
Abstract
Both gene flow and historical events influence the genetic diversity of natural populations. One way to understand their respective impact is to analyze population genetic structure at large spatial scales. We studied the distribution of genetic diversity of 17 populations of Vouacapoua americana (Caesalpiniaceae) in French Guiana, using nine microsatellite loci. Low genetic diversity was observed within populations, with a mean allelic richness and gene diversity of 4.1 and 0.506, respectively, which could be due to low effective population size and/or past bottlenecks. Using the regression between Fst/(1−Fst), estimated between pairs of populations, and the logarithm of the geographical distance, the spatial genetic structure can partly be explained by isolation-by-distance and limited gene flow among populations. This result is in agreement with the species' biology, including seed and pollen dispersal by rodents and insects, respectively. In contrast, no clear genetic signal of historical events was found when examining genetic differentiation among populations in relation to biogeographical hypotheses or by testing for bottlenecks within populations. Our conclusion is that nuclear spatial genetic structure of V. americana, at the geographic scale of French Guiana, is better explained by gene flow rather than by historical events.
Similar content being viewed by others


Introduction
Historical events and gene flow/genetic drift are the main evolutionary factors explaining the geographical distribution of neutral genetic diversity within species. Under constant gene flow and demographic equilibrium, spatial genetic structure among populations can be predicted by several theoretical models (eg Wright, 1931; Slatkin, 1993; Rousset, 1997). However, historical events, such as fragmentation or rapid expansion of a species distribution, can modify these genetic patterns. In some instances, genetic drift and reduction of gene flow during periods of contraction can create high genetic divergence between isolated populations (Latta and Mitton, 1997). Furthermore, recolonization by a few founders during periods of expansion can produce population bottlenecks and loss of genetic diversity (Nei et al, 1975; Cornuet and Luikart, 1996). These historical events can be detected by comparing genetic diversity among several geographical groups of populations, as performed for numerous tree species in temperate areas (eg Latta and Mitton 1997; Le Corre et al, 1997; Taberlet et al, 1998). Compared to other plants, tree species are generally characterized by high genetic diversity within populations and low differentiation among them, a genetic pattern that has been interpreted as a consequence of large effective population size and extensive gene flow (Hamrick et al, 1992). However, the question of whether this pattern occurs in Neotropical tree species remains open, because studies of populations separated by more than a few kilometres are rare in tropical rainforest (Loveless, 1992). Low density, clustering of individuals, and putative extinction/recolonization in Neotropical tree species could promote stronger spatial genetic structure than in temperate tree species.
Low density and clustering of individuals at fine spatial scales, which are common characteristics of many Neotropical tree species (Condit et al, 2000), may result in limited effective population size, and lead to reduced genetic diversity (Wright, 1931), as well as significant genetic differentiation among populations (Cardoso et al, 1998, Dutech et al, 2002). Moreover, Neotropical rainforest ecosystems are characterized by rapid short-term environmental changes, such as tree fall gap-disturbances (Hubbell et al, 1999) and periods of drought (Bawa and Markham, 1995), sometimes associated with fires (Tardy, 1998). Local extinction may therefore be frequent, especially for species with low density, low growth rate, or low seed dispersal. Such factors should be considered because theoretical studies have shown that genetic differentiation among populations of a metapopulation may increase under these conditions, especially when there are few and/or genetically related founders (eg Wade et al, 1994). High gene flow can counteract these predictions, but seed and pollen dispersal is thought to predominantly occur over small distances for various tropical tree species (Loveless, 1992).
The history of Neotropical tree populations should also be taken into account when analyzing genetic diversity at larger spatial scales. Biogeographical and historical data suggest ancient contraction–expansion of rainforest in several areas of the Amazonian Basin and the Guianas during the Pleistocene and Holocene periods (20 000–1000 years before present (BP); Granville, 1982; Hooghiemstra and van der Hammen, 1998; Pennington et al, 2000). A decrease in precipitation throughout the Pleistocene and Holocene periods would have led to contraction of rainforest that was previously limited to areas of higher altitude, and expansion of dry forests in other areas (Hooghiemstra and van der Hammen, 1998). These events should have produced population extinction–recolonization processes over a large area. Genetic diversity within species that were sensitive to climatic changes and that slowly recolonized forest should therefore have been affected. However, most genetic studies have been conducted on geographically close populations (ie a few kilometres apart) and at latitudes where tree species often occur at lower densities than temperate tree species (Loveless, 1992; but see Gillies et al, 1999). Such populations may be vulnerable to historical events, as has been suggested by recent studies in French Guiana based on the analysis of cpDNA diversity in Dicorynia guianensis (Caron et al, 2000) and Vouacapoua americana (Dutech et al, 2000b). In V. americana, homogeneous patches of cpDNA haplotypes, occurring over several thousand hectares along the coast of French Guiana, strongly suggest large extinction–recolonization events in the past (Dutech et al, 2000b).
The goal of this study is to evaluate the respective roles of the ecological traits mentioned above (ie low density, spatial clustering, and gene flow mediated by animals) and historical factors (expansion and contraction of rainforest) in shaping the nuclear genetic diversity of the Neotropical tree species V. americana in French Guiana. The primary dispersal agents of this tree are two rodent species (Dasyprocta leporina and Myoprocta exilis), which predominantly bury seeds up to 10 m away from the maternal trees (Forget, 1990). The floral syndrome (small-sized, fragrant and yellow flowers) indicates small insects as the likely pollinators. The average adult density is generally lower than nine individuals/ha (Dutech et al, 2002), and individuals have a clustered distribution (Forget et al, 1999). From these traits, one can predict low gene flow, low genetic diversity within populations and substantial differentiation among populations. Furthermore, V. americana is a typical, shade-tolerant species of mature tropical rainforests of the eastern Guiana shield, with low adult growth rate (Gourlet-Fleury and Houllier, 2000). Its genetic structure should therefore contain the signature of past biogeographical dynamics (if any) of the rainforest in French Guiana. According to biogeographical and historical hypothesis (Granville, 1982; Tardy, 1998), we assume that several V. americana populations, mainly located in the coastal area, could have experienced local extinction–recolonization during the Pleistocene and Holocene periods. In contrast, three other areas in the central, the north-western and the north-eastern part of the country, mainly located in areas of higher altitude, could have been weakly disturbed by past climatic changes or fire events (Granville, 1982; Tardy, 1998). The former group should be more strongly marked by bottleneck events, while high genetic divergence is expected between the three forest refugial areas.
Our goals were to evaluate whether: (i) neutral nuclear genetic diversity within populations is low, as may be inferred from ecological traits and putative bottleneck events; (ii) indirect estimates of gene flow among populations better explain spatial nuclear genetic structure in French Guiana compared with biogeographical hypotheses; and (iii) bottlenecks can be detected, especially in coastal populations that appear to have been strongly disturbed in the past and may have suffered more population reductions.
Material and methods
Sampling
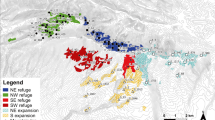
A total of 17 populations of V. americana were sampled throughout French Guiana (Table 1). The choice of sampling sites was based on three criteria: (i) accessibility, which disqualified most of southern French Guiana, (ii) even distribution over northern French Guiana, and (iii) sampling areas with supposedly different biogeographical histories and levels of past disturbance. These areas were defined according to phytogeographical studies in French Guiana, which indicated that the Sinnamary River (Figure 1) might be a biogeographical barrier for many species (Granville, 1982). It has also been shown that the flora of central French Guiana exhibits high species richness and endemism, differing from areas located northwards (Granville et al, 1996). Based on these data, the sampled populations fall into three biogeographical groups that could have genetically diverged during the Pleistocene and Holocene periods: Centre of French Guiana, East and West of the Sinnamary River (referred to, respectively, as Centre, North-east and North-west; Table 1 and Figure 1). We also sampled populations that had been differentially affected by historical events during the Pleistocene and Holocene periods (Granville, 1982; Tardy, 1998). One to three populations sampled in each of the three areas were assumed to have been weakly disturbed, six populations were located close to these forest refugial areas and five populations were located on the coast where rainforest was highly disturbed (Granville, 1982; see Table 1 and Figure 1).

Geographical location of the 17 populations of V. americana studied in French Guiana. Rivers are indicated in italics. The numbers refer to populations (Table 1). The three most important refuges according to Granville (1982) and Tardy (1998) are indicated by the darker areas and the coastal area is shown in white. Limits between the three biogeographical goups are indicated by bold lines.
In total, 21–24 individuals (mean=23.6) were sampled per population. To reduce the chance of common maternal origin, sampled individuals were separated by a minimum of 50 m. Samples were collected as either leaves of large trees (greater than 10 cm diameter at breast height) where a gun and small shots were used, or leaves of seedlings were sampled; in some populations both sampling methods were used (Table 1). In populations sampled as seedlings, individuals were sampled under different trees. We assumed that the genetic diversity of these seedlings is representative of that of adult populations. Leaves were stored in plastic bags up to 3 days after collection and were then stored at −80°C in the laboratory.
DNA analysis
Total DNA was extracted in liquid nitrogen using a CTAB buffer (Dutech et al, 2000b). Nine microsatellite loci isolated from the genome of V. americana, and composed of at least 10 noninterrupted (AG) repeats, were analyzed using the polymerase chain reaction (PCR) method following Dutech et al (2000a). PCR products were separated by electrophoresis in denaturing polyacrylamide sequencing gels and stained with silver nitrate.
Statistical analyses
Diversity within populations
For each population and locus, we estimated the observed number of alleles (A), the observed heterozygosity (Ho), and the expected heterozygosity (He). Departures from Hardy–Weinberg equilibrium at each locus and genotypic disequilibria between pairs of loci in each population were tested using exact tests (Raymond and Rousset, 1995a). Hardy–Weinberg equilibrium and genotypic disequilibria were tested, respectively, over all loci in each population and over all populations, using Fisher's method. Wright's Fis was estimated in each population following Weir and Cockerham (1984). All calculations and tests above were performed using Genepop version 3.1d (Raymond and Rousset, 1995b).
Genetic differentiation among populations
Exact tests of genetic differentiation were performed per locus and pair of populations. Fst was estimated over all populations and between each pair of populations using the method of Weir and Cockerham (1984). ρst, an analog of Fst taking into account the difference in size between microsatellite alleles (see Rousset, 1996), was also estimated according to Michalakis and Excoffier (1996). We compared the average estimates of Fst and ρst over all populations across loci using Wilcoxon's signed ranks test. Isolation-by-distance was tested by examining the relationship between geographical and genetic differentiation among populations, using Rousset's (1997) method. Assuming a two-dimensional habitat and isolation-by-distance, Rousset showed that the ratio Fst/(1−Fst), estimated between pairs of populations, is linearly related to the logarithm of the Euclidean geographical distance separating these populations. The significance of the regression of Fst/(1−Fst) against distance was assessed using a permutation method and the Spearman rank correlation coefficient. If gene flow is the main factor explaining the genetic differentiation among populations and under the hypothesis of migration-genetic drift equilibrium, the inverse of the regression slope gives an estimate of the product between 4π, the variance of dispersal between parent and offspring (σ2), and the density of reproductive individuals (D) (Rousset, 1997). All analyses were performed using Genepop version 3.1d (Raymond and Rousset, 1995b) except for the difference of pairwise Fst from zero which was tested by a permutation method (with 100 permutations of individuals among populations) using Arlequin version 2.0 (Schneider et al, 1999).
As mentioned above, three geographical groups were defined a priori (see Table 1). An analysis of molecular variance (AMOVA; Excoffier et al, 1992) was performed to estimate the correlation of genetic diversity at different levels of geographical subdivision: among geographical groups, among populations within groups, and within populations. This analysis was performed using Arlequin version 2.0 (Schneider et al, 1999). The significance of genetic differentiation estimates among populations and among geographical groups was evaluated using a permutation method (1000 permutations of individuals or populations depending on the differentiation estimates tested; see Schneider et al, 1999).
A hierarchical classification of populations was also constructed to analyze the geographical distribution of genetic variation. The chord distance (Cavalli-Sforza and Edwards, 1967) was computed between all pairs of populations and the populations were clustered using the neighbor-joining method. The chord distance assumes that genetic divergence among populations is caused by genetic drift, although not constant and equal population size (Felsenstein, 1993). This analysis was conducted using Phylip version 3.57c (Felsenstein, 1993). The robustness of clustering was evaluated using a bootstrap method with resampling (100 replicates) among loci within populations.
Detection of historical events
According to biogeographical and historical studies in French Guiana (Granville, 1982; Tardy, 1998), some populations of V. americana have experienced bottlenecks, especially those far from areas of higher altitude, that is mainly on the coast (Figure 1). Cornuet and Luikart (1996) developed a test for detecting a recent bottleneck or expansion in populations, using genetic data. The test is based on the difference between the expected heterozygosity (gene diversity) and the heterozygosity estimated from the number of alleles expected in a random-mating population of constant size and at mutation – genetic drift equilibrium. A bottleneck in the past produces a loss of rare alleles, while the expected heterozygosity is less affected (Nei et al, 1975). The heterozygosity estimated from allele number is therefore lower than the expected heterozygosity (heterozygosity excess; Cornuet and Luikart, 1996). The temporal dynamics of the difference between these two heterozygosities after the bottleneck depends on the mutation model assumed. There are equivocal reports about whether the stepwise mutation model (SMM) or the infinite allele model (IAM) describes more accurately the mutation process at microsatellite loci. Since we have no information to select either of these models in V. americana, we performed the tests for both models. A standardized difference between the two heterozygosities was computed for each locus (Cornuet and Luikart, 1996). A Wilcoxon's signed ranks test was then performed to evaluate whether the difference across loci significantly differs from zero. The tests were performed using Bottleneck version 1.2.02 (available at http://www.ensam.inra.fr/URLB). Using this method, bottleneck signature can be detected up to two and four Ne (effective population size) generations after the event (see Cornuet and Luikart, 1996). The low growth rate of V. americana (Gourlet-Fleury and Houllier, 2000) and the diameter at first reproduction (20 cm diameter at breast height; Caron et al, 2004b) suggest an approximate generation time of 100 years. Demographic events that occurred during the Holocene can therefore be detected. Coastal populations (highly disturbed; see above and Table 1) should return more significant tests of bottlenecks than other populations.
Results
Variability and genetic structure within populations
The mean number of alleles per population was 4. 1 (maximum=5.1 in Dorlin; minimum=3.2 in Voltaire; Table 1). Locus Wac1 was highly polymorphic with 24 alleles over all populations studied; no other locus exhibited more than 10 alleles (Table 2). When Wac1 was excluded, the mean number of alleles per population (3.0; range=2.3–3.7) was significantly lower than when Wac1 was included (Wilcoxon signed ranks test, N=17, P<0.001). Allelic richness was positively correlated with both the number of repeat units and the maximum number of uninterrupted repeat units found in the cloned sequence (Spearman rank correlation coefficient over the nine loci: r=0.77 and 0.71, P=0.014 and 0.031, respectively). The mean observed heterozygosity per population (Ho) and gene diversity (He) were 0.467 and 0.506, respectively. The smallest Ho and He values were observed in the Voltaire population (0.304 and 0.340, respectively) and the highest Ho and He values in the two central populations (0.546 in Saül and 0.522 in Dorlin, respectively; Table 1). Significant deviations (at the 5% level) from Hardy–Weinberg expectations were detected in nine out of 150 tests performed and concerned seven out of nine loci. Significant deviations for each locus were not observed in more than two populations (data not shown); three tests were significant in both Nouragues and St Eugène (Wac1, Wac10, and Wac13 in Nouragues and Wac7, Wac12, and Wac16 in St Eugène). No single-locus test was significant over all populations (Fisher's method, P>0.05) and tests in each population over all loci indicated a highly significant heterozygote deficiency only in the Nouragues and St Eugène populations (Fisher's method, P<0.01). Fis estimates per population and locus, as well as estimates per population over all loci, were generally low and not significantly different from zero (Tables 1 and 2).
Genotypic disequilibrium was tested in 589 pairs of loci-population combinations. The hypothesis of no disequilibrium was rejected at the 5% level in 25 cases only, which is less than what is expected by chance at this level of significance. No pairs of loci showed a significant disequilibrium at the 5% level when considering all populations (Fisher's method).
Population differentiation
The exact test of pairwise differentiation was conducted in 1224 locus-population combinations. All pairs of populations showed a significant differentiation for at least two loci and about 80% of the pairs were differentiated at more than five out of nine loci analyzed (data not shown). Using locus Wac1, all pairs of populations could be differentiated except one (Tortue and St Georges). The total number of alleles per locus was significantly correlated with the number of population pairs that a locus could differentiate (Spearman rank correlation coefficient, r=0.95, P=10−4). All pairs of populations were significantly differentiated, when all loci were considered (P<0.01).
The pairwise Fst estimates between populations ranged between 0.008 (St Georges-Saül) and 0.195 (Lysis-Lucifer). All pairwise Fst were significantly different from zero except for St Georges-Saül and St Georges-Tortue (100 permutations). The Fst and ρst estimates over all populations were 0.08 and 0.05, respectively (see Table 2), and were not significantly different (Wilcoxon signed ranks test; N=9, P=0.11).
The ratio of Fst/(1−Fst) increased with the logarithm of distance (Figure 2). The null hypothesis of no isolation-by-distance was rejected (Spearman rank correlation test, P<0.02). The equation of the regression was Fst/(1−Fst)=0.0199 log (distance in kilometres)−0.0052, which produced an estimate of Dσ2 slightly lower than four individuals. The AMOVA showed that differentiation both among the three geographical groups and among populations within groups was highly significant (P<0.001; Table 3). However, the percentage of total variance explained by the geographical groups is about three times lower than the variance explained by the differentiation among populations within groups (2.3 and 6.6%, respectively, Table 3).

Relationship between R=Fst/(1−Fst) and the logarithm of the distance (in km) in V. americana in French Guiana. Pairwise Fst are estimated following Weir and Cockerham (1984). The regression is y=0.0199x–0.0052.
Tree reconstruction (Figure 3) produced bootstrap values higher than 75% for two groups of populations only, that is the central populations and two north-western populations (Voltaire and Lucifer). Other groups were clustered with much lower bootstrap values indicating little geographic cohesion within the north-eastern and north-western groups.

Hierarchical classification of the 17 V. Americana populations studied using the chord distance and the neighbor-joining method. The numbers at the tree tips refer to population reference number (Table 1). Circles, bold characters, and underlined characters refer to the three biogeographical groups: central, north-western, and north-eastern populations, respectively. The number near the nodes are bootstrap values (in %). The scale of the genetic distance is given in the upper right corner.
Tests for demographic bottleneck
Under the SMM, three populations out of 17 exhibited a significant deficit of heterozygosity when considering all loci (St Georges, Tibourou, and St Eugène; Wilcoxon signed ranks test, P<0. 01, =0.02, and =0.02, respectively; Table 4). Such a deficit can be detected under the SMM, either after a recent expansion, or just before reaching equilibrium after a bottleneck that can take approximately four Ne generations (see Cornuet and Luikart, 1996). Two north-eastern populations (Kaw and Lysis) exhibited an excess of heterozygosity under the IAM which may indicate a recent bottleneck (P=0.02 and 0.01, respectively; Table 4). No population assumed to have been highly disturbed in the past (Table 1) exhibited a significant signal of past expansion or bottleneck.
Discussion
Relationship between ecological traits, populations dynamics, and diversity within populations
Studies using microsatellite loci in tree populations often reveal high expected heterozygosity (He) and large numbers of alleles per locus (Na). For example, Na and He were higher than 17 and 0.84, respectively, in two European oak species (Streiff et al, 1998) and in a Brazilian tree species (Caryocar brasiliense; Collevatti et al, 2001). However, our results indicate much lower population diversity in V. americana, with mean Na and He smaller than 5 and 0.6, respectively. The method used to isolate microsatellite loci in this species (based on an enrichment protocol; Dutech et al, 2000a) could be biased towards loci with small numbers of repeats and low variability. The number of repeats and variability were indeed correlated, as in other studies of microsatellite variation (for a review, see Jarne and Lagoda, 1996). Limited genetic diversity was also found for V. americana, relative to four other tree species sampled in Paracou, when using randomly amplified polymorphic DNAs markers and amplified fragment length polymorphism markers (Caron et al, 2004a). These congruent results therefore suggest that low genetic diversity most likely reflects biological traits and the evolutionary history of V. americana, rather than locus sampling bias.
The mating system strongly affects the amount of variation within populations, with selfing species often showing low variation (Hamrick et al, 1992). Independent data indicate that the selfing rate in V. americana is low (<0.05 in two populations from St Eugène; Chauvet, 2001). This estimate is consistent with our low estimates of Fis, and the fact that most populations did not deviate from Hardy–Weinberg expectations. Mating system, therefore, does not explain low genetic diversity within V. americana populations, which might rather be explained by small effective population size (Wright, 1931). Several arguments favor this explanation. First, the adult density of V. americana populations is often lower than nine individuals per hectare. Second, low seed and pollen dispersal and clustering of individuals (Forget et al, 1999; Dutech et al, 2002) should mainly limit reproduction to few adults from the same patch. Third, only a proportion of adult trees in a patch may reproduce. Indeed, recruitment would occur in a few vacant sites, mainly located at the outer limit of dense patches of adults or sometimes even further away (S Traissac, unpublished data). Trees located at these limits may therefore be favored, which in turn should limit the number of maternal trees. Finally, recent bottlenecks can also decrease the genetic diversity within populations, and will be considered below.
Genetic differentiation among populations and gene flow
One result of our study is the relatively low differentiation (overall Fst estimate of 0. 08) among populations of V. americana separated by up to 300 km. This is slightly lower than the mean value estimated in tropical tree species using allozymes (0.11; Loveless, 1992), and more comparable to values generally observed in woody perennial, outcrossed species (Hamrick et al, 1992). A possible explanation is that gene flow among populations, especially mediated by pollen, is higher in V. americana than previously assumed. However, inferring gene flow from a global Fst estimate should be done with extreme caution, because several factors influence this parameter besides gene flow (eg models of genetic differentiation, mutation model, sampled geographical scale, or sampling variance; see Whitlock and McCauley (1999) for numerous criticisms). Actually, the overall Fst estimated in this study does not take account of the strong differentiation that can exist among patches of individuals within populations, as has been shown at Paracou and Nouragues (Dutech et al, 2002).
The relationship between Fst estimated per pair of populations and geographic distance, which is less influenced by sampling design and mutation model (Rousset, 1996, 1997), provides a better insight into factors affecting population differentiation. A significant pattern of isolation-by-distance was detected at the scale of the whole study. This result suggests that gene flow is an important factor that contributes to shaping genetic differentiation among populations of V. americana. Furthermore, based on the regression slope, the estimate of Dσ2, less than four individuals, may indicate either low density, and/or restricted gene flow in V. americana. The available ecological data are compatible with this interpretation. First, the average density of adults is low for most V. americana populations (fewer than nine individuals/ha). Second, insect pollination and seed dispersal by rodents suggest that gene flow is mainly limited to a few hundreds of metres around parental trees. Inference of gene flow from our data should therefore be taken with some caution. Firstly, because of the clustering of V. americana, the average density cannot accurately be estimated at the spatial scale of French Guiana. Secondly, the regression method would probably be more efficient at shorter geographical distances, when historical events are less likely to influence the current genetic differentiation (Rousset, 1997). Thirdly, recent variation in local density of populations affects the distribution of variability among populations, and may interfere with interpretations based on models assuming demographic equilibrium (Rousset, 1997). Despite these limitations, theoretical studies have shown that the regression method for inferring gene flow is robust to local and temporal variation of effective population size (Leblois, 2000). Moreover, the analysis of spatial genetic structure within the Paracou and Nouragues populations suggested that most pollination events occurred over distances less than 100 m, and confirmed restricted seed dispersal (Dutech et al, 2002). These congruent results strongly support the hypothesis that low gene flow and isolation-by-distance explain an important part of the spatial genetic structure of V. americana in French Guiana.
Effect of historical factors on genetic diversity of V. americana in French Guiana
Although the AMOVA showed that 2.3% of the genetic variation occurred among the three assumed biogeographical groups, the data in general suggest that historical factors have had very little influence on the nuclear spatial genetic structure of V. americana in French Guiana. A larger genetic variation (ie 6.6% of the total variance) occurred among populations within biogeographical groups and few populations tended to cluster in the genetic distance analysis. Furthermore, the absence of a difference between ρst and Fst estimates favors this interpretation, since such a result is expected when genetic differentiation is approximately independent of the mutation process (ie high migration rates and/or recent time of divergence among populations; Rousset, 1996). The weak genetic divergence among biogeographical groups could be explained by short periods of isolation and/or low but constant pollen flow. This assumption is in agreement with the description (based on the presence of charcoals in soil) of several short periods of drought that produced rapid contraction–expansion of rainforest in French Guiana during the Holocene period (10 000–1000 years BP; Tardy, 1998).
Our data provide scant evidence of genetic bottlenecks within populations. Tests were only significant in a few populations and not for the same mutation model. Moreover, they were significant in populations located within or near areas that are assumed to have been weakly disturbed in the past, and in contradiction to expectations. The power to detect founder effects may be limited by the low number of loci used, unrealistic mutation models or migration among populations (Cornuet and Luikart, 1996). In addition, recent local population extinction–recolonization in V. americana, independent of the rainforest history, could explain the detection of bottlenecks for populations located in areas assumed to have been weakly disturbed during the Pleistocene and Holocene. These recent extinction–recolonization events are suggested by several observations of V. americana populations mostly composed of large and dead trees, and indicate a recent and significant decrease in the local adult population size (O Brunaux, personal communication and personal observations). Studies combining demography and genetics at a reduced geographic scale could shed more light on these aspects. There is also no clear explanation for the dependence of test results on the mutation models (see Table 4). A striking result is that under the SMM, significant excess of heterozygosity was detected in three populations, but under the IAM model, deficit was detected in two other populations. These asymmetrical results may not only be related to differences in demographic dynamics among populations independently of the contraction–expansion of the rainforest, but also to the statistical power to detect demographic changes under the two mutation models (Cornuet and Luikart, 1996).
The results obtained on nuclear spatial genetic structure strongly contrast with preliminary results of the distribution of cpDNA haplotypes, which suggested that V. americana populations have experienced historical bottlenecks (Dutech et al, 2000b). Two nonexclusive hypotheses are proposed to explain this pattern: ancient regressions of populations followed by recolonization from isolated populations, and reduction of range area during the Holocene period. The difference recorded in genetic diversity between nuclear loci and the chloroplast genome may be explained by the slow return of the chloroplast genome to a mutation–drift equilibrium after a bottleneck, because of its clonal evolution and low mutation rates. Consequently, genetic drift in small residual populations during the decline of a species and a bottleneck experienced during recolonization can be detected after a larger number of generations in the chloroplast genome than in the nuclear genome. The difference of evolution between nuclear and organelle genomes has already been extensively discussed (eg, Fay and Wu, 1999; Comes and Abbott, 2000), and probably occurs in V. americana populations.
Although sampling was limited to the half of North French Guiana (ie populations separated by a maximum of 300 km), this study is one of the rare studies in the Neotropics comparing several populations at this spatial scale. It provides a clearer perspective on the associated effects of gene flow and past climatic changes on the genetic structure of V. americana populations in French Guiana. The low nuclear genetic divergence among biogeographical groups therefore indicates that the V. americana populations were probably weakly isolated over short periods of times during post glacial climatic changes in the tropics (between 20 000 years BP and now). This might have prevented significant genetic drift within populations and promoted low but constant gene flow among populations. Of course, studies at spatial scales larger than French Guiana would be useful for comparing different biogeographical areas suspected to have been more strongly isolated in South America during the Pleistocene period (see eg Granville, 1982). In this perspective, studying V. americana populations from northeastern Brazil, as well as populations sampled in species from various plant families, would provide a clearer view about the effects of past climatic changes on the species of Neotropical rainforest ecosystems.
References
Bawa KS, Markham A (1995). Climate change and tropical forests. Trends Ecol Evol 10: 348–349.
Cardoso MA, Provan J, Powell W, Ferreira PCG, De Oliveira DE (1998). High genetic differentiation among remnant populations of the endangered Caesalpinia echinata Lam. (Leguminosae-Caesalpinioideae). Mol Ecol 7: 601–608.
Caron H, Dumas S, Marque G, Messier C, Bandou E, Petit RJ et al (2000). Spatial and temporal distribution of chloroplast DNA polymorphism in a tropical tree species. Mol Ecol 9: 1089–1098.
Caron H, Bandou E, Kremer A (2004a). Multilocus assessment of level of genetic diversity in tropical trees in Paracou stand. In: Gourlet-Fleury S, Guehl J-M, Larossinie O (eds) Ecology and Management of a Neotropical Rain forest: The Lessons of PARACOU (French Guiana), Elsevier: Paris (in press).
Caron H, Dutech C, Bandou E (2004b). Reproductive phenology and mating system of trees in Paracou stand. In: Gourlet-Fleury S, Guehl J-M, Larossinie O (eds) Ecology and Management of a Neotropical Rain forest: The Lessons of PARACOU (French Guiana), Elsevier: Paris (in press).
Cavalli-Sforza LL, Edwards AWF (1967). Phylogenetic analysis: models and estimation procedures. Am J Hum Gen 19: 233–257.
Chauvet S (2001). Effets de la fragmentation forestière sur les interactions plantes-animaux: conséquences pour la régénération végétale. PhD Thesis,. Université P. & M. Curie, Paris VI.
Collevatti RG, Grattapaglia D, Hay JD (2001). Population genetic structure of the endangered tropical tree species Caryocar brasiliensis, based on variability at microsatellite loci. Mol Ecol 10: 349–356.
Comes HP, Abbott RJ (2000). Random amplified polymorphic DNA (RAPD) and quantitative trait analyses across a major phylogeographical break in the Mediterranean ragwort Senecio gallicus Vill. (Asteraceae). Mol Ecol 9: 61–76.
Condit R, Ashton PS, Baker P, Bunyavejchewin S, Gunatilleke S, Gunatilleke N et al (2000). Spatial patterns in the distribution of tropical tree species. Science 288: 1414–1418.
Cornuet JM, Luikart G (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Dutech C, Amsellem L, Billotte N, Jarne P (2000a). Characterization of (GA)n microsatellite loci using an enrichment protocol in the neotropical tree species Vouacapoua americana. Mol Ecol 9: 1433–1434.
Dutech C, Maggia L, Joly HI (2000b). Chloroplast diversity in Vouacapoua americana (Caesalpiniaceae) a neotropical forest tree. Mol Ecol 9: 1427–1432.
Dutech C, Seiter J, Petronelli P, Joly HI, Jarne P (2002). Evidence of low gene flow in a neotropical tree species in two forest stands of French Guiana. Mol Ecol 11: 725–738.
Excoffier L, Smouse PE, Quattro JM (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131: 479–491.
Fay JC, Wu C-I (1999). A human population bottleneck can account for the discordance between patterns of mitochondrial versus nuclear DNA variation. Mol Biol Evol 16: 1003–1005.
Felsenstein J (1993). PHYLIP (Phylogeny Inference Package), version 3.5c. Department of Genetics, University of Washington: Seattle.
Forget P-M (1990). Seed dispersal of Vouacapoua americana (Caesalpiniaceae) by caviomorph rodents in French Guiana. J Trop Ecol 6: 459–468.
Forget P-M, Mercier F, Collinet F (1999). Spatial patterns of two rodent-dispersed rainforest trees, Carapa procera (Meliaceae) and Vouacapoua americana (Caesalpiniaceae) at Paracou, French Guiana. J Trop Ecol 15: 301–313.
Gillies ACM, Navarro C, Lowe AJ, Newton AC, Hernàndez M, Wilson J et al (1999). Genetic diversity in Mesoamerican populations of mahogany (Swietenia macrophylla) assessed using RAPDs. Heredity 83: 722–732.
Gourlet-Fleury S, Houllier F (2000). Modelling diameter increment in a lowland evergreen rain forest in French Guiana. For Ecol Manage 131: 269–289.
Granville J-J de (1982). Rain forest and xeric flora refuges in French Guiana. In: Prance GT (ed) Biological Diversification in the Tropics, Columbia University Press: New York pp 137–158.
Granville J-J de, Cremers G, Hoff M (1996). L'endémisme en Guyane française. In: Guillaumet JL, Belin M, Puig H (eds) Phytogéographie tropicale – Réalités et Perspectives, ORSTOM Editions: Bondy pp 101–113.
Hamrick JL, Godt MJW, Sherman-Broyles SL (1992). Factors influencing levels of genetic diversity in woody plant species. New For 6: 95–124.
Hooghiemstra H, van der Hammen T (1998). Neogene and Quaternary development of the neotropical rain forest: the forest refugia hypothesis, and a literature overview. Earth-Sci Rev 44: 147–183.
Hubbell SP, Foster RB, O'Brien ST, Harms KE, Condit R, Weschsler B et al (1999). Light-gap distrubances, recruitment limitation and tree diversity in a neotropical forest. Science 283: 554–557.
Jarne P, Lagoda PJL (1996). Microsatellites, from molecules to populations and back. Trends Ecol Evol 11: 395–444.
Latta RG, Mitton JB (1997). A comparison of population differentiation across four classes of gene marker in limber pine (Pinus flexilis James). Genetics 146: 1153–1163.
Le Corre V, Dumolin-Lapègue S, Kremer A (1997). Genetic variation at allozyme and RAPD loci in sessile oak Quercus petraea (Matt.) Liebl.: the role of history and geography. Mol Ecol 6: 519–529.
Leblois R (2000). Etude de l'influence des facteurs mutationnels et démographiques sur l'estimation de paramètres démographiques à partir de génotypes individuels. Diplôme d'Etudes Approfondies, Université Montpellier II.
Loveless MD (1992). Isozyme variation in tropical trees: patterns of genetic organization. New For 6: 67–94.
Michalakis Y, Excoffier L (1996). A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142: 1061–1064.
Nei M, Maruyama T, Chakraborty R (1975). The bottleneck effect and genetic variability in populations. Evolution 29: 1–10.
Pennington RT, Prado DA, Pendry C (2000). Neotropical seasonally dry forests and Pleistocene vegetation changes. J Biogeogr 27: 261–273.
Raymond M, Rousset F (1995a). An exact test for population differentiation. Evolution 49: 1280–1283.
Raymond M, Rousset F (1995b). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Rousset F (1996). Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics 142: 1357–1362.
Rousset F (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Schneider S, Kuffer JM, Roessli D, Excoffier L (1999). Arlequin V 2.000, a software for population genetics data analysis, University of Geneva, Geneva.
Slatkin M (1993). Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47: 264–269.
Streiff R, Labbé T, Bacilieri R, Steinkellner H, Glössl J, Kremer A (1998). Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol 7: 317–328.
Taberlet P, Fumagalli L, Wust-Saucy A, Cosson J-F (1998). Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7: 453–464.
Tardy C (1998). Paléoincendies naturels, feux anthropiques et environnements forestiers de Guyane Française du tardiglaciaire à l'holocène récent: Approches chronologique et anthracologique. PhD Thesis,. Université Montpellier II.
Wade MJ, McKnight ML, Shaffer HB (1994). The effects of kin-structured colonization on nuclear and cytoplasmic genetic diversity. Evolution 48: 1114–1120.
Weir BS, Cockerham CC (1984). Estimating F-statistics for the analysis of population structure. Evolution 8: 1358–1370.
Whitlock MC, McCauley DE (1999). Indirect measures of gene flow and migration: Fst≠1/(1+4Nm). Heredity 82: 117–125.
Wright S (1931). Evolution in Mendelian populations. Genetics 16: 97–159.
Acknowledgements
We thank L Maggia for hosting CD in his laboratory in Kourou during the completion of this work, L Teillier (ONF- Guyane), P-M Forget (Station des Nouragues – CNRS/MNHN), S Chauvet (Station de St Eugène – MNHN), D Fournier (Azarco- Guyane Française), J-F Milian (CBJ-France), and M Boudrie for help with collection of plant material, E Bandou (INRA-Guyane), C Bidal for technical help in the laboratory, J-F Molino, V Sork and two anonymous reviewers for their helpful comments on the manuscript. C Dutech is supported by a PhD fellowship from the Ministère Français de la Recherche, de la Technologie et de l'Enseignement. This work was supported by a grant from Cirad (Centre International de la Recherche en Agronomie pour le Développement)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dutech, C., Joly, H. & Jarne, P. Gene flow, historical population dynamics and genetic diversity within French Guianan populations of a rainforest tree species, Vouacapoua americana. Heredity 92, 69–77 (2004). https://doi.org/10.1038/sj.hdy.6800384
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800384
Keywords
This article is cited by
-
Population genetic structure and demographic history of the timber tree Dicorynia guianensis in French Guiana
Tree Genetics & Genomes (2024)
-
Genetic structure and demographic history of Allium mongolicum based on SSR markers
Plant Systematics and Evolution (2022)
-
Morphological and genetic diversity of shea tree (Vitellaria paradoxa) in the savannah regions of Ghana
Genetic Resources and Crop Evolution (2017)
-
Local habitat condition rather than geographic distance determines the genetic structure of Tamarix chinensis populations in Yellow River Delta, China
Tree Genetics & Genomes (2016)
-
Structure and genetic diversity of Ixora margaretae an endangered species
Tree Genetics & Genomes (2013)
