
Abstract
Seed dormancy is a key adaptive trait under polygenic control in many plants. We introduced the chromosomal regions containing the dormancy QTLs qSD1, qSD7-1, and qSD12 from an accession of weedy rice into a nondormant genetic background to examine component genetic effects and their interactions with time of afterripening (DAR). A BC4F2 plant, which was heterozygous for the three loci, was selected to develop the BC4F3 population. Single point analysis detected only qSD7-1 and qSD12 (R2=38–72%) at 10, 30, and 50 DAR in the population. However, multiple linear regression analysis detected genetic effects of the three QTLs and their trigenic epistasis, an environmental effect of DAR (E), and interactions of E with qSD12 and with the qSD1 × qSD7-1 and qSD7-1 × qSD12 epistases. The linear model demonstrates that QTL main effects varied with DAR, and that some epistasis or epistasis-by-DAR interactions partially counteract the main effects. The three QTLs were isolated as single Mendelian factors from the BC4F3 population and estimated for component genic effects based on the BC4F4 populations. Isolation improved estimation of the qSD1 effect and confirmed the major effect of qSD12. The qSD1 and qSD12 loci displayed a gene-additive effect. The qSD7-1, which was further narrowed to a chromosomal region encompassing the red pericarp color gene Rc, displayed gene additive and dominant effects.
Similar content being viewed by others

Introduction
The polygenic nature of seed dormancy determines the continuous distribution over time for germination in natural and experimental populations (Johnson, 1935). A complete linkage map enables genome-wide scanning for polygenes or quantitative trait loci (QTLs) and manipulation of individual alleles for experimentation or breeding. Many dormancy QTLs have been identified from model plants and major cereal crops. For example, dormancy QTLs are distributed over all five chromosomes (chr) in Arabidopsis (Arabidopsis thaliana) (Van der Schaar et al, 1997; Alonso-Blanco et al, 2003; Clerkx et al, 2004) and 11 of the 12 chr in cultivated (Oryza sativa) (Wan et al, 1997; Lin et al, 1998; Dong et al, 2002; Miura et al, 2002), wild (O. rufipogon) (Cai and Morishima, 2000; Thomson et al, 2003), and weedy (O. sativa) (Gu et al, 2004) rice. Dormancy QTLs in barley (Oberthur et al, 1995; Li et al, 2003; Prada et al, 2004), sorghum (Lijavetzky et al, 2000), and wheat (Anderson et al, 1993; Kato et al, 2001; Mares and Mrva, 2001; Groos et al, 2002; Osa et al, 2003; Kulwal et al, 2004) have been identified to seek gene resources to impart resistance to preharvest sprouting (PHS) and to manipulate germination programs in the malting process. Additional research has been aimed at isolation of individual dormancy alleles from donor parents to determine gene effects, interactions with environmental factors, and breeding potential.
Several dormancy QTL regions have been isolated in introgression or near isogenic lines for Arabidopsis, barley, and rice (Han et al, 1999; Alonso-Blanco et al, 2003; Gao et al, 2003; Takeuchi et al, 2003). Fine mapping these regions suggests: (1) a dormancy QTL may consist of a gene cluster (Han et al, 1999); (2) some dormancy QTLs may lose their effect in a nondormant genetic background (Alonso-Blanco et al, 2003); (3) some QTLs are especially sensitive to the environment during seed development (Gao et al, 2003); and (4) an influence of flowering time on dormancy may be due to tight linkage between loci for both traits (Takeuchi et al, 2003). Some of the introgression lines were used to estimate gene-additive effects for the QTL. Many genetic analyses suggest some dormancy genes may also consist of gene-dominant effects (Johnson, 1935; Takahashi, 1962; Bhatt et al, 1983; Buraas and Skinnes, 1984). These observations must be confirmed with the populations segregating for individual QTLs, or Mendelian factors. The major obstacle to the confirmation is that most dormancy QTLs contributed relatively little to phenotypic variance such that it is difficult to distinguish a component gene effect from environmental error.
Dormancy is released by afterripening, which is defined by a period of seed exposure to a set of environmental conditions after maturation and separation from the parent plant (Simpson, 1990). Environmental conditions that facilitate afterripening vary by species. For example, rice normally requires afterripening under warm, dry conditions (Leopold et al, 1988) and Arabidopsis responds best to cool, moist conditions (Koornneef and Karssen, 1994). Rice species (Oryza ssp) differ in patterns of afterripening under warm, dry conditions (Veasey et al, 2004). Some dormancy QTLs in cultivated rice lost their effect upon drying (Lin et al, 1998). Dormancy QTLs identified from weedy rice could be grouped into relatively early, constant, and late expression categories during afterripening (Gu et al, 2004), which was similar to that from Arabidopsis (Alonso-Blanco et al, 2003). Thus, time of afterripening is an important environmental factor affecting expression of dormancy genes.
Previous research identified six dormancy QTLs from an accession of weedy rice (Gu et al, 2004). Subsequent research transferred five dormancy QTLs into a nondormant breeding line by phenotypic and marker-assisted selections (Gu et al, 2005). In this research, we synchronized the genetic background of three dormancy QTLs to develop a genetic model involving the genotypic (G) and genotype-by-environmental (time of afterripening) (G × E) interaction effects, and then isolated individual loci as single Mendelian factors to estimate component gene effects.
Materials and methods
Developing heterozygotes for qSD1, qSD7-1, and qSD12
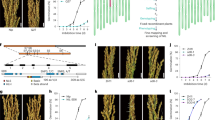
Phenotypic and marker-assisted selections were used to develop a trigenic heterozygote for the loci qSD1, qSD7-1, and qSD12, where the dormancy alleles were derived from the weedy rice SS18-2 (Gu et al, 2003). Initially, five generations of single-plant selection for dormancy and recurrent backcrossing introduced several SS18-2-derived chr segments into the genetic background of EM93-1, a nondormant, early maturation breeding line. Then, the framework linkage map (Gu et al, 2004) was used to scan for the introgression segments in selected BC4F1 plants, and to select a genotype carrying dormancy alleles at all three QTLs from a BC4F2 population (Gu et al, 2005). Finally, we selected BC4F2 plant #51, as it was heterozygous for the qSD1-, qSD7-1-, and qSD12-containing regions, and the remaining chr or chr segments were identical to EM93-1 (Figure 1). BC4F2 plant #51 was self-pollinated to generate the BC4F3 population to develop a genetic model involving the three QTLs, the time (days) of afterripening (DAR), and their interactions.

Graphic genotypes for chromosomes 1, 7, and 12 of BC4F2 plant #51. Empty or solid bars denote the EM93-1- or SS18-2-derived chromosomes or chromosomal segments, respectively, which were determined by rice microsatellite (RM) markers at the tick mark positions on the framework linkage map (Gu et al, 2004); chromosomes not shown were identical to EM93-1. QTLs for seed dormancy (qSD) are placed to the left of segments.
Molecular markers in the three QTL regions (Figure 1) were used to select single-locus heterozygotes from the BC4F3 population. The selected plants carried only a single dormancy allele at one of the three QTLs, as judged by its nearest and flanking markers (Table 1). For each QTL, we selected two BC4F3 plants that differed in length of the introgressed QTL segment, but shared the region encompassing the nearest markers (Table 1). These six BC4F3 heterozygotes were self-pollinated to generate BC4F4 segregation populations to estimate gene additive and dominant effects for individual QTLs.
Plant cultivation and dormancy assessment
Plants were grown in pots (28 cm diameter × 25 cm height) filled with a mixture of clay soil and SUNSHINE medium (Sun Gro® Horticulture Canada Ltd, Seba Beach, AB) and watered daily. The six BC4F4 populations were maintained in one greenhouse. Day/night temperatures were set at 29/21°C and the daylength set for 14 h. Seeds were harvested at 40 days after flowering, which was tagged by emergence of the first panicle in the plant. Seeds were cleaned by removal of empty or immature spikelets and air-dried in the greenhouse for 3 days to about 12% moisture. Dried seeds were sealed in plastic containers and stored at −20°C to prevent afterripening.
The degree of dormancy was measured with percent germination. Prior to germination, seeds from the BC4F3 population were afterripened at the room temperature (about 25°C) for 10, 30, and 50 days; the time interval was determined based on germination for the BC4F2 population (Gu et al, 2005). Seeds from the six BC4F4 populations were afterripened for 1 or 7, and 14 days. A shorter period of afterripening was applied to the BC4F4s because more that 80% of the BC4F3 parental plant seeds germinated following <30 DAR (Table 1). There were three replications per experiment, with each replication consisting of approximately 50 seeds in a 9-cm Petri dish lined with a Whatman No.1 filter paper and wetted with 10-ml deionized water (Roberts, 1961; Gu et al, 2003). Germination was conducted at 30°C and 100% relative humidity in the dark for 7 days. Germination was evaluated visually by protrusion of the radicle or coleoptile from the hull by ≥3 mm. Germination percentage (y) was transformed by sin−1(y)−0.5 and the mean averaged over the three replications at each DAR was used for QTL analysis.
Genotypic identification and QTL effect analyses
The BC4F3 and BC4F4 plants were genotyped with microsatellite (RM) markers on the SS18-2-derived segments (Figure 1). Genomic DNA was prepared from young leaves. The DNA was extracted, markers amplified by polymerase chain reaction (PCR), and the PCR products displayed using the methods previously described (Gu et al, 2004). The partial linkage map was adjusted with MAPMAKER/EXP 3.0 (Lincoln et al, 1992).
Dormancy QTL epistases and qSD12 × DAR were detected in the primary segregation population (Gu et al, 2004). There are four factors (ie, qSD1, qSD7-1, qSD12, and time of afterripening) and their interactions regulating phenotypic variation in germination in the BC4F3 population because the genetic background of three QTLs has been synchronized (Figure 1). Thus, multiple-linear regression was used to estimate the relative importance of component QTL genotypic (G) and G × E interaction effects using the model:

where, yij is the mean germination for the ith BC4F3 plant in the jth afterripening treatment (j=1, 2, and 3 corresponding to 10, 30, and 50 DAR, respectively); m is the mean of the model; x1, x2, and x3 are the variables for codominant markers nearest to qSD1, qSD7-1, and qSD12, respectively, where EM93-1-type homozygous, heterozygous, and SS18-2-type homozygous genotypes for each marker were scored as −1, 0, and 1, respectively; x12, x13, and x23 are the digenic interactions between variables x1 and x2, x1 and x3, and x2 and x3, respectively; x123 is the trigenic interaction among the three QTLs; xt is the variable for afterripening treatment; x1t, x2t, x3t, x12t, x13t, x23t, and x123t are the interactions between time of afterripening and the variables x1, x2, x3, x12, x13, x23, and x123, respectively; b1, b2, b3, b12, b13, b23, b123, bt, b1t, b2t, b3t, b12t, b13t, b23t, and b123t are the partial regression coefficients corresponding to the regressor variables x1, x2, x3, x12, x13, x23, x123, xt, x1t, x2t, x3t, x12t, x13t, x23t, and x123t, respectively; eij is the random error in the experiment conditions; and N (N=213–221) is the number of BC4F3 plants that were evaluated for germination at a DAR. Analysis was performed by the SAS procedure REG (SAS Institute, 1999). The variables retained in the final model were determined by a stepwise selection at a significant level of 5%.
One-way ANOVA was used to estimate the contribution of individual QTLs to phenotypic variance in germination at different DAR in BC4F3 and BC4F4 populations. The analysis was based on the linear model in which a phenotypic value was partitioned into mean, genotypic, and error (also including genetic effects that are not explained by the QTL for the BC4F3 population) components. The contribution (R2) of each QTL was calculated as the proportion of component type III sum-of-square (SS) to the corrected total SS. ANOVA was performed with the SAS procedure GLM (SAS Institute, 1999).
Component additive (a) and dominant (d) effects of a dormancy QTL were estimated based on the BC4F4 populations. Each BC4F4 segregated for one QTL-containing region. Thus, the method described by Kearsey and Pooni (1996) is well suited for the estimations:


where, MEE, MSS, and MES are means of the EM93-1-type homozygous, SS18-2-type homozygous, and heterozygous genotypes, respectively, for the marker locus nearest to the QTL. SE for the parameters a and d were estimated as:


where, the s2EE, s2SS, and s2ES are variances of the means MEE, MSS, and MES, respectively. Significance of the estimates for a and d were determined by Student's t-test.
Results
A genetic model for three dormancy QTLs and time of afterripening
The BC4F3 population skewed to low and high germination at 10 and 50 DAR, respectively, and distributed over the whole germination scale at 30 DAR (Figure 2). One-way ANOVA detected only the qSD7-1 and qSD12 loci (Figure 2). These two QTLs displayed divergent responses to time of afterripening. For example, qSD7-1 accounted for 18 and 5%, while qSD12 explained 38 and 72% of the phenotypic variance at 10 and 50 DAR, respectively. Locus qSD1 was not detected in this single point analysis, indicating that there must be something masking its main effect under the experimental conditions.

Frequency distribution of the BC4F3 population for percentage germination at 10 (•), 30 (open columns), and 50 (○) days of afterripening (DAR). Sample size (N), and proportion of the phenotypic variance explained by qSD7-1 and qSD12 (R2) are shown in parentheses.
The multiple linear regression analysis detected eight variables significantly contributing to variation in germination (Table 2). The linear relationship of the eight variables accounted for 82% of the total variance. These variables can be grouped into three categories, that is, genotypic, environmental, and G × E effects. The genotypic effect includes the main effect of the three QTLs and their trigenic epistatic effect. As indicated by sign and magnitude of the partial regression coefficients (b), substitution of a nondormancy allele with a dormant allele at any of the three QTLs tended to reduce germination, with the effect (b2, b3=−0.12 to −0.13) of the qSD7-1 and qSD12 dormancy alleles being much greater than that (b1=−0.04) of the qSD1 dormancy allele. In contrast to the QTL main effects, interaction among the three QTLs tended to promote germination, although the magnitude was relatively small (b123=0.06).
The environmental effect in the experimental conditions was caused by differences in DAR. An increase in time of afterripening under warm, dry conditions greatly promoted germination of dormant seeds (bt=0.32), or dramatically shifted the germination distribution (Figure 2). The G × E effect includes the interactions of time of afterripening with: (1) the major locus qSD12, (2) the qSD7-1 × qSD12 epistasis, and (3) the qSD7-1 × qSD1 epistasis (Table 2). The qSD12 × E interaction (b3t=−0.15) accounted for a high proportion of the great increase in the qSD12 major effect with progression of afterripening (Figure 2). The digenic epistases of qSD7-1 × qSD12 (b23=−0.05) and qSD1 × qSD7-1 (b12=0.01) displayed a decreasing and an increasing effect on germination with time of afterripening during the 50 DAR. These component G × E interactions explained the asymmetry distributions of germination (Figure 2).
Single-locus models for dormancy QTLs
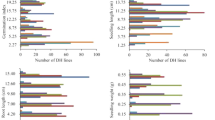
The isolated qSD1, qSD7-1, and qSD12 had significant effects on germination (Table 3). The three QTLs differed in gene component effects. Locus qSD1 accounted for about 7 and 12% of the phenotypic variances, respectively, at 7 and 14 DAR in both BC4F4 (66) and (159) populations. The gene-additive component explained the genetic effect of qSD1, as its dominant effect was not significantly different relative to experimental error (Table 3).
The locus qSD7-1 accounted for 11–16% of the phenotypic variances at 1–14 DAR in the BC4F4 (12) and (114) populations. Both gene additive and dominant effects were important for qSD7-1, based on the codominant marker RM5672 (Table 3). The BC4F4 (12) and (114) populations also segregated for the red pericarp color gene Rc, a dominant marker (Figure 3b). The Rc locus was about 2 cM from RM5672 and contributed 13–18% to phenotypic variance at 1–14 DAR in the two populations. The slightly larger contribution to phenotypic variance of the Rc locus compared with the RM5672 locus, and the significant dominant effect for qSD7-1 suggest that Rc is closer to qSD7-1 than to RM5672.

Distribution of germination at 7 or 14 days of afterripening (DAR) for populations segregating for qSD1 (a), qSD7-1 (b), and qSD12 (c). The bars depicted genotypes for the marker nearest a QTL. RM220 and RM270 are codominant markers, and Rc is a dominant marker gene for red pericarp/testa color.
The locus qSD12 accounted for about 67 and 50% of the phenotypic variances at 7 and 14 DAR, respectively, in the BC4F4 (48) and (145) populations. This is a major dormancy QTL as compared with qSD1 and qSD7-1. A gene additive effect explained the genetic effect of this major QTL; the component dominant effect was insignificant or relatively minor (Table 3).
Discussion
The model developed based on the BC4F3 population includes all three dormancy QTLs and their epistasis, time of afterripening, and interactions between time of afterripening and QTL or epistasis (Table 2). This multiple linear model provides insight into how a set of dormancy genes regulates germination of seeds afterripened under warm, dry conditions. The genetic effect of three dormancy loci can be partitioned into linear and nonlinear components. The linear components delay or repress germination. The nonlinear component may partially counterbalance the linear effects on germination. In the BC4F3 genetic background, only trigenic epistasis was significant, and other significant epistases involved interactions with time of afterripening (G × E). In several ways, epistases and G × E interactions indicate the complexity in regulation of germination or detection of dormancy genes. First, only part of a QTL effect is independent of time of afterripening, a substantial remaining amount involved epistases and/or G × E interactions (Table 2). Second, some epistatic and G × E interaction effects enhance dormancy, while others may partially counteract a QTL main effect (Gu et al, 2004; Table 2). Finally, the main effect of a QTL, especially one with a relatively small effect, could be masked by gene or gene-by-E interactions. For example, qSD1 was detected in the synchronized genetic background, that is, the two BC4F4 populations (Table 3), but not in the primary segregation population (Gu et al, 2004), one of the BC4F2 populations (Gu et al, 2005), and the BC4F3 population (Figure 2).
We isolated three dormancy QTLs as Mendelian factors. The three genotypes for a QTL, no matter how large a QTL's effect on germination, displayed overlapping rather than discrete distributions, as estimated by nearest markers (Figures 3a–c). If possible rare recombinants between a QTL and its nearest marker are ignored, the continuous variation within each marker genotype must have arisen from environmental variation, because the genetic background was synchronized. The environmental (within-genotype) variance appeared inversely proportional to the component genetic variance in percentage germination in populations segregating for a single dormancy gene. For example, genetic and environmental variances accounted for a smaller (14%) and larger (86%) amount, respectively, of the total variance in germination at 14 DAR in the BC4F4 (159) population segregating for qSD1, and a larger (68%) and smaller (32%) amount, respectively, of the total variance in germination at 7 DAR in the BC4F4 (145) population segregating for qSD12 (Table 3). The inverse proportionality occurred even in isogenic lines because total variance in different populations can reach a similar level by adjusting the time of afterripening (Figure 3) when dormancy is quantified by percent germination (Gu et al, 2003). It was reported that heritability for dormancy in rice cultivars was 0.12–0.44 (Chang and Yen, 1969), and the range of within-parental line variation in germination was similar to that of the F2 population cultivated under field conditions (Chang and Tagumpay, 1973). The low heritability for dormancy or large environmental variation in dormancy makes it very difficult to manipulate individual dormancy alleles. For example, we were unable to isolate and distinguish the three QTLs without use of linked markers, as the selected BC4F3 plants had similar germination (Table 1). Thus, a marker-assisted selection technique will be important to integrate dormancy genes into new varieties to impart resistance to PHS.
We purposely selected two BC4F3 plants for each QTL that differed in interval length (Table 1) to narrow the region flanking dormancy alleles. The Rc locus was mapped between RM180 and RM5672 (Figure 1). Both BC4F3 plants #12 and #114 carry the qSD7-1 and Rc loci according to segregation data from their BC4F4 populations (Figure 3b), but plant #114 has a much shorter SS18-2-derived chr segment (between RM180 and RM241) than plant #12 (between RM4896 and RM214) (Table 1 and Figure 1). The Rc locus contributed a few percent more to phenotypic variances in germination than the RM5672 and RM180 loci in the BC4F4 (12) and (114) populations, suggesting that qSD7-1 locates on the same 4 cM chr segment as Rc. In addition, Rc is dominant; the qSD7-1 locus also had a dominant effect (Table 3). The tight linkage and the gene-dominant effect suggest that Rc and qSD7-1 might be the same locus. Red grain color gene has long been associated with dormancy in wheat and the association has been hypothesized as a pleiotropic effect of the homoelogous red color genes (R1–R3) (Nilsson-Ehle, 1914; Gfeller and Svejda, 1960; Flintham, 2000). Map-based cloning of the qSD7-1-Rc region will test the hypothesis using rice.
References
Alonso-Blanco C, Bentsink L, Hanhart CJ, Vries HBE, Koornneef M (2003). Analysis of natural allelic variation at seed dormancy loci of Arabidopsis thaliana. Genetics 164: 711–729.
Anderson JA, Sorrells ME, Tanksley SD (1993). RFLP analysis of genomic regions associated with resistance to pre-harvest sprouting in wheat. Crop Sci 33: 453–459.
Bhatt GM, Ellison FW, Mares DJ (1983). Inheritance studies in dormancy in three wheat crosses. In: Kruger JE, LaBerge DE (eds) Third International Symposium on Pre-Harvest Sprouting in Cereals. Westview Press: Boulder, Colorado. pp 274–278.
Buraas T, Skinnes H (1984). Genetic investigations on seed dormancy in barley. Hereditas 101: 235–244.
Cai HW, Morishima H (2000). Genomic regions affecting seed shattering and seed dormancy in rice. Theor Appl Genet 100: 840–846.
Chang TT, Tagumpay O (1973). Inheritance of grain dormancy in relation to growth duration in 10 rice crosses. SABRAO Newsl 5: 87–94.
Chang TT, Yen ST (1969). Inheritance of grain dormancy in four rice crosses. Bot Bul Acad Sin 10: 1–9.
Clerkx EJ, El-Lithy ME, Vierling E, Ruys GJ, Blankestijn-De Vries H, Groot SP et al (2004). Analysis of natural allelic variation of Arabidopsis seed germination and seed longevity traits between the accessions Landsberg erecta and Shakdara, using a new recombinant inbred line population. Plant Physiol 135: 432–443.
Dong Y, Tsozuki E, Kamiunten H, Terao H, Lin D, Matsuo M et al (2002). Identification of quantitative trait loci associated with pre-harvest sprouting resistance in rice (Oryza sativa L.). Field Crops Res 81: 133–139.
Flintham JE (2000). Different genetic components control coat-imposed and embryo-imposed dormancy in wheat. Seed Sci Res 10: 43–50.
Gao W, Clancy JA, Han F, Prada D, Kleinhofs A, Ullrich SE (2003). Molecular dissection of a dormancy QTL region near the chromosome 7 (5H) L telomere in barley. Theor Appl Genet 107: 552–559.
Gfeller F, Svejda F (1960). Inheritance of post-harvest seed dormancy and kernel colour in spring wheat lines. Can J Plant Sic 40: 1–6.
Groos C, Gay G, Perretant MR, Gervais L, Bernard M, Dedryver F et al (2002). Study of the relationship between pre-harvest sprouting and grain color by quantitative trait loci analysis in a white × red grain bread-wheat cross. Theor Appl Genet 104: 39–47.
Gu X-Y, Chen Z-X, Foley ME (2003). Inheritance of seed dormancy in weedy rice. Crop Sci 43: 835–843.
Gu X-Y, Kianian SF, Foley ME (2004). Multiple loci and epistases control genetic variation for seed dormancy in weedy rice (Oryza sativa). Genetics 166: 1503–1516.
Gu X-Y, Kianian SF, Foley ME (2005). Phenotypic selection for dormancy introduced a set of adaptive haplotypes from weedy into cultivated rice. Genetics 21 June [Epub ahead of print].
Han F, Ullrich SE, Clancy JA, Romagosa I (1999). Inheritance and fine mapping of a major barley seed dormancy QTL. Plant Sci 143: 113–118.
Johnson LPV (1935). The inheritance of delayed germination in hybrids of Avena fatua and A. sativa. Can J Res 13: 367–387.
Kato K, Nakamura W, Tabiki T, Miura H (2001). Detection of loci controlling seed dormancy on group 4 chromosomes of wheat and comparative mapping with rice and barley genomes. Theor Appl Genet 102: 980–985.
Koornneef M, Karssen CM (1994). Seed dormancy and germination. In: Koornneef M, Karssen CM (eds) Arabidopsis. Cold Springs Harbor Laboratory Press: Cold Springs. pp 313–334.
Kulwal PL, Singh R, Balyan HS, Gupta PK (2004). Genetic basis of pre-harvest sprouting tolerance using single-locus and two-locus QTL analyses in bread wheat. Funct Integr Genomics 4: 94–101.
Kearsey MJ, Pooni HS (1996). The Genetical Analysis of Quantitative Traits. Chapman & Hall: London.
Leopold AC, Glenister R, Cohn MA (1988). Relationship between water content and afterripening in red rice. Plant Physiol 74: 659–662.
Li CD, Tarr A, Lance RCM, Harasymow S, Uhlmann J, Westcot S et al (2003). A major QTL controlling seed dormancy and pre-harvest sprouting/α-amylase in two-rowed barley (Hordeum vulgare L.). Aust J Agric Res 54: 1303–1313.
Lijavetzky D, Martinez MC, Carrari F, Hopp HE (2000). QTL analysis and mapping of pre-harvest sprouting resistance in sorghum. Euphytica 112: 125–135.
Lin SY, Sasaki T, Yano M (1998). Mapping quantitative trait loci controlling seed dormancy and heading date in rice. Theor Appl Genet 96: 997–1003.
Lincoln S, Daly M, Lander E (1992). Constructing Genetic Maps With MAPERMAKER/EXP 3.0 (3rd edn). Whitehead Institute: Cambridge, MA.
Mares DJ, Mrva K (2001). Mapping quantitative trait loci associated with variation in grain dormancy in Australian wheat. Aust J Agric Res 52: 1257–1265.
Miura K, Lin SY, Yano M, Nagamine T (2002). Mapping quantitative trait loci controlling seed longevity in rice (Oryza sativa L.). Theor Appl Genet 104: 981–986.
Nilsson-Ehle H (1914). Zur kenntnis der mit der keimungsphysiologie des weizens in zusammenhang stehenden inneren faktoren. Z Pflanzenzüchtung 2: 153–187.
Oberthur L, Blake TK, Dyer WE, Ullrich SE (1995). Genetic analysis of seed dormancy in barley (Hordeum vulgare L.). J Quant Trait Loci 1: 5.
Osa M, Kato K, Mori M, Shindo C, Torada A, Miura H (2003). Mapping QTLs for seed dormancy and the Vp1 homologue on chromosome 3A in wheat. Theor Appl Genet 106: 1491–1496.
Prada D, Ullrich SE, Molina-Cano JL, Cistue L, Clancy JA, Romagosa I (2004). Genetic control of dormancy in a Triumph/Morex cross in barley. Theor Appl Genet 109: 62–70.
Roberts EH (1961). Dormancy in rice seed. I. The distribution of dormancy period. J Exp Bot 12: 319–329.
SAS Institute Inc. (1999). SAS/STAT User's Guide, Version 8, Cary, NC, USA.
Simpson GM (1990). Seed Dormancy in Grass. Cambridge University Press: Cambridge. pp 3–113.
Takahashi N (1962). Physiocogenetical studies on germination of rice seeds with special reference to its genetical factors (in Japanese, with English summary). Bull Inst Agric Res Tohoku Univ 14: 1–87.
Takeuchi Y, Lin SY, Sasaki T, Yano M (2003). Fine linkage mapping enables dissection of closely linked quantitative trait loci for seed dormancy and heading in rice. Theor Appl Genet 107: 1174–1180.
Thomson MJ, Tai TH, McClung AM, Lai XH, Hinga ME, Lobos KB et al (2003). Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet 107: 479–493.
Veasey EA, Karasawa MG, Santos PP, Rosa MS, Mamani E, Oliveira GC (2004). Variation in the loss of seed dormancy during after-ripening of wild and cultivated rice species. Ann Bot (Lond) 94: 875–882.
Van der Schaar W, Alonso-Blanco WC, Leon-Kloosterziel KM, Jansen RC, Van Ooijen JW, Koornneef M (1997). QTL analysis of seed dormancy in Arabidopsis using recombinant inbred lines and MQM mapping. Heredity 79: 190–200.
Wan J, Nakazaki T, Kawaura K, Ikehashi H (1997). Identification of marker loci for seed dormancy in rice (Oryza sativa L.). Crop Sci 37: 1759.
Acknowledgements
We thank B Hoffer, T Nelson, and C Kimberlin for technical assistance. Funding for this work was provided by USDA-National Research Initiative (0200668).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gu, XY., Kianian, S. & Foley, M. Isolation of three dormancy QTLs as Mendelian factors in rice. Heredity 96, 93–99 (2006). https://doi.org/10.1038/sj.hdy.6800757
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800757
Keywords
This article is cited by
-
Antagonistic control of seed dormancy in rice by two bHLH transcription factors
Nature Genetics (2022)
-
Characterization of and genetic variation for tomato seed thermo-inhibition and thermo-dormancy
BMC Plant Biology (2018)
-
Substitution mapping of QTLs controlling seed dormancy using single segment substitution lines derived from multiple cultivated rice donors in seven cropping seasons
Theoretical and Applied Genetics (2017)
-
Identification of markers linked to genes for sprouting tolerance (independent of grain color) in hard white winter wheat (HWWW)
Theoretical and Applied Genetics (2016)
-
Mapping and characterization of seed dormancy QTLs using chromosome segment substitution lines in rice
Theoretical and Applied Genetics (2012)
