs grown to 70% confluence were serum deprived for 24 h, then treated with 10−7 mol/L Ang II following pretreatment with or without valsartan (antagonist of AT-1 receptor) and PD123319 (antagonist of AT-2 receptor) (Sigma)3, 12 in DMEM containing 2% FBS for the indicated time periods.
Transfection and infection
The cultured VSMCs grown to 60% confluence were serum deprived for 24 h and then transfected with rat KLF5-specific siRNA (5′-AACCCGGAUCUGGAGAAGCGA-3′) or non-specific NS-siRNA (5′-GCGCGCUUUGUAGGAUUCG-3′)13 using the Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer's protocol. Twenty hours after transfection, the VSMCs were treated with Ang II (10−7 mol/L) or vehicle for 24 h. Alternatively, VSMCs were infected with Ad-KLF5 or empty Ad for 48 h and then treated with Ang II. Cells were then collected for assays, which are described below.
MTT assay
After treatment, the viability of VSMCs cultured in 96-well plates was measured using MTT (3-[4,5-dimehyl-2-thiazolyl]-2, 5-diphenyl-2H-tetrazolium bromide). In brief, after cells were collected and incubated in medium containing 2 mg/mL MTT reagent (Sigma Chemical Co) at 37 °C for 4 h, the formazan crystals converted from tetrazolium salts by viable cells were dissolved in DMSO (200 μL/well) and the absorbance was measured at 570 nm using a microplate reader to reflect cell viability. The experiment was repeated three times.
Cell number assay
Cell count experiments were performed as described previously14. Briefly, a suspension of VSMCs (0.5×105 cells/mL) was prepared and treated with or without Ang II (10−7 mol/L) for different time periods. Cells were counted in a hemocytometer using a light microscope.
Flow cytometric analysis
Cells were plated in 100-mm dishes at a density of 3×106 per dish. The medium was removed and pooled with trypsinized adherent cells. Cells were washed and resuspended in cold PBS at a density of (1–3)×106 cells/mL, and then 500 μL of propidium iodide (PI) buffer (0.1 mg/mL PI in PBS) and 500 μL of RNase A (2 mg/mL) were added, respectively. After 30 min incubation at room temperature in the dark, cells were filtered through a nylon mesh filter and subjected to flow cytometry.
RNA isolation and reverse transcription (RT)-PCR
Total RNA was isolated from VSMCs using the TRIzol reagent, according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Reverse transcription was performed with the Superscript First Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). The cDNA was then used as a template for PCR using specific primers for cyclin D1 (forward, 5′-ACCAATCTCCTCAACGACC-3′; reverse, 5′-TTGTTCTCATCCGCCTCT-3′) and β-actin (forward, 5′-CAGGGTGTGATGGTGGG-3′; reverse, 5′-GGAAGAGGATGCGGCAG-3′). The amplified products were separated on a 2% agarose gel containing ethidium bromide, and the band intensities were quantified using the Image J software provided by the NIH.
Western blot and immunoprecipitation
Lysates from VSMCs were prepared with lysis buffer (1% Triton X-100, 150 mmol/L NaCl, 10 mmol/L Tris-HCl, pH 7.4, 1 mmol/L EDTA, 1 mmol/L EGTA, pH 8.0, 0.2 mmol/L Na3VO4, 0.2 mmol/L phenylmethylsulfonyl fluoride and 0.5% NP-40). Equal amounts of protein (60∼100 μg) were separated via 8% SDS-PAGE and transferred to a PVDF membrane. Membranes were blocked with 5% BSA for 2 h at room temperature and then incubated with anti-KLF5 (1:400), anti-cyclin D1 (1:300), anti-PCNA (1:400), anti-SM22α (1:400) (Santa Cruz), anti-AT-1 (1:400), anti-ERK (1:400), anti-phospho-ERK (1:400), anti-JNK (1:200), anti-phospho-JNK (1:200), anti-p38 MAPK (1:400), anti-phospho-p38 MAPK (1:400) or anti-β-actin (1:1000) antibodies (Cell Signaling) overnight, and then with the HRP-conjugated secondary antibody (1:10 000) for 2 h. Bands were detected using the enhanced chemiluminescence (ECL) detection system. The experiments were repeated three times. Immunoprecipitation was performed as described previously15. Briefly, the cell extract was first pre-cleared with protein A-agarose. The supernatant was incubated with 1–2 μg of anti-phospho-Ser or anti-KLF5 antibodies and subsequently incubated with protein A-agarose overnight at 4 °C. Protein A-agarose-antigen-antibody complexes were then analyzed via Western blot as described above. The experiments were repeated at least three times.
Chromatin immunoprecipitation (ChIP) assay
VSMCs were treated with Ang II (10−7 mol/L) for 1 h and fixed with 1% formaldehyde. ChIP assays were then performed using 2 μg of anti-KLF5 and anti-c-Jun antibodies (Santa Cruz) as described previously16. An aliquot of the cell lysate was used to isolate total DNA. PCR amplification of the immunoprecipitated DNA was performed using the following primers: KLF5, 5′-CACCTTATCGGCTCACACAT-3′ and 5′-GAGACACGATAGGCTCTTTG-3′ (-868∼-1094); 5′-GGATAAACCGGTCACTGTAA-3′ and 5′-CCAGACGAGCCCTAAGTTTT-3′ (-538∼-801); 5′-CGCTCTTTCCCAGCTTAGA-3′ and 5′-CAACTTCAACAAAACTCCCCT-3′ (-10∼-245); AP-1, 5′-CGCTCTTTCCCAGCTTAGA-3′ and 5′-CAACTTCAACAAAACTCCCCT-3′ (-10∼-245).
Luciferase assay
COS-7 cells were grown to 70% confluence and then transfected in triplicate with pGL3-CD1-luc, pEGFP-KLF5, pcDNA3.1-c-Jun or pGL3-Basic, along with pRL-TK in the absence of serum medium. Twenty-four hours after transfection, cells were treated with Ang II (10−7 mol/L) for 24 h. Cell lysates were harvested and then the luciferase activity was measured with the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI) according to the manufacturer's manual. The relative luciferase activities compared with the luciferase activities of pRL-TK were presented as means±SEM.
Statistical analysis
The experiments were repeated three times with similar results. The data shown are derived from replicated measurements obtained from a single VSMC culture. Data are presented as means±SEM. Statistical analyses were carried out with the SPSS 11.5 statistical software package (Chicago, IL). The primary statistical test used was the one-way ANOVA. P<0.05 was considered statistically significant.
Results
Ang II promotes cell cycle progression to stimulate growth of VSMCs
Ang II functions as a growth factor in VSMCs17. Thus, we tested the effects of Ang II on VSMC growth. The results from MTT, cell number and flow cytometric analyses, were used to indirectly and directly determine cell viability, were expected to reflect the regulation of cell growth by Ang II. The results of the MTT and cell number assays showed that Ang II treatment caused a concentration-dependent stimulation of VSMC growth at 10−8 mol/L and higher. Significant stimulation was observed from the dose of 10−7 mol/L Ang II (Figure 1A, 1B). These results indicate that Ang II exerts a growth-stimulating effect on VSMCs. Furthermore, the proliferative effect of Ang II was verified by evaluating the fate of Ang II-treated VSMCs with regard to cell cycle progression. Cells were synchronized via 24-h serum starvation and then induced by treatment with Ang II to re-enter the cell cycle. Flow cytometric analysis was performed after propidium iodide staining. After 24 h of treatment, Ang II significantly reduced the proportion of cells in the G0/G1-phase, and increased the proportion of S-phase cells in a dose-dependent fashion (10−8-10−6 mol/L; Figure 1C). The level of proliferating cell nuclear antigen (PCNA), a marker of cell growth, paralleled the number of cells that re-entered the cell cycle, while the level of SM22α (a marker of differentiated VSMCs) inversely correlated with this number (Figure 1D). These results suggest that Ang II promotes cell cycle progression to stimulate growth of VSMCs.

Ang II promotes cell cycle progression to stimulate growth of VSMCs. (A and B) VSMC proliferation induced by Ang II was tested using MTT (A) or cell number (B) assays. bP<0.05, cP<0.01 vs PBS control. (C) Cell cycle of VSMCs was tested by flow cytometric analysis. bP<0.05, cP<0.01 vs PBS control. (D) Expression of PCNA and SM22α in VSMCs was detected by Western blotting. β-actin was used as an internal control.
KLF5 is involved in Ang II-induced cyclin D1 expression and VSMC proliferation
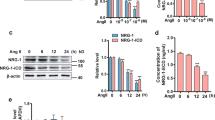
Cyclin D1 is a growth sensor that is induced by growth factors and mitogens to accelerate G1 progression18. To investigate the observed G1-phase progression after Ang II treatment, we first examined the effect of Ang II on cyclin D1 expression. In VSMCs, the amounts of cyclin D1 protein detected via Western blot analysis gradually increased with increasing doses of Ang II (Figure 2A). To determine the mechanism of cyclin D1 induction, total RNA was isolated from VSMCs, and RT-PCR was performed. Ang II (10−7 mol/L) induced the expression of cyclin D1 at the mRNA level in a time-dependent manner, with a maximum of a 4-fold increase at 12 h, and maintenance at this level at least for 24 h, with concurrent changes in the level of cyclin D1 protein expression (Figure 2).

KLF5 is involved in Ang II-induced cyclin D1 expression and VSMC proliferation. (A) cyclin D1 expression induced by Ang II was detected by Western blotting (left and middle) and RT-PCR analysis (right), respectively. The graphs represent the relative level of the protein and mRNA of four independent experiments. bP<0.05, cP<0.01 vs PBS control. (B) KLF5 protein was detected by Western blotting. β-actin was used as an internal control. The graphs represent the relative level of KLF5 protein of four independent experiments. bP<0.05, cP<0.01 vs PBS control. (C and D) Expression of KLF5 and cyclin D1 was detected by Western blotting. β-actin was used as an internal control. (E and F) Cell proliferation of VSMCs was analyzed by MTT assay (E) and flow cytometry (F). bP<0.05, cP<0.01 vs Ad or NS-siRNA.
KLF5 has previously been shown to increase cell growth and cell cycle progression in NIH3T3 cells by accelerating both the G1/S transition and entry into mitosis8, 9. To investigate whether KLF5 is involved in Ang II-induced cyclin D1 expression and cell proliferation, the level of KLF5 protein in VSMCs was detected via Western blot analysis following Ang II treatment. As shown in Figure 2B, Ang II increased KLF5 protein levels in a dose-dependent manner, which correlated with Ang II-induced cell proliferation and cyclin D1 expression. The level of KLF5 protein began to increase at 3 h after Ang II treatment, peaked at a maximum of 4-fold at 12 h, and declined thereafter, consistant with cyclin D1 expression profiles.
To confirm the role of KLF5 in Ang II-induced cyclin D1 expression and cell proliferation, VSMCs were infected with Ad-KLF5 to overexpress KLF5 or with KLF5-specific siRNA to knockdown endogenous KLF5 expression (Figure 2C). Overexpression of KLF5 increased endogenous cyclin D1 protein levels (Figure 2C). As shown in Figure 2D, in response to Ang II, the level of KLF5 protein was significantly attenuated in VSMCs transfected with KLF5-specific siRNA as compared to cells transfected with non-specific siRNA (NS-siRNA). Similarly, KLF5-specific siRNA abolished Ang II–induced cyclin D1 protein expression. However, these changes were not observed in the cells treated with NS-siRNA. MTT and flow cytometric analysis showed that KLF5 overexpression resulted in increased proliferation, and a reduction in the proportion of cells in the G0/G1-phase, compared with empty vector controls. In addition, knockdown of KLF5 expression dramatically reduced cell growth and the number of S-phase cells, even after Ang II treatment (Figure 2E, F), indicating that KLF5 is critical for Ang II-mediated VSMC proliferation. Taken together, KLF5 is required for both Ang II-induced cyclin D1 expression and VSMC proliferation.
Ang II induces KLF5 activation via the ERK 1/2 and p38 MAPK pathways
Protein phosphorylation plays a major role in modulating the activity of transcription factors19. As Ang II induced both KLF5 and cyclin D1 expression in same profile manner, KLF5-mediated cyclin D1 expression may result from KLF5 activation but not its expression. For this, the phosphorylation status of KLF5 was tested via cross-immunoprecipitation using anti-phospho-Ser and anti-KLF5 antibodies. As shown in Figure 3A, Ang II effectively increased the level of phosphorylated KLF5, and effect that was significant at 0.5 h and peaked at 1 h. In contrast, Ang II treatment had little influence on total KLF5 protein levels. Ang II regulates cell growth through ERK 1/2, which is a key mediator of growth20. ERK 1/2 mediates the transmission of signals elicited by extracellular stimuli acting on plasma membrane proteins to the nucleus by phosphorylating and activating a variety of transcription factors. KLF5 has been shown to be phosphorylated by ERK 1/29. To determine whether ERK 1/2 is involved in KLF5 phosphorylation in VSMCs stimulated by Ang II, the phosphorylation of three MAPKs (ERK 1/2, JNK, and p38 MAPK) in response to Ang II treatment was analyzed via Western blot, using specific anti-phospho-kinase antibodies. After treatment of VSMCs with Ang II, phosphorylated ERK 1/2 and phosphorylated p38 MAPK increased and reached a maximum at 15 min, and then decreased gradually within 30 min after treatment, while phosphorylated JNK levels did not change (Figure 3B). Importantly, blockade of ERK activity with PD98059 and p38 MAPK using SB203580 depressed both Ang II–induced KLF5 phosphorylation and cyclin D1 protein expression (Figure 3C), indicating that either ERK 1/2 or p38 MAPK are involved in Ang II-induced KLF5 activation. To address the role of either AT-1 or AT-2 receptors in Ang II-induced KLF5 phosphorylation and cell proliferation, VSMCs were pretreated by AT-1 antagonist valsartan and AT-2 antagonist PD123319, respectively, and then stimulated by Ang II. The KLF5 phosphorylation induced by Ang II was abolished in valsartan-pretreated cells with decrease in cell proliferation (Figure 3D), suggesting that AT-1 receptor triggers the mechanism by which KLF5 mediates Ang II-induced VSMC proliferation.

Ang II induces KLF5 activation via the ERK 1/2 and p38 MAPK pathways. (A) Phosphorylated KLF5 was immunoprecipitated with anti-phospho-Ser and then analyzed by Western blotting. The graphs represent the relative level of p-KLF5 of four independent experiments. bP<0.05, cP<0.01 vs 0 h. (B) Phosphorylation of ERK, JNK and p38 MAPK in VSMCs was analyzed by Western blotting. The graphs represent the relative activity of these kinases of four independent experiments. bP<0.05, cP<0.01 vs 0 min. (C) KLF5 phosphorylation and cyclin D1 expression were analyzed by immunoprecipitation and Western blotting, respectively. β-actin was used as an internal control. The graphs represent the relative level of p-KLF5 and cyclin D1 protein of four independent experiments. cP<0.01 vs no inhibitors. (D) The expression and phosphorylation of KLF5 and AT-1 were analyzed by immunoprecipitation and Western blotting, respectively. β-actin was used as an internal control. VSMC proliferation was tested using MTT. cP<0.01 vs control.
KLF5 mediates Ang II-induced cyclin D1 expression via its functional interaction with AP-1
To study the involvement of the KLF5 in cyclin D1 gene activation, a cyclin D1 promoter fragment spanning positions -1745 to +134 was analyzed via sequence comparison. These data showed that the cyclin D1 promoter contains multiple KLF5 and AP-1 regulatory elements that may be involved in the transcriptional regulation of gene expression (Figure 4A). To further characterize this, the pGL3-CD1-Luc construct containing the cyclin D1 promoter (from -1745 to +134) was transfected into COS-7 cells and the promoter activity was assayed following Ang II treatment. Ang II increased cyclin D1 promoter activity significantly over the level observed in the untreated cells, and cotransfection of KLF5 with the c-Jun expression vectors significantly increased cyclin D1 promoter activity 5-fold over that seen with the reporter alone following Ang II treatment (Figure 4B). Further, the ability of KLF5 to bind to the promoter of cyclin D1 gene was assessed via a ChIP assay. As shown in Figure 4C, Ang II significantly increased KLF5 and c-Jun binding to the cyclin D1 promoter. The two distal KLF5 binding elements of the cyclin D1 gene promoter were amplified by PCR in the immunoprecipitates pulled down by the anti-KLF5 antibody. However, a DNA fragment containing two AP-1 binding sites and one proximal KLF5 element (-10∼-245) was amplified only in pellets immunoprecipitated using anti-c-Jun, but not anti-KLF5 antibody (Figure 4C). These observations were consistent with the results of the reporter gene assay, suggesting that there is a functional interaction between KLF5 and c-Jun in transactivating expression of cyclin D1 gene.

KLF5 mediates Ang II-induced cyclin D1 expression via interaction with AP-1. (A) Schematic representation of the KLF5 and AP-1 binding sites in the cyclin D1 promoter regions. (B) COS-7 cell lysates co-transfected using pGL3-CD1-Luc with expression vectors expressing KLF5 and/or c-Jun were subjected to luciferase activity assays. The graphs are expressed as the relative luciferase activity (n=3). bP<0.05, cP<0.01 vs empty vector control. (C) The chromatin fragments were immunoprecipitated (IP) with anti-KLF5 or anti-c-Jun antibodies, and then the cyclin D1 promoter regions containing KLF5 (−538∼−801 and −868∼−1094) or AP-1 (−10∼−245) binding sequences were amplified by PCR. The graphs represent the relative binding activity of KLF5 or AP-1 to DNA elements of four independent experiments. cP<0.01 vs 0 h.
Discussion
The major purpose of the present study was to examine the action mechanism of KLF5 during Ang II-induced cyclin D1 expression and VSMC proliferation. We demonstrated the direct effect of Ang II on the induction of cyclin D1 expression and the stimulation of VSMC growth. We also showed that Ang II increased the activity of KLF5 by inducing both its expression and phosphorylation via the ERK 1/2 and p38 MAPK pathways activated by AT-1. Evidence for a functional interaction between KLF5 and c-Jun was provided by ChIP and the reporter gene assays. After Ang II treatment or KLF5 and c-Jun overexpression, there is a increased activity of cyclin D1 promoter, with increased binding ability of KLF5 and AP-1 on cyclin D1 promoter. Our results support a physiological role for Ang II as a positive regulator of KLF5.
It has been demonstrated that the molecular and cellular actions of Ang II in cardiovascular are almost exclusively mediated by AT-1 receptor and AT-1 regulate cell growth through the ERK 1/2 pathway21 which is required for mediating Ang II-induced proliferation in rat aortic SMCs22. In the present study, we also showed that AT-1 receptor mediated Ang II-induced KLF5 activation and cell proliferation by ERK 1/2 signaling pathway.
The role of KLF5 in the vasculature is first suggested when it is isolated as a transcription factor that binds the promoter of the embryonic smooth muscle myosin heavy chain SMemb gene23. In vascular tissues, KLF5 is abundantly expressed in embryonic SMCs and is downregulated with vascular development, but notably, it is reinduced in proliferating neointimal SMCs in response to vascular injury24. The previous study has shown that KLF5 and c-Jun cooperatively inhibit p21 expression in Ang II-stimulated VSMCs10, indicating the mechanism by which KLF5 promotes cell proliferation. Recently, KLF5 has been demonstrated direct binding to the promoter and up-regulated gene expression of cyclin D1 in cardiovascular cells25. Meanwhile, our findings also indicate a novel mechanism by which KLF5 mediates Ang II-induced cyclin D1 expression and proliferation of VSMCs via functional interaction with c-Jun.
The activity of KLF5 is regulated by a variety of transcriptional regulators and nuclear receptors through phosphorylation and/or acetylation19, 26, 27. Ang II has been shown to activate the ERK 1/2 pathway21. Moreover, KLF5 is activated via an ERK 1/2-dependent mechanism under certain conditions28. We show that Ang II induces both KLF5 and cyclin D1 expression in same profile manner, and cyclin D1 protein expression depends on KLF5 phosphorylation via ERK and p38 MAPK signaling pathway, suggesting that KLF5-mediated cyclin D1 expression may result from KLF5 activation but not its expression.
In summary, KLF5, as a target of Ang II-mediated signaling, plays an important role in the expression of cyclin D1 and the proliferation of VSMCs. The AT-1 receptor mediates the activation of KLF5 via ERK 1/2 and p38 MAPK pathways in Ang II-induced VSMCs. KLF5 activates the transcription of cyclin D1 gene via functional interaction with c-Jun in Ang II-induced VSMC proliferation.
Author contribution
Mei HAN and Jin-kun WEN designed research and revised the manuscript; Yu LIU performed research and analyzed data; Li-hua DONG and Bin ZHENG performed research.
References
Kim S, Iwao H . Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 2000; 52: 11–34.
Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T . Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem 2001; 276: 7957–62.
Gao D, Niu X, Ning N, Hao G . Regulation of angiotensin II-induced krüppel-like factor 5 expression in vascular smooth muscle cells. Biol Pharm Bull 2006; 29: 2004–8.
Schmitz U, Ishida T, Ishida M, Surapisitchat J, Hasham MI, Pelech S, et al. Angiotensin II stimulates p21-activated kinase in vascular smooth muscle cells: role in activation of JNK. Circ Res 1998; 82: 1272–8.
Kusuhara M, Takahashi E, Peterson TE, Abe J, Ishida M, Han J, et al. p38 Kinase is a negative regulator of angiotensin II signal transduction in vascular smooth muscle cells: effects on Na+/H+ exchange and ERK1/2. Circ Res 1998; 83: 824–31.
Suzuki T, Muto S, Miyamoto S, Aizawa K, Horikoshi M, Nagai R . Functional interaction of the DNA-binding transcription factor Sp1 through its DNA-binding domain with the histone chaperone TAF-I. J Biol Chem 2003; 278: 28758–64.
Bateman NW, Tan D, Pestell RG, Black JD, Black AR . Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem 2004; 279: 12093–101.
Nandan MO, Chanchevalap S, Dalton WB, Yang VW . Kruppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett 2005; 579: 4757–62.
Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW . Krüppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene 2004; 23: 3404–13.
He M, Han M, Zheng B, Shu YN, Wen JK . Angiotensin II stimulates KLF5 phosphorylation and its interaction with c-Jun leading to suppression of p21 expression in vascular smooth muscle cells. J Biochem 2009; 146: 683–91
Han M, Wen JK, Zheng B, Cheng Y, Zhang C . Serum deprivation results in redifferentiation of human umbilical vascular smooth muscle cells. Am J Physiol Cell Physiol 2006; 291: C50–8.
Li AY, Han M, Zheng B, Wen JK . Roscovitine inhibits ERK1/2 activation induced by angiotensin II in vascular smooth muscle cells. FEBS Lett 2008; 582: 243–8.
Chanchevalap S, Nandan MO, McConnell BB, Charrier L, Merlin D, Katz JP, et al. Kruppel-like factor 5 is an important mediator for lipopolysaccharide-induced proinflammatory response in intestinal epithelial cells. Nucleic Acids Res 2006; 34: 1216–23.
Xiao F, Puddefoot JR, Barker S, Vinson GP . Mechanism for aldosterone potentiation of angiotensin II-stimulated rat arterial smooth muscle cell proliferation. Hypertension 2004; 44: 340–5.
Li JJ, Han M, Wen JK, Li AY . Osteopontin stimulates vascular smooth muscle cell migration by inducing FAK phosphorylation and ILK dephosphorylation. Biochem Biophys Res Commun 2007; 356: 13–9.
Wang C, Han M, Zhao XM, Wen JK . Kruppel-like factor 4 is required for the expression of vascular smooth muscle cell differentiation marker genes induced by all-trans retinoic acid. J Biochem 2008; 144: 313–21.
Weiss D, Sorescu D, Taylor WR . Angiotensin II and atherosclerosis. Am J Cardiol 2001; 87: 25C–32C.
Knudsen KE, Diehl JA, Haiman CA, Knudsen ES . Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene 2006; 25: 1620–8.
Zhang Z, Teng CT . Phosphorylation of Kruppel-like factor 5 (KLF5/IKLF) at the CBP interaction region enhances its transactivation function. Nucleic Acids Res 2003; 31: 2196–208.
Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K, Tamaki T . Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smooth muscle cells. Hypertens Res 2001; 24: 251–61.
Gervais M, Dugourd C, Muller L, Ardidie C, Canton B, Loviconi L, et al. Akt down-regulates ERK1/2 nuclear localization and angiotensin II-induced cell proliferation through PEA-15. Mol Biol Cell 2006; 17: 3940–51.
Dugourd C, Gervais M, Corvol P, Monnot C . Akt is a major downstream target of PI3-kinase involved in angiotensin II-induced proliferation. Hypertension 2003; 41: 882–90.
Watanabe N, Kurabayashi M, Shimomura Y, Kawai-Kowase K, Hoshino Y, Manabe I, et al. BTEB2, a Kruppel-like transcription factor, regulates expression of the SMemb/Nonmuscle myosin heavy chain B (SMemb/NMHC-B) gene. Circ Res 1999; 85: 182–91.
Suzuki T, Aizawa K, Matsumura T, Nagai R . Vascular implications of the Kruppel-like family of transcription factors. Arterioscler Thromb Vasc Biol 2005; 25: 1135–41.
Suzuki T, Sawaki D, Aizawa K, Munemasa Y, Matsumura T, Ishida J, et al. Kruppel-like factor 5 shows proliferation-specific roles in vascular remodeling, direct stimulation of cell growth, and inhibition of apoptosis. J Biol Chem 2009; 284: 9549–57.
Miyamoto S, Suzuki T, Muto S, Aizawa K, Kimura A, Mizuno Y, et al. Positive and negative regulation of the cardiovascular transcription factor KLF5 by p300 and the oncogenic regulator SET through interaction and acetylation on the DNA-binding domain. Mol Cell Biol 2003; 23: 8528–41.
Matsumura T, Suzuki T, Aizawa K, Munemasa Y, Muto S, Horikoshi M, et al. The deacetylase HDAC1 negatively regulates the cardiovascular transcription factor Kruppel-like factor 5 through direct interaction. J Biol Chem 2005; 280: 12123–9.
Zhang H, Bialkowska A, Rusovici R, Chanchevalap S, Shim H, Katz JP, et al. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Kruppel-like factor 5. J Biol Chem 2007; 282: 15541–9.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 30670845 and 30770787), the “973” Program of China (No 2008CB517402) and the Hebei Province Natural Science Foundation (No C2006000814 and C2005000722).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Y., Wen, Jk., Dong, Lh. et al. Krüppel-like factor (KLF) 5 mediates cyclin D1 expression and cell proliferation via interaction with c-Jun in Ang II-induced VSMCs. Acta Pharmacol Sin 31, 10–18 (2010). https://doi.org/10.1038/aps.2009.185
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.185
Keywords
This article is cited by
-
Effects of KLF11 on Vascular Smooth Muscle Cells and its Underlying Mechanisms in Intracranial Aneurysm
Biochemical Genetics (2024)
-
Krüppel-like factor 5 accelerates the pathogenesis of Alzheimer’s disease via BACE1-mediated APP processing
Alzheimer's Research & Therapy (2022)
-
Long Non-coding RNAs: Potential Players in Cardiotoxicity Induced by Chemotherapy Drugs
Cardiovascular Toxicology (2022)
-
Genome Wide Association Study Identifies L3MBTL4 as a Novel Susceptibility Gene for Hypertension
Scientific Reports (2016)
-
Pioglitazone Ameliorates Smooth Muscle Cell Proliferation in Cuff-Induced Neointimal Formation by Both Adiponectin-Dependent and -Independent Pathways
Scientific Reports (2016)
