
Abstract
Background:
Expression of protein kinase C alpha (PKCα) is elevated in prostate cancer (PCa); thus, we have studied whether the development of tumourigenesis in prostate epithelial cell lines modifies the normal pattern of choline (Cho) metabolite release on PKC activation.
Methods:
Normal and tumourigenic human prostate epithelial cell lines were incubated with [3H]-Cho to label choline phospholipids. Protein kinase C was activated with phorbol ester and blocked with inhibitors. Choline metabolites were resolved by ion-exchange chromatography. Phospholipase D (PLD) activity was measured by transphosphatidylation. Protein expression was detected by western blotting and/or RT–PCR. Choline uptake was measured on cells in monolayers over 60 min.
Results:
Normal prostate epithelial cell lines principally released phosphocholine (PCho) in contrast to tumourigenic lines, which released Cho. In addition, only with normal cell lines did PKC activation stimulate Cho metabolite release. Protein kinase C alpha expression varied between normal and tumourigenic cell lines but all showed a PKCα link to myristoylated alanine-rich C kinase substrate (MARCKS) protein. The five cell lines differed in Cho uptake levels, with normal PNT2C2 line cells showing highest uptake over 60 min incubation. Normal and tumourigenic cell lines expressed mRNA for PLD1 and PLD2, and showed similar levels of basal and PKC-activated PLD activity.
Conclusions:
The transition to tumourigenesis in prostate epithelial cell lines results in major changes to Cho metabolite release into the medium and PKC signalling to phosphatidylcholine turnover. The changes, which reflect the metabolic and proliferative needs of tumourigenic cells compared with untransformed cells, could be significant for both diagnosis and treatment.
Similar content being viewed by others


Main
Conventional protein kinase C alpha (PKCα) is linked to the regulation of cell proliferation, motility, survival, apoptosis, differentiation, metastasis and multidrug resistance (Gutcher et al, 2003; Gavrielides et al, 2004; Koivunen et al, 2006; Larsson, 2006). Protein kinase C alpha expression is reduced in many cancers (Koivunen et al, 2006; Griner and Kazanietz, 2007; Mackay and Twelves, 2007; Ali et al, 2009). However, early prostate adenocarcinomas (PCa) show increased PKCα protein (Cornford et al, 1999; Koren et al, 2004; Lahn et al, 2004). Rat prostatic tumour cell lines also show elevated PKCα expression over controls (Powell et al, 1994). Androgen-independent human prostatic epithelial carcinoma lines PC3 and DU145 express PKCα mRNA and protein more prominently than does the androgen-sensitive LNCaP line (Krongrad and Bai, 1994; Powell et al, 1996), although only LNCaP cells undergo PKCα-mediated apoptosis when stimulated with phorbol esters (O’Brian 1998; Gutcher et al, 2003; Gonzalez-Guerrico et al, 2005). However, PKCα mediates the apoptosis induced by activation of Toll-like receptor 3 in both LNCaP and PC3 lines (Paone et al, 2008). Protein kinase C alpha can affect the growth-inhibiting effects of transforming growth factor-β in PC3 cells (Lamm et al, 1997), as well as epidermal growth factor receptor transactivation and activation of Erk1/2 (Stewart and O’Brian, 2005). Protein kinase C alpha is a proposed therapeutic target in androgen-independent PCa (O’Brian, 1998); therefore, it is important to understand how elevated PKCα expression as observed in PCa influences downstream targets which are also implicated in tumourigenesis; for example, phospholipase D (PLD) (Cockcroft, 2001; Foster, 2009).
Phospholipase D expression and activity are elevated in several human tumours and neoplastic cell lines (Foster and Xu, 2003; Foster 2006, 2009), resulting in increased cell proliferation and prevention of cell-cycle arrest and apoptosis (Joseph et al, 2002; Zhong et al, 2003; Foster, 2006). These effects occur partly through the increased formation of phosphatidic acid (PtdOH), which modulates the activity of Raf and mammalian target of rapamycin (mTOR), both regulators of cell proliferation (Guertin and Sabatini, 2007; Foster, 2007a, 2007b). Mammalian target of rapamycin is also implicated in signals that suppress apoptosis in cancer cells enabling their survival and proliferation under stress conditions (Foster, 2009). Enhanced PLD activity results in elevated levels of Cho metabolites, especially phosphocholine (PCho), in breast and prostate cancer cells (Ackerstaff et al, 2001; Glunde et al, 2006) where PCho levels may be a useful biomarker of malignant disease (Eliyahu et al, 2007; Beloueche-Babari et al, 2009). Choline kinase (CK) activity is increased in many tumours (Ramirez de Molina et al, 2002; Gallego-Ortega et al, 2009) and is a link to cell-cycle regulation (Ramirez de Molina et al, 2008) through MAPK and PI3K/AKT signalling (Chua et al, 2009; de Souza et al, 2009; Yalcin et al, 2010).
Stimulation of phosphatidylcholine (PtdCho) turnover in cells (Kiss, 1990) results in the release of Cho metabolites into the medium (Mufson et al, 1981; Hii et al, 1991; van Blitterswijk et al, 1991; Troyer et al, 1992; Morreale et al, 1997) wherein PCho can promote the mitogenic activity of insulin and growth factors (Cuadrado et al, 1993; Tomono et al, 1995; Chung et al, 1997). In this study, we show that tumourigenesis in human prostate epithelial cell lines alters the nature and control of Cho metabolites released into the medium on PKC activation.
Materials and methods
Cell culture
PNT1A, PNT2C2 and LNCaP prostate epithelial cell lines (up to passages 80, 150 and 50, respectively) were cultured in RPMI1640 (Gibco, Invitrogen Ltd, Paisley, Scotland, UK) with 10 mM HEPES, 2 mM glutamine and 10% fetal bovine serum (FBS) (R10). PC3 cells up to passage 50 were grown in Ham's F12 medium (Lonza, Slough, Berkshire, UK) with 7% FBS (F7). P4E6 line cells (Maitland et al, 2001) were cultured in KSFM medium (Gibco) with epidermal growth factor and pituitary additives+2% FBS (K2). For passage/experimentation, cells were rinsed with Tris-saline (TS) and released with Tris-trypsin (TT) for 10 min. Trypsin was inactivated with R10, cells pelleted by centrifugation and resuspended in normal growth medium for counting.
[3H]-Choline headgroup release into the medium
A total of 7.5 × 104 cells were seeded in triplicate into wells of 24-well plates in normal growth medium (see above) and cultured overnight. For LNCaP cells, wells were coated with poly-L-lysine (20 μg ml−1 in water) to aid adhesion. At 80–90% confluency, the medium was replaced with RPMI1640, F12 or KSFM containing 1% FBS and 0.5 μCi [3H]-choline (Perkin-Elmer, Beaconsfield, Buckinghamshire, UK) per well for 30 h to label Cho phospholipids to equilibrium. With this low level of serum, cells were just becoming confluent when used, and thus significant changes to enzyme activity because of contact inhibition or cell-cycle effects were avoided. Radioactive medium was then removed and cells were incubated for 60 min at 37°C with serum-free medium. Cells were gently rinsed twice more with warm (37°C) serum-free medium and 0.5 ml serum-free medium containing 1 mM choline chloride and 1 mM phosphocholine plus PKC activators/inhibitors (see Figure legends) was added per well. Aliquots (25 μl) of medium were removed from wells at T=0 and then as appropriate. A volume of 25 μl fresh medium was added back to wells to maintain volume. Aliquots of medium were centrifuged at 13 000 r.p.m. to pellet any cell debris. Duplicate 10 μl aliquots were added to 96-well Top Count plates for scintillation counting with 75 μl Microscint-20 (Perkin-Elmer Ltd.). Mean c.p.m. values from triplicate wells were calculated with s.d. (n=6). In experiments to reduce PKCα protein content, cells in triplicate wells were chronically treated with 250 nM TPA for the last 9 h of [3H]-choline labelling and then used as above.
Vesicle release
A volume of 400 μl of culture medium was removed from wells at the end of 3 h Cho release assays and triplicate 10 μl aliquots were taken for Top-Count scintillation counting as above. The remaining medium was transferred to a Beckman Eppendorf tube and centrifuged for 5 min at 13 000 r.p.m. to sediment any cell debris. Again, triplicate 10 μl aliquots of the low-speed supernatant were removed for counting. The remaining medium was then centrifuged at 100 000 g for 30 min at 4 °C to sediment exosomes and/or other vesicles (Nilsson et al, 2009). Aliquots (10 μl) of supernatant medium from this step were also counted.
Distribution of [3H]-Cho in intracellular Cho metabolites and lipids on labelling
Cells were seeded into wells of 24-well plates in triplicate and labelled with 0.5 μCi [3H]-Cho for 30 h as above. The labelling medium was removed and cells were rinsed three times with ice-cold PBS before extraction with 0.5 ml methanol, 0.5 ml chloroform:methanol, (1 : 2 v/v) and twice with 0.5 ml chloroform : methanol (1 : 1v/v). Solvent extracts were pooled, chloroform added to a final ratio of chloroform : methanol 2 : 1 and solvent/aqueous phases separated by the addition of 0.1 M KCl. Aliquots of each phase were taken in triplicate for scintillation counting. Choline metabolites in 400 μl top phase or medium from release experiments were diluted to 5 ml in distilled water and added to Dowex-50WH+ ion exchange resin columns to resolve GPCho (glycerylphosphorylcholine), PCho and Cho (Cook and Wakelam, 1989; Kiss et al, 1994). Radioactivity in triplicate 0.5 ml aliquots of each fraction was measured by scintillation counting to calculate total dpm/fraction.
Western blotting
A total of 1 × 105 cells were seeded into 24-well plates in normal growth medium and cultured overnight. The medium was removed and cells were rinsed with Tris/saline and solubilised in 100 μl warm (37°C) 2 × Laemmli sample buffer (Sigma-Aldrich, Poole, Dorset, UK) containing protease and phosphatase inhibitor pellets (Roche Diagnostics Ltd, Burgess Hill, West Sussex, UK). Extracts were heated at 100°C for 10 min. Equal volumes of cell extracts were resolved by SDS–PAGE on 12.5% gels for western blotting (Dawson et al, 2003) on Immobilon P (Millipore, Dundee, Scotland, UK). Protein kinase C alpha, actin and GAPDH blots were blocked in 5% Marvel/Tris buffered saline-0.2% Tween 20 (TBST). Phospho-MARCKS blots were blocked in 4% bovine serum albumin (BSA)/TBST. A mouse monoclonal antibody against the C-terminal V5 region of PKCα was prepared by Professor Nigel Groome (Oxford Brookes University) and used at 1 : 100. A MARCKS phospho (pS159/163) rabbit monoclonal antibody (Epitomics, InSight Biotechnology Ltd, Wembley, Middlesex, UK) was used at 1 : 1000. A polyclonal anti-actin antibody (Sigma-Aldrich) was used at 1 : 1000. Polyclonal antibodies to PKCδ and PKCɛ (Cell Signaling, New England Biolabs, Hitchin, Hertfordshire, UK) were used at 1 : 1000. An anti-GAPDH antibody (Abcam, Cambridge, Cambridgeshire, UK) was used at 1 : 2000. Detection was by ECL, and X-ray film was pre-flashed for densitometry using Image J. Protein kinase C alpha and p-MARCKS blots were stripped and reprobed for GAPDH or actin as loading controls.
Reverse transcriptase–PCR
Cells were grown in 75 cm2 flasks, rinsed and total RNA was extracted using a Qiagen RNeasy mini kit (Qiagen, Crawley, West Sussex, UK) and QIA shredder. RNA was quantified spectrophotometrically and 1 μg taken for cDNA synthesis using the Invitrogen SuperScript II RT (Invitrogen Ltd, Paisley, Scotland, UK) protocol. This was used to prepare a master mix with appropriate water controls for PCR. Conditions for amplification were 94°C for 0.5 min, 54°C for 0.5 min, 72°C for 1 min for 35 cycles. Primers were from Eurogentec Ltd (Southampton, UK). Sequences for hPLD1 and hPLD2 primers were as described by Gibbs and Meier (2000). Primers for PKCα and actin were as described by Myklebust et al (2000).
Transphosphatidylation
A total of 5 × 105 cells per well were cultured in duplicate in six-well plates in 2 ml normal growth medium to near confluency as mentioned above. Cells were rinsed and labelled for 6 h with 1 μCi [3H]-myristic acid (Amersham, GE Health Care, Chalfont St Giles, Buckinghamshire, UK) in 1 ml serum-free medium. Cells were then incubated for 30 min in fresh serum-free medium, which was then replaced with 1 ml fresh serum-free medium containing 0.3% n-butanol (Morris et al, 1997) and either 1 μ M 4α-phorbol, 1 μ M 12-O-tetradecanoylphorbol 13-acetate (TPA) or 1 μ M TPA+1 μ M Ro31-8220. Cells were incubated for 30 min at 37 °C, rinsed and lipids recovered with 1 ml methanol, followed by 1 ml each of 1 : 2 chloroform : methanol and 1 : 1 chloroform : methanol. Chloroform was added to combined extracts and a two-phase system was generated with 0.1 M KCl. The chloroform phase was evaporated, redissolved in 200 μl C/M (2 : 1v/v) and triplicate 10 μl aliquots were taken into scintillation vials for total counts. Triplicate 25 μl aliquots were applied to oxalate-impregnated silicic acid TLC plates and overlayed with authentic PtdBut, PtdOH and PtdCho standards (Lipid Products, Nutfield, UK). Plates were developed in chloroform : methanol : acetic acid (9 : 1 : 1, v/v) and lipids detected with iodine. After removal of iodine, PtdBut, PtdOH and PtdCho adsorbent areas were scraped into vials for scintillation counting. Means of triplicate values were calculated, and dpm in PtdBut expressed as a percentage of PtdCho d.p.m.
Myristoylated alanine-rich C kinase substrate phosphorylation
Cells seeded into 24-well plates as above were cultured overnight in normal growth medium before transfer into RPMI1640, F12 or KSFM containing 1% FBS for 24 h. At 9 h before experiments, some wells were treated with 250 nM TPA to downregulate PKCα. Subsequently, cells were rinsed with serum-free medium for 60 min and then stimulated for 0, 15 and 30 min with 1 μ M TPA. Cells were rinsed and immediately solubilised in warm 2 × Laemmli sample buffer (Sigma-Aldrich) containing protease and phosphatase inhibitors (Roche) for western blotting as above.
Choline uptake
A total of 1 × 105 cells were seeded with six replicates into BD amine (BD Biosciences, Oxford, UK) 24-well plates in their normal growth medium and cultured overnight. Cells were rinsed with serum-free medium and incubated for 60 min in the same medium. Cells were then rinsed once with TS and twice with Cho-free uptake buffer (Muller et al, 2009) at 37°C. This was finally replaced with 250 μl uptake buffer containing 10 μ M Cho plus 1 μCi [3H]-Cho per well. Cells were incubated for 60 min at 37°C and uptake stopped by addition of 750 μl ice-cold phosphate-buffered saline (PBS). Cells were rinsed three times with ice-cold PBS, solubilised in 250 μl 1% SDS/0.2 M NaOH (Wang et al, 2007) and triplicate 25 μl aliquots taken for scintillation counting. Use of BD amine plates ensured that LNCaP cells remained attached during rinsing.
Statistical treatment
Statistical significance was determined by the Student's two-tailed t-test or by a one-way Anova and the Tukey HSD test.
Results
Protein kinase C alpha expression by prostate epithelial cell lines
Reverse transcriptase–PCR with the same amount of total RNA from each cell line taken for reverse transcription confirmed that all lines express PKCα mRNA. The amplified band for PKCα was most prominent in PC3 cells and weakest in the P4E6 line (results not shown). Western blotting of equal cell numbers revealed that all lines express PKCα protein (Figure 1A). Expression of GAPDH protein by the five cell lines was almost uniform (Figure 1B and E). Actin expression was low in LNCaP cells compared with other lines (Figure 1C and E), making it an unsatisfactory loading control for comparison between the different cell lines used, although satisfactory for the same cell line. Bands in the PKCα blot were thus normalised to GAPDH (Figure 1D) to reveal differences in PKCα protein expression.

(A) Western blot detection of PKCα protein expression in PNT2C2, PNT1A, P4E6, LNCaP and PC3 prostate epithelial cell lines as described in Materials and Methods. The PKCα blot shown was stripped and reprobed for (B) GAPDH or (C) actin. Bands were quantified using Image J. (D) PKCα protein content of prostate epithelial cell lines normalised to GAPDH protein. (E) Comparison of GAPDH and actin protein content of prostate cell lines showing low actin content of LNCaP cells. Blots shown are typical of several repeats. Positions of 100, 75, 50 and 37 kDa markers are shown.
Prostate cell lines express PKCδ, PKCɛ, PKCζ and MARCKS
Western blotting revealed that all five prostate cell lines express PKCδ, PKCɛ, PKCζ and MARCKS protein (results not shown).
Myristoylated alanine-rich C kinase substrate phosphorylation
TPA (1 μ M) stimulation of cells for 15 and 30 min increased MARCKS phosphorylation in PNT1A, P4E6 and LNCaP lines (Figure 2). Constitutively phosphorylated MARCKS was detected in PNT2C2 and PC3 cells, and this phosphorylation was not obviously increased by addition of TPA. Cells that had been exposed to 250 nM TPA for 9 h to downregulate PKCα showed almost complete suppression of MARCKS phosphorylation on restimulation with TPA, except for the PC3 line where MARCKS phosphorylation was at a reduced level (Figure 2).

Phorbol ester stimulation of prostate cell lines results in MARCKS phosphorylation. Prostate cells were cultured, stimulated with TPA for 15 or 30 min (15, 30) and proteins resolved for western blotting to detect phospho-MARCKS as described above. Some cells were pretreated for 9 h with 250 nM TPA to downregulate PKCα protein before restimulation with 1 μ M TPA for 30 min (30D). In all lines, downregulation of PKCα protein results in significantly reduced MARCKS phosphorylation. Blots were stripped and reprobed for actin as a loading control. A repeat experiment gave similar results. Positions of 80, 75, 50 and 37 kDa markers are indicated.
Prostate cell lines express mRNA for both PLD1 and PLD2
Reverse transcriptase–PCR with equal quantities of total RNA from each cell line taken for reverse transcription revealed that all five prostate epithelial cell lines express mRNA for PLD1 and PLD2 (results not shown). PLD1 mRNA expression was most prominent in PC3 cells. Phospholipase D2 mRNA expression was prominent in P4E6 and PC3 lines.
Phospholipase D activity
All cell lines showed basal (unstimulated) PtdBut formation (Figure 3) in the transphosphatidylation reaction indicating PLD activity. PtdBut formation was increased 2- to 2.5-fold when PNT2C2, PNT1A, P4E6 and LNCaP cell lines were treated with TPA to activate PKC, and by about three-fold in PC3 cells. In all cell lines, TPA-stimulated PtdBut formation was reduced to basal level by inclusion of the PKC inhibitor Ro31-8220 (Figure 3).

PNT2C2, PNT1A, P4E6, LNCaP and PC3 prostate cell lines show basal and TPA-stimulated PLD activity in the transphosphatidylation reaction. Cells were stimulated for 30 min with 4α-phorbol to measure basal (B) PLD activity, 1 μ M TPA (T) or 1 μ M TPA+1 μ M Ro31-8220 (TR). Lipids were extracted and resolved as described in Materials and Methods. Formation of PtdBut is expressed as % PtdBut d.p.m./PtdCho d.p.m. Results are ±s.e.m., n=3. ***P<0.0001 compared with basal and TPA/Ro31-8220-treated cells.
[3H]-label is not released into the medium as vesicles or exosomes
Neither low- nor high-speed centrifugation (see Materials and Methods section) reduced levels of radioactivity in 3 h media from PNT2C2, PNT1A, P4E6 and PC3 lines (Figure 4). Radioactivity in medium from LNCaP cells decreased on low-speed centrifugation because of sedimentation of cells that had detached during the incubation. No further decrease in radioactivity occurred when LNCaP medium was centrifuged at 100 000 g (Figure 4).

Prostate cell lines do not release [3H]-label into the medium as vesicles. Levels of radioactivity released into the medium from PNT2C2, PNT1A, P4E6 and PC3 cells did not decrease on low- or high-speed centrifugation as described. Results shown are for medium from cells stimulated with TPA for 3 h before centrifugation (1) and after low-speed (2) and high-speed (3) and are ±s.d., n=3.
Only PNT2C2 and PNT1A line cells show a consistent phorbol ester-stimulated release of [3H]-label into the medium
Prostate cell lines labelled with [3H]-choline were stimulated with 4α-phorbol (basal) or TPA, and the medium collected to measure released radioactivity. Results of [3H]-label released in 6 h incubations are shown in Figure 5A–E and are typical of several repeats. In fact, Cho metabolite release by all cell lines increased linearly well beyond 6 h, but normal release experiments usually only extended to 3 h (Figure 6A and B) to avoid PKC downregulation caused by longer-term phorbol ester treatment. LNCaP cells were particularly difficult to work with in these experiments as they adhered poorly to plastic (even after polylysine treatment), leading to variable levels of Cho release (Figures 5D and 6A). A reproducible TPA-stimulated release of [3H]-label to the medium was only observed with PNT2C2 and PNT1A cell lines. This release effect was always greater in PNT2C2 cells than in the PNT1A line. Routinely, TPA had little or no stimulatory effect on the release of [3H]-label from P4E6 and LNCaP lines in incubations up to 3 h; however, by 6 h a significant stimulation of label release by TPA was observed, especially from LNCaP cells (Figure 5C and D). In the results shown in Figure 5E, for PC3 cells, TPA induced a small but significant release of [3H]-label to the medium at 3 and 6 h over basal. This was not a consistent effect, however, as shown by the results for TPA stimulation of PC3 cells in inhibitor experiments (Figure 6A and B). Dimethylsulphoxide (DMSO), solvent for phorbol esters and inhibitors, had no effect on [3H]-label release into the medium (results not shown). Levels of [3H]-label released into the medium by P4E6 and PC3 lines were consistently lower than levels released by PNT2C2, PNT1A and LNCaP lines, although equal cell numbers were seeded initially; cells were labelled identically and 60 min uptake rates (Figure 8) were the same as those in PNT1A cells. The three media used contained unlabelled choline chloride at 21 μ M (RPMI, KSFM) and 100 μ M (F12).

(A–E) Prostate epithelial cell lines show different patterns of [3H]-label release into the medium on stimulation with 1 μ M 4α-phorbol [  ] or 1 μ M TPA [
] or 1 μ M TPA [  ] for up to 6 h as described in Materials and Methods. Results are means±s.d. (n=6). Repeat experiments (n=3) showed similar trends. For PNT2C2 and PNT1A lines **P<0.001, ***P<0.0001 against basal values. For P4E6, LNCaP and PC3 lines *P<0.05, **P<0.01, ***P<0.001 against basal values.
] for up to 6 h as described in Materials and Methods. Results are means±s.d. (n=6). Repeat experiments (n=3) showed similar trends. For PNT2C2 and PNT1A lines **P<0.001, ***P<0.0001 against basal values. For P4E6, LNCaP and PC3 lines *P<0.05, **P<0.01, ***P<0.001 against basal values.

(A) Protein kinase C inhibitors reduce phorbol ester-stimulated release of [3H] label into the medium from PNT2C2 and PNT1A cells. PNT2C2, PNT1A, LNCaP and PC3 cell lines labelled with [3H]-choline were stimulated as described in Materials and Methods section with 1 μ M 4α-phorbol (basal, B), 1 μ M TPA (TP), 1 μ M TPA+1 μ M Ro31-8220 (Ro), 1 μ M TPA+1 μ M GF-109203X (GF) or 1 μ M TPA+1 μ M Go6976 (Go) and release of [3H]-label into the medium was monitored at 3 h. In both PNT2C2 and PNT1A cells, PKC inhibitors reduced release to basal values. Results are ±s.d. (n=6). For PNT2C2 and PNT1A cells, *P<0.01, **P<0.001, ***P<0.0001 against the basal value. For LNCaP cells **P< 0.01 against the basal value. (B) D609 and hemicholinium-3 effects on [3H]-label release by prostate cell lines. PNT2C2, PNT1A, P4E6, LNCaP and PC3 cells labelled with [3H]-choline were stimulated as described in Materials and Methods with 1 μ M 4α-phorbol (basal, B), 1 μ M TPA (T), 1 μ M TPA+200 μ M HC-3 (H) or 1 μ M TPA+100 μ M D609 (D) and release of [3H]-choline metabolites to the medium was monitored at 3 h. Results are ±s.d. (n=6). For PNT2C2 cells ***P<0.0001 against the basal value. For PNT1A cells **P<0.001 against the TPA value, ***P<0.0001 against basal value. For LNCaP *P<0.05 against the basal value, **P<0.01 against the TPA value.

Comparison of Cho uptake by non-tumourigenic and tumourigenic prostate cell lines in monolayer culture. Results are shown as d.p.m. per 25 μl from 1 × 105 cells solubilised in 250 μl over 60 min at 37°C as described in Materials and Methods section and are ±s.d. (n=6).
Inhibition of phorbol ester-stimulated [3H]-radioactivity release
Ro31-8220 and GF109203X reduced TPA-stimulated release of [3H]-choline metabolites from PNT2C2 and PNT1A cells to basal values as shown in results from a 3-h incubation (Figure 6A). With LNCaP cells, GF109203X reduced [3H]-label release to the medium to below basal values. Go6976, a PKC inhibitor supposedly selective for α and β1 isoforms, had only a small inhibitory effect on [3H]-label release from PNT2C2 cells and was without significant effect on label release from PNT1A cells.
Effects of hemicholinium-3 and D609
When monitored in a 3-h incubation (Figure 6B), D609 at 200 μ M almost completely inhibited the TPA-stimulated release of [3H]-label from PNT2C2 and PNT1A cells. The choline transporter inhibitor hemicholinium 3 (HC-3) at 100 μ M was without effect on [3H]-label released from PNT2C2 cells, and induced a partial but significant inhibition of label release from PNT1A cells. In the experiment shown (Figure 6B), activation of PKC with TPA stimulated a slight increase in label release from LNCaP cells, but this was not a consistent effect. Hemicholinium 3 reduced this stimulated release to basal levels. In this experiment, effects of TPA and inhibitors on PC3 cells were not significantly different from basal values.
Nature of the choline metabolites released into the medium
Medium from basal and phorbol ester-stimulated cells at 3 h time points was resolved into GPCho(GPC), PCho and Cho fractions on Dowex-50WH+ ion exchange resin columns (Figure 7). Results for each Cho metabolite are expressed as a % of the total Cho metabolites released (i.e., GPCho+PCho+Cho). Phosphocholine was the major metabolite released into the medium by basal and phorbol ester-stimulated PNT2C2 cells, whereas Cho was the major metabolite detected in media from basal and stimulated P4E6, LNCaP and PC3 lines. PNT1A cells were intermediate in that PCho and Cho each accounted for about equal proportions of the [3H]-label released. In the results shown for PC3 cells, Cho accounted for a higher proportion of the metabolites released on TPA stimulation (68%) compared with unstimulated cells (45%). With all other lines, the proportions of GPCho, PCho and Cho were the same in unstimulated and TPA-stimulated cells. Thus, in PNT2C2 cells, TPA treatment increased Cho metabolite release, but the proportions of GPCho : PCho :Cho were the same as from unstimulated cells.

Tumourigenic prostate cell lines release mainly Cho into the medium as the principal metabolite, whereas PCho is released by non-tumourigenic PNT2C2 cells. GPC (GPCho), PCho and Cho in medium from 3 h time points from basal ( ) and TPA-treated (
) and TPA-treated ( ) cells were fractionated as described in Materials and Methods section. Results for individual metabolites are expressed as percentage of the total GPCho+PCho+Cho d.p.m. measured±s.e.m. (n=4).
) cells were fractionated as described in Materials and Methods section. Results for individual metabolites are expressed as percentage of the total GPCho+PCho+Cho d.p.m. measured±s.e.m. (n=4).
[3H]-Choline uptake by prostate cell lines
PNT2C2 cells at 1 × 105 cells per well showed a greater uptake of [3H]-Cho label over 60 min at 37°C (Figure 8) compared with the other four cell lines where levels taken up were more comparable under identical conditions.
[3H]-Choline distribution in Cho metabolites and phospholipids after labelling
Over the 30-h labelling period, PNT1A, P4E6, LNCaP and PC3 cells contained more label into choline phospholipids than into Cho metabolites (Figure 9A). This was the opposite in PNT2C2 cells where the label detected in Cho metabolites was higher than in Cho phospholipids. These results also confirm the uptake data (Figure 8), indicating that PNT2C2 cells incorporate more label than the other cell lines under similar conditions. After labelling, most radioactivity was detected in PCho in all cell lines, especially in PNT2C2, LNCaP and PC3 lines; the least label was detected in Cho. Surprisingly, quite a high proportion of label was in GPCho, especially in P4E6 cells.

(A) Distribution of [3H]-Cho in lipid  and aqueous
and aqueous  phases of prostate cell lines immediately after labelling cells as described in Materials and Materials and Materials and Methods. Results are d.p.m.±s.d. (n=3). (B) Distribution of [3H]-Cho label in GPC(GPCho), PCho and Cho in prostate epithelial cell lines immediately after labelling as described in Materials and Methods. Results for individual metabolites are expressed as percentage of the total GPC+PCho+Cho dpm±s.d. (n=3).
phases of prostate cell lines immediately after labelling cells as described in Materials and Materials and Materials and Methods. Results are d.p.m.±s.d. (n=3). (B) Distribution of [3H]-Cho label in GPC(GPCho), PCho and Cho in prostate epithelial cell lines immediately after labelling as described in Materials and Methods. Results for individual metabolites are expressed as percentage of the total GPC+PCho+Cho dpm±s.d. (n=3).
Chronic phorbol ester treatment of PNT2C2 and PNT1A cells downregulates PKCα protein and reduces choline headgroup release
Chronic exposure of PNT2C2 and PNT1A cells to 250 nM TPA for 9 h markedly reduced the PKCα protein content of cells (Figure 10A). Reprobing for actin indicated that approximately equal levels of total cell protein had been resolved, in agreement with previous results. TPA activation of PKC stimulated Cho metabolite release into the medium from both PNT2C2 and PNT1A lines. This effect was reduced to basal levels in PKCα-depleted PNT1A cells and by about 60% in PKCα-depleted PNT2C2 cells (Figure 10B). In contrast, TPA treatment of PC3 cells failed to stimulate significant Cho metabolite release over basal levels (Figure 10B), showing the variable effect of TPA on Cho metabolite release from PC3 cells.

(A) Chronic treatment of PNT1A and PNT2C2 line cells with TPA downregulates PKCα. A total of 7.5 × 104 cells were seeded into wells of 24-well plates, cultured for 24 h and treated with 250 nM TPA for the times shown, up to 9 h. Protein kinase C alpha was then resolved as described in Materials and Methods section. Blots were stripped and reprobed for actin as a loading control. Positions of 100, 75, 50 and 37 kDa markers are shown. (B) Reducing PKCα protein in PNT2C2 and PNT1A cells decreases the release of Cho metabolites into the medium. Cells were cultured and labelled with [3H]-Cho as in Methods. Cells were treated for 3 h with 4α-phorbol (basal, B), with 1 μ M TPA (TPA) or with 1 μ M TPA following downregulation of PKCα by treatment with 250 nM TPA for 9 h (DR). Results are mean c.p.m. per 10 μl medium±s.d., n=4. As usual, PC3 line cells showed no significant TPA-stimulated release of choline headgroups. For PNT2C2 cells, TPA ***P<0.0001 against the basal value, for DR ***P<0.0001 against TPA value. For PNT1A cells **P<0.001 against basal, ***P<0.0001 against TPA value.
Discussion
Protein kinase C alpha expression
We studied five cell lines to span the non-tumourigenic to metastatic extremes of PCa. PNT2C2- and PNT1A-immortalised cell lines were derived from normal prostate epithelia (Cussenot et al, 1991; Berthon et al, 1995). The P4E6-immortalised line was derived from an early prostate tumour (Maitland et al, 2001). The widely studied tumourigenic LNCaP and PC3 cell lines differ in their apoptotic response to PKCα activation, formation of metastases and regulation of the PI3K–PKB pathway (Sharrard and Maitland, 2007). We focused on PKCα because it regulates PLD (Cockcroft, 2001), which is linked to tumourigenesis (Foster, 2009). When normalised to GAPDH, PKCα protein expression varied considerably between the five cell lines, being weakest in P4E6 cells derived from an early prostate tumour (Figure 1D). This was surprising as PKCα expression is reportedly increased in PCa (Cornford et al, 1999; Koren et al, 2004; Lahn et al, 2004) and, in agreement, was 2–4 times higher in tumourigenic LNCaP and PC3 cell lines compared with P4E6 cells. This observation with P4E6 cells derived from an early tumour may indicate that PKCα protein expression is reduced in early PCa and that expression increases in later metastatic disease. However, the immortalised PNT2C2 and PNT1A cell lines from normal prostate epithelia express PKCα protein at the same level as the tumourigenic LNCaP and PC3 cell lines. A study of PKCα expression in primary prostate epithelial cells from normal and tumour tissue is in progress to determine whether immortalisation influences PKCα expression.
Protein kinase C alpha signalling to MARCKS
Protein kinase C alpha regulates cell spreading and motility through the F-actin-binding protein MARCKS (Uberall et al, 1997; Larsson, 2006). Protein kinase C activation stimulates MARCKS phosphorylation in PNT1A, LNCaP and P4E6 cells (Figure 2), indicating that a phorbol ester-PKCα-MARCKS pathway is active even in the P4E6 line with its weaker PKCα protein content. Surprisingly, some MARCKS was constitutively phosphorylated in unstimulated PNT2C2 and PC3 cells. Novel PKCɛ, a MARCKS kinase in fibroblasts (Uberall et al, 1997; Rombouts et al, 2008), might contribute to MARCKS phosphorylation in these prostate cell lines. However, PKCα protein turns over much more rapidly than PKCɛ on chronic exposure of cells to phorbol ester (Olivier and Parker, 1992) and is barely detectable in PNT1A and PNT2C2 cells exposed to TPA for 9 h (Figure 10A). Myristoylated alanine-rich C kinase phosphorylation in prostate cell lines depleted in PKCα protein was reduced, indicating that PKCα is the major link to MARCKS. Phosphorylation of MARCKS causes its release from the plasma membrane exposing PI(4,5)P2 and regulating local F-actin organisation for cell spreading and focal adhesion formation (Larsson, 2006). Thus, constitutively phosphorylated MARCKS in PC3 cells could contribute to the increased motility and invasiveness shown by this line compared with LNCaP cells (Lang et al, 2002). MicroRNA-21 (miR-21), which is overexpressed in PCa (Krichevsky and Gabriely, 2009), targets MARCKS, promoting resistance to apoptosis and increased invasiveness (Li et al, 2009). MicroRNA-21 expression is higher in PC3 cells than in the LNCaP line (Li et al, 2009) contributing to their greater invasiveness.
Phospholipase D activation by PKCα
Phospholipase D activity is elevated in many cancers and transformed cell lines (Foster and Xu, 2003; Foster, 2006); thus, we were surprised that levels of basal and PKC-stimulated PLD activity (Figure 3) were similar between the five cell lines. Protein kinase C alpha is specifically linked to activation of PLD1 (Kim et al, 1999; Cockcroft, 2001) and PLD2 (Chen and Exton, 2004); results show that a PKCα link to PLD is active in cell lines derived from both normal and tumourigenic epithelia, including P4E6 cells derived from an early tumour. Activation of PKCα increases PLD activity more in PC3 cells probably because this cell line expresses PKCα, PLD 1 and PLD2 prominently. Standard PLD assay conditions with 30 mM 1-butanol can interfere with the interaction between PKCα and PLD1, leading to a reduction in measured PLD1 activity (Hu and Exton, 2005) This might account for why basal PLD activity is similar in normal and tumourigenic cell lines (Figure 3). Myristoylated alanine-rich C kinase, which functions as a reversible source of plasma membrane PI(4, 5)P2 (Larsson, 2006), is a key regulator of the PKCα–PLD pathway (Sundaram et al, 2004).
[3H]-label release
The centrifugation results (Figure 4) indicate that [3H]-label is not released into the medium from cell lines in any membrane-bound prostasome or exosome form (Whiteside, 2005; Nilsson et al, 2009). Differences in levels of basal [3H]-label release from the five cell lines (Figure 5A–E) must reflect a variation in the initial [3H]-Cho uptake into cells by Cho transporters (Michel et al, 2006), as well as in Cho metabolism and rates of PtdCho synthesis and turnover. Our uptake results (Figure 8) indicate that PNT2C2 cells, which release the highest levels of Cho metabolites, also show the greatest uptake of Cho over 60 min. The uptake results also confirm that LNCaP cells import Cho more rapidly than do PC3 cells, as noted by Baba et al (2007) and Muller et al (2009). Choline transporter expression has been partially defined for LNCaP and PC3 cells (Hara et al, 2006; Baba et al, 2007; Muller et al, 2009). According to Muller et al (2009), Cho uptake into LNCaP and PC3 cells involves a selective Cho transporter (Michel et al, 2006). Our finding that TPA-stimulated Cho metabolite release from PNT2C2 cells is not HC-3 sensitive suggests that a CTL1 family member (Michel et al, 2006) is not involved in the release mechanism. Choline metabolite release from PNT1A cells is partially HC-3 sensitive, suggesting a contribution by a CTL1-type component (Figure 6B). Bakovic (personal communication) comments ‘CTL1 could efflux free Cho as it regulates an ATP-independent, passive transport depending on Cho concentration gradient and that CTL1, as well as OCTs are probably not involved in PCho and GPCho transport, though the efflux of such metabolites has not been tested’ (Michel et al, 2006). Our results in Figure 9B indicate that most Cho taken up by all the cell lines is converted into PCho by CK as found by Hara et al (2006) for PC3 cells. After labelling, only non-tumourigenic PNT2C2 cells had label preferentially in water-soluble Cho metabolites (mainly PCho, Figure 9B) compared with phospholipids, perhaps indicating slower membrane turnover compared with tumourigenic cell lines. Phosphocholine levels in LNCaP and PC3 cells have been measured at about 0.8 and 1.2 mM, respectively, compared with 0.1 mM for senescent normal prostate epithelial cells (Ackerstaff et al, 2001; Glunde et al, 2006).
Stimulated Cho metabolite release
Unstimulated prostate cell lines release GPCho, PCho and Cho into the medium in varying proportions. However, the main Cho metabolite released changes from PCho to Cho with the transition to tumourigenesis (Figure 7). Significantly, tumourigenic cell lines fail to show a consistent PKC-stimulated release of Cho metabolites (Figures 5A–E, 6A and B) compared with the marked stimulation shown by PNT2C2 cells derived from normal epithelia. PNT1A cells, also from normal prostate epithelia, occupy an intermediate position in that PCho and Cho are released in about equal proportions, whereas PKC activation stimulates Cho metabolite release more weakly than is detected with PNT2C2 cells. Other cell types, for example, fibroblasts, are known to release PCho into the medium on ATP stimulation (Chung et al, 1997). The TPA-stimulated release of Cho metabolites from PNT2C2 and PNT1A cells is reduced to basal levels by the widely used PKC inhibitors Ro31-8220 and GF109203X at 1 μ M concentration (Figure 6A). Neither inhibitor is specific for PKC; however, the MAPKAP kinase-1β and p70S6 kinase also inhibited by GF109203X and Ro31-8220 (Alessi, 1997) are not directly involved in PtdCho turnover and Cho metabolism. These inhibitor results indicate a PKC involvement in the TPA-stimulated release pathway and are also in agreement with the observations in Figure 3 that Ro31-8220 inhibits TPA-stimulated PLD activity in all the cell lines. PNT lines depleted in PKCα protein (Figure 10A) show reduced Cho metabolite release on restimulation (Figure 10B), further supporting the PKCα link to PLD. Therefore, in all cell lines, a TPA–PKCα–PLD pathway stimulates turnover of PtdCho to generate PtdOH and Cho. However, PCho is the main metabolite released by PNT2C2 cells, and thus Cho formed by PLD action must be converted into PCho before release. PNT1A cells release both PCho and Cho, indicating that the two non-tumourigenic cell lines differ in Cho metabolism, Cho transporter expression and PtdCho turnover. Phosphocholine may be released as a secondary signal (Cuadrado et al, 1993; Chung et al, 1997; Kiss and Mukherjee, 1997).
Phosphocholine release from PNT2C2 cells
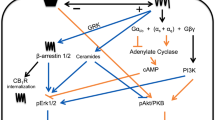
In HeLa cells, basal turnover of PtdCho occurs through phosphatidylcholine-specific phospholipase C (PC-PLC), DAG kinase and lipid phosphate phosphatases, and does not involve PLD (Hii et al, 1991). Therefore, PCho released by PNT cell lines could be formed by PC-PLC activity, as is observed in normal and ovarian epithelial cancer cells (Spadaro et al, 2008) or in phorbol ester- or PDGF-stimulated fibroblasts (Podo et al, 1996; van Dijk et al, 1997). Involvement of a PC-PLC would explain the inhibition of Cho metabolite release from PNT lines by 100 μ M D609 (Figure 6B), initially reported as a PC-PLC inhibitor (Muller-Decker, 1989). However, D609 can inhibit PLD and a group IV PLA2 (Kiss and Tomoro, 1995; van Dijk et al, 1997; Kang et al, 2008), as well as sphingomyelin synthase (Luberto and Hannun, 1998). In epithelial ovarian cancer cells and NK cells, D609 has no effect on PLD or sphingomyelin synthase (Cecchetti et al, 2007; Spadaro et al, 2008). Intriguingly, PCho could also be released by the two PNT cell lines following translocation of a PC-PLC enzyme to the external surface of the plasma membrane (Ramoni et al, 2001, 2004). At this site, hydrolysis of PtdCho in the outer lipid leaflet would result in a direct release of PCho into the medium (Figure 11). As an example, exogenous B. cereus PC-PLC hydrolyses PtdCho in the outer lipid leaflet of fibroblasts (van Dijk et al, 1997). Such a translocation of PC-PLC may be regulated by PKC (Figure 11) as TPA can stimulate PC-PLC movement to the plasma membrane in fibroblasts (Ramoni et al, 2004). Hemicholinium 3 at 200 μ M, an inhibitor of CK (Jimenez et al, 1995) and of high- and medium-affinity Cho transporters (Michel et al, 2006), has no effect on TPA-stimulated Cho metabolite release from PNT2C2 cells. This supports the possibility that PC-PLC translocated to the cell surface releases PCho directly into the medium (Figure 11). Hemicholinium 3 partially inhibits PCho/Cho release from TPA-stimulated PNT1A cells, suggesting that Cho release via a CTL1 family transporter is blocked, whereas PCho release is unaffected.

Summary of Cho metabolite formation and release from non-tumourigenic PNT2C2 and tumourigenic P4E6, LNCaP and PC3 cell lines. PNT2C2 cells: basal PC-PLC generates PCho, some of which is reused for PtdCho synthesis for membrane biogenesis and PtdCho turnover. DAG from PC-PLC activity might sustain long-term PKC activity and generates PtdOH via DAG kinase to regulate MAPK, mTOR, PI3K/Akt signalling for cell proliferation. Phosphocholine is released to function as an external secondary signal promoting growth factor signalling: PCho transporters are not identified. Phosphocholine may be released into the medium directly by PC-PLC translocated to the external cell surface. Protein kinase C isoforms may regulate PC-PLC translocation, which would increase on addition of phorbol ester. Protein kinase C activation by phorbol ester (TPA) stimulates PLD, increasing PCho formation, and may upregulate PC-PLC activity. In tumourigenic cell lines, PLD and CKα activities are upregulated to maintain high PCho levels for PtdCho formation for increased membrane biogenesis and PtdCho turnover for PtdOH formation. PtdOH promotes cell proliferation and malignant cell survival through MAPK, PI3K/Akt and mTOR pathways. PC-PLC activity is upregulated, increasing PCho formation. Increased Cho uptake as reported in some malignant cells (but not observed in these cell lines) would further increase PCho levels. Mechanisms of PCho release are downregulated to maintain high intracellular PCho levels in cancer cells. Phorbol ester (TPA) activation of PKC does not stimulate Cho formation by PLD and its release into the medium as it is rapidly phosphorylated to PCho by CKα.
Choline metabolite release from tumourigenic cell lines
Our results show that, although PKC stimulates PLD activity in P4E6, LNCaP and PC3 cell lines (Figure 3), there is no consistent increase in Cho metabolite release into the medium (Figures 5C–E and 9). If the new Cho formed is not released, it must be rapidly converted into PCho by CK as detected in PC3 and LNCaP cells (Ackerstaff et al, 2001; Glunde et al, 2006; Hara et al, 2006) and in other malignant cells and cancers (Glunde et al, 2006). Choline kinase activity is upregulated in tumour-derived cell lines (Ramirez de Molina et al, 2002), probably accounting for the rapid conversion of Cho to PCho in tumourigenic prostate lines and why Cho is not released on PLD activation. Phospholipase D expression and activity is also increased in several cancers and malignant cell lines (Foster and Xu, 2003), which could further increase Cho formation. Such an increase was not detected in these tumourigenic prostate cell lines perhaps because of the butanol inhibition effects discussed above. Choline transport into tumourigenic cells may be increased (Figure 11), as has been observed in several cancer cell lines (Katz-Brull et al, 2002; Yoshimoto et al, 2004; Iorio et al, 2005), although we did not observe this with the cell lines studied here (Figure 8). Protein kinase C may also influence CK activity directly (Macara, 1989; Choi et al, 2005), further explaining why Cho is not released into the medium on TPA activation of PKC. PC-PLC activity may also be upregulated in tumourigenic prostate lines (Figure 11), as detected in ovarian and breast cancer cells (Glunde et al, 2004; Iorio et al, 2005, 2010), further increasing PCho formation. Elevated levels of PCho in neoplastic cells promote growth factor-induced mitogenic signalling to Raf-1 and MAP kinases (Cuadrado et al, 1993; Jimenez et al, 1995; Yalcin et al, 2010) and will also maintain flow through the Kennedy pathway, increasing PtdCho synthesis for membrane biogenesis (Figure 11) and cell proliferation, as well as to compensate for endosome formation for growth factor signalling (Vieira et al, 1996; Li et al, 1997). The α isoform of CK also affects cell-cycle regulation promoting both cell survival and proliferation (Ramirez de Molina et al, 2008; Chua et al, 2009). PtdOH generated in transformed cells also regulates cell proliferation and survival pathways via mTOR and Raf (Foster and Xu, 2003; Foster, 2007a, 2007b; Ramirez de Molina et al, 2008; Foster, 2009). An increase in cytidylyltransferase (CT) activity in tumourigenic prostate cells, as has been detected in some breast cancer lines (Eliyahu et al, 2007), would further increase PCho utilisation for PtdCho synthesis. A coupling between CT and PLD turnover of PtdCho, which might further stimulate PtdCho synthesis, has been reviewed (Cornell and Northwood, 2000). As PCho levels are elevated in neoplastic cells and transformed cell lines, it is relevant to ask why this metabolite is not released into the medium from tumourigenic prostate epithelial cells as occurs with normal PNT cell lines. Our findings with these cell lines infer that tumourigenesis in prostate epithelia results in the downregulation of normal mechanisms of PCho release into the medium so that high intracellular levels of PCho are maintained to enhance mitogen pathway signalling and PtdCho synthesis for increased cell proliferation and survival, as summarised in Figure 11.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ackerstaff E, Pflug BR, Nelson JB, Bhujwalla ZM (2001) Detection of increased choline compounds with proton nuclear magnetic resonance spectroscopy subsequent to malignant transformation of human prostatic epithelial cells. Cancer Res 61: 3599–3603
Alessi D (1997) The protein kinase C inhibitors Ro318220 and GF 109203X are equally potent inhibitors of MAPKAP kinase-1β (Rsk-2) and p70S6 kinase. FEBS Lett 402: 121–123
Ali AS, Ali S, El-Rayes BF, Philip PA, Sarkar FH (2009) Exploitation of protein kinase C: useful target for cancer therapy. Cancer Treat Rev 35: 1–8
Baba S, Engles J, Wahl R (2007) Choline uptake and RNA expression profile in cancerous and normal prostate cell lines: No significant role of choline transporter in PET tracer uptake. J Nucl Med 48 (Supp 2): 335P
Beloueche-Babari M, Peak JC, Jackson LE, Tiet M-Y, Leach MO, Eccles SA (2009) Changes in choline metabolism as potential biomarkers of phospholipase Cγ1 inhibition in human prostate cancer cells. Mol Cancer Ther 8: 1305–1311
Berthon P, Cussenot O, Hopwood L, Le Duc A, Maitland NJ (1995) Functional expression of SV40 in normal human prostatic epithelial and fibroblastic cells: differentiation pattern of non-tumorigenic cell lines. Int J Oncol 6: 333–343
Cecchetti S, Spadaro F, Lugini L, Podo F, Ramoni C (2007) Functional role of phosphatidylcholine-specific phospholipase C in regulating CD16 membrane expression in natural killer cells. Eur J Immunol 37: 2912–2922
Chen J-S, Exton JH (2004) Regulation of phospholipase D2 activity by protein kinase Cα. J Biol Chem 279: 22076–22083
Choi M-G, Kurnov V, Kersting MC, Sreenivas A, Carman GM (2005) Phosphorylation of the yeast choline kinase by protein kinase C. J Biol Chem 280: 26105–26112
Chua BT, Gallego-Ortega D, Ramirez de Molina AR, Ullrich A, Lacal JC, Downward J (2009) Regulation of Akt (ser-473) phosphorylation by choline kinase in breast carcinoma cells. Mol Cancer 8: 131
Chung T, Crilly KS, Anderson WH, Mukherjee JJ, Kiss Z (1997) ATP-dependent choline phosphate-induced mitogenesis in fibroblasts involves activation of pp70 S6 kinase and phosphatidylinositol 3-kinase through an extracellular site. J Biol Chem 272: 3064–3072
Cockcroft S (2001) Signalling roles of mammalian phospholipase D1 and D2. Cell Mol Life Sci 58: 1674–1687
Cook SJ, Wakelam MJO (1989) Analysis of the water-soluble products of phosphatidylcholine breakdown by ion-exchange chromatography. Biochem J 263: 581–587
Cornell RB, Northwood IC (2000) Regulation of CTP:phosphocholine cytidylyltransferase by amphitropism and relocalisation. Tr Biochem Sci 25: 441–447
Cornford P, Evans J, Dodson A, Parsons K, Woolfenden A, Neoptolemos J, Foster CS (1999) Protein kinase C isoenzyme patterns characteristically modulated in early prostate cancer. Am J Pathol 154: 137–144
Cuadrado A, Carnero A, Dolfi F, Jimenez B, Lacal JC (1993) Phosphorylcholine: a novel second messenger essential for mitogenic activity of growth factors. Oncogene 8: 2959–2968
Cussenot O, Berthon P, Faille A, Berger R, Mowszowicz I, Teillac P, LeDuc A, Calvo F (1991) Immortalisation of human adult normal prostate epithelial cells by liposomes containing SV40. J Urol 143: 881–886
Dawson J, Hotchin N, Lax S, Rumsby M (2003) Lysophosphatidic acid induces process retraction in CG-4 line oligodendrocytes and oligodendrocyte precursor cells but not in differentiated oligodendrocytes. J Neurochem 87: 947–957
de Souza PL, Russell PJ, Kearsley J (2009) Role of the Akt pathway in prostate cancer. Curr cancer Drug targets 9: 163–175
Eliyahu G, Kreizman T, Degani H (2007) Phosphocholine as a biomarker of breast cancer: molecular and biochemical studies. Int J Cancer 120: 1721–1730
Foster DA (2006) Phospholipase D survival signals as a therapeutic target in cancer. Curr Signal Transduction Ther 1: 295–303
Foster DA (2007a) Regulation of mTOR by phosphatidic acid? Cancer Res 67: 1–4
Foster DA (2007b) Phosphatidic acid signalling to mTOR: signals for the survival of cancer cells. Biochim Biophys Acta 1791: 949–955
Foster DA (2009) Phosphatidic acid signalling to mTOR: Signals for the survival of human cancer cells. Biochim Biophys Acta 1791: 949–955
Foster DA, Xu L (2003) Phospholipase D in cell proliferation and cancer. Mol Cancer Res 1: 789–800
Gallego-Ortega D, Ramirez de Molina AR, Ramos MA, Valdes-Mora F, Barderas MG, Sarmentero-Estrata J, Lacal JC (2009) Differential roles of human choline kinase α and β enzymes in lipid metabolism: implications in cancer onset and treatment. PLoS ONE 4: e7819
Gavrielides MV, Frijhoff AF, Conti CJ, Kazanietz MG (2004) Protein kinase C and prostate carcinogenesis:Targeting the cell cycle and apoptotic mechanisms. Curr Drug Targets 5: 431–443
Gibbs TC, Meier KE (2000) Expression and regulation of phospholipase D isoforms in mammalian cell lines. J Cell Physiol 182: 77–87
Glunde K, Ackerstaff E, Mori N, Jacobs MA, Bhujwalla ZM (2006) Choline phospholipid metabolism in cancer: consequences for molecular pharmaceutical interventions. Mol Pharm 3: 496–506
Glunde K, Jie C, Bhujwalla ZM (2004) Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res 64: 4270–4276
Gonzalez-Guerrico AM, Meshki J, Xiao L, Benavides F, Conti CJ, Kazanietz MG (2005) Molecular mechanisms of protein kinase C-induced apoptosis in prostate cancer cells. J Biochem Mol Biol 38: 639–645
Griner EM, Kazanietz MG (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 7: 281–294
Guertin DA, Sabatini DM (2007) Defining the role of mTOR in cancer. Cancer Cell 12: 9–21
Gutcher I, Webb PR, Anderson NG (2003) The isoform-specific regulation of apoptosis by protein kinase C. Cell Mol Life Sci 60: 1061–1070
Hara T, Bansal A, DeGrado TR (2006) Choline transporter as novel target for molecular imaging of cancer. Mol Imaging 5: 498–509
Hii CST, Edwards YS, Murray AW (1991) Phorbol ester-stimulated hydrolysis of phosphatidylcholine and phosphatidylethanolamine by phospholipase D in HeLa cells: evidence that the basasl turnover of phosphoglycerides does not involve phospholipase D. J Biol Chem 266: 20238–20243
Hu T, Exton JH (2005) 1-butanol interferes with phospholipase D1 and protein kinase Cα association and inhibits phospholipase D1 basal activity. Biochem Biophys Res Commun 327: 1047–1051
Iorio E, Mezzanzanica D, Alberti P, Spadaro F, Ramoni C, D’Ascenzo S, Millimaggi D, Pavan A, Dolo V, Canevari S, Podo F (2005) Alterations of choline phospholipid metabolism in ovarian tumour progression. Cancer Res 65: 9369–9376
Iorio E, Ricci A, Bagnoli M, Pisanu ME, Castellano G, Vito MD, venturini E, Glunde K, Bhujwalla ZM, Mezzanzanica D, Canevari S, Podo F (2010) Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res 70: 2126–2135
Jimenez B, del Peso L, Montaner S, Esteve P, Lacal JC (1995) Generation of phosphorylcholine as an essential event in the activation of Raf-1 and MAP-kinases in growth factor-induced mitogenic stimulation. J Cell Biochem 57: 141–149
Joseph T, Bryant A, Wooden R, Kerkhoff E, Rapp UR, Foster DA (2002) Phospholipase D overcomes cell cycle arrest induced by high intensity Raf signalling. Oncogene 21: 3651–3658
Kang MS, Jung SY, Jung KM, Kim SK, Ahn KH, Kim DK (2008) D609, an inhibitor of phosphatidylcholine-specific phospholipase C, inhibits Group IV cytosolic phospholipase A2. Mol Cells 26: 481–485
Katz-Brull R, Seger D, Rivenson-Segal D, Rushkin E, Degani H (2002) Metabolic markers of breast cancer: Enhanced choline metabolism and reduced choline-ether-phospholipid synthesis. Cancer Res 62: 1966–1970
Kim Y, Kim J-E, Lee SD, Lee TG, Kim JH, Park JB, Han JM, jang SK, Suh P-G, Ryu SH (1999) Phospholipase D1 is located and activated by protein kinae C α in the plasma membrane in 3Y1 fibroblast cells. Biochim Biophys Acta 1436: 319–330
Kiss Z (1990) Effects of phorbol ester on phospholipid metabolism. Prog Lipid Res 29: 141–166
Kiss Z, Mukherjee JJ (1997) Phosphocholine and sphingosine-1-phosphate synergistically stimulate DNA synthesis by a MAP kinase-dependent mechanism. FEBS Letts 412: 197–200
Kiss Z, Tomono M, Anderson WB (1994) Phorbol ester selectively stimulates phospholipase D-mediated hydrolysis of phosphatidylethanolamine in multidrug resistant MCF-7 human breast carcinoma cells. Biochem J 302: 649–654
Kiss Z, Tomoro M (1995) Compound D609 inhibits phorbol ester-stimulated phospholipase D activity and phospholipase C-mediated phosphatidylethanolamine hydrolysis. Biochim Biophys Acta 1259: 105–108
Koivunen J, Aaltonen V, Peltonen J (2006) Protein kinase C (PKC) family in cancer progression. Cancer Letts 235: 1–10
Koren R, Ben Meir D, Langzam L, Dekel Y, Konichezky M, Baniel J, Livne PS, Gal R, Sampson SR (2004) Expression of protein kinase C isoenzymes in benign hyperplasia and carcinoma of prostate. Oncol Rep 11: 321–326
Krichevsky AM, Gabriely G (2009) miR-21: a small multi-faceted RNA. J Cell Mol Med 13: 39–53
Krongrad A, Bai G (1994) c-fos promoter insensitivity to phorbol ester and possible role of protein kinase C in androgen-independent cancer cells. Cancer Res 54: 6073–6077
Lahn M, Sundell K, Gleave M, Ladan F, Su C, Li S, Ma D, Paterson BM, Bumol TF (2004) Protein kinase C-α in prostate cancer. BJU Int 93: 1076–1081
Lamm MGL, Long DD, Goodwin SM, Lee C (1997) Transforming growth factor-β1 inhibits membrane association of protein kinase C-α in a human prostate cancer cell line, PC3. Endocrinol 138: 4657–4664
Lang SH, Hyde C, Reid IN, Hitchcock IS, Hart CA, Bryden AAG, Villette J-M, Stower MJ, Maitland NJ (2002) Enhanced expression of vimentin in motile prostate cell lines and in poorly differentiated and metastatic prostate carcinoma. The Prostate 52: 253–263
Larsson C (2006) Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal 18: 276–284
Li G, D’Souza-Schorey C, Barbieri MA, Cooper JA, Stahl PD (1997) Uncoupling of membrane ruffling and pinocytosis during Ras signal transduction. J Biol Chem 272: 10337–10340
Li T, Li D, Sha J, Sun P, Huang Y (2009) MicroRNA-21 directly targets MARCKS and promotes apoptosis resistance and invasion in prostate cancer cells. Biochem Biophys Res Commun 383: 280–285
Luberto C, Hannun YA (1998) Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. J Biol Chem 273: 14550–14559
Macara IG (1989) Elevated phosphocholine concentration in Ras-transformed NIH 3T3 cells arises from increased choline kinae activity, not from phosphatidylcholine breakdown. Mol cell Biol 9: 325–328
Mackay HJ, Twelves CJ (2007) Targeting the protein kinase C family: are we there yet? Nat Rev Cancer 7: 554–562
Maitland NJ, Macintosh CA, Hall J, Sharrard M, Quinn G, Lang S (2001) In vitro models to study cellular differentiation and function in human prostate cancers. Radiation Res 155: 133–142
Michel V, Yuan Z, Ramsubir S, Bakovic M (2006) Choline transport for phospholipid synthesis. Exp Biol Med 231: 490–504
Morreale A, Mallon B, Beale G, Watson J, Rumsby M (1997) Ro31-8220 inhibits protein kinase C to block the phorbol ester-stimulated release of choline and ethanolamine metabolites from C6 glioma cells: p70 S6 kinase and MAPKAP kinase-1β do not function downstream of PKC in activating PLD. FEBS Lett 417: 38–42
Morris AJ, Frohman MA, Engebrecht J (1997) Measurement of phospholipase D activity. Anal Biochem 252: 1–9
Mufson RA, Okin E, Weinstein IB (1981) Phorbol esters stimulate the rapid release of choline from prelabelled cells. Carcinogenesis 2: 1095–1102
Muller SA, Holzapfel K, Seidl C, Treiber U, Krause BJ, Senekowitsch-Schmidtke R (2009) Charactyerisation of choline uptake in prostate cancer cells following bicalutamide and docetaxel treatment. Eur J Nucl Med Mol Imaging 36: 1434–1442
Muller-Decker K (1989) Interruption of TPA-induced signals by an antiviral and antitumoral xanthate compound: inhibition of a phospholipase C-type reaction. Biochem Biophys Res Comm 162: 198–205
Myklebust JH, Smeland EB, Josefsen D, Sioud M (2000) Protein kinase C-α isoform is involved in erythropoietin-induced erythroid differentiation of CD34+ progenitor cells from human bone marrow. Blood 95: 510–518
Nilsson J, Skog J, Nordstrand A, Baranov V, Mincheva-Nilsson L, Breakefield XO, Widmark A (2009) Prostate cancer-derived urine exosomes: a novel approach to biomarkers for prostate cancer. Brit J Cancer 100: 1603–1607
O’Brian CA (1998) Protein kinase C-α: A novel target for the therapy of androgen-independent prostate cancer? Oncol Rep 5: 305–309
Olivier AR, Parker PJ (1992) Identification of multiple PKC isoforms in Swiss 3T3 cells: Differential down-regulation by phorbol ester. J Cell Physiol 152: 240–244
Paone A, Starace D, Galli R, Padula F, De Cesaris P, Filippini A, Ziparo E, Riccioli A (2008) Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKCα dependent mechanism. Carcinogenesis 29: 1334–1342
Podo F, Ferretti A, Knijn A, Zhang P, Ramoni C, Barletta B, Pini C, Baccarini S, Pulciani S (1996) Detection of phosphatidylcholine-specific phospholipase C in NIH 3T3 fibroblasts and their H-ras transformants: NMR and immunochemical studies. Anticancer Res 16: 1399–1412
Powell CT, Brittis NJ, Stec D, Hug H, Heston WDW, Fair WR (1996) Persistant membrane translocation of protein kinase C α during 12-0-tetradecanoylphorbol-13-acetate-induced apoptosis of LNCaP numan prostate cancer cells. Cell Growth Differ 7: 419–428
Powell CT, Fair WR, Heston WDW (1994) Differential expression of protein kinase C isozyme messenger RNAs in Dunning R-3327 rat prostatic tumours. Cell Growth Differ 5: 143–149
Ramirez de Molina AR, Gallego-Ortega D, Sarmentero-Estrada J, Lagares D, del Pulgar TG, bandres E, Garcia-Foncillas J, Lacal JC (2008) Choline kinase as a link connecting phospholipid metabolism and cell cycle regulation:implications for cancer therapy. Int J Biochem Cell Biol 40: 1753–1763
Ramirez de Molina AR, Rodriguez-Gonzalez A, Gutierrez R, Martinez-Pineiro L, Sanchez JJ, Bonilla F, Rosell R, Lacal JC (2002) Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate and colorectal cancers. Biochem Biophys Res Comm 296: 580–583
Ramoni C, Spadaro F, Barletta B, Dupuis ML, Podo F (2004) Phosphatidylcholine-specific phospholipase C in mitogen-stimulated fibroblasts. Exp Cell Res 299: 370–382
Ramoni C, Spadaro F, Menegon M, Podo F (2001) Cellular localisation and functional role of phosphatidylcholine-specific phospholipase C in NK cells. J Immunol 167: 2642–2650
Rombouts K, Lottini B, Caligiuri A, Liotta F, Mello T, Carloni V, Marra F, Pinzani M (2008) MARCKS is a downstream effector in platelet-derived growth factor-induced cell motility in activated human hepatic stellate cells. Exp Cell Res 314: 1444–1454
Sharrard RM, Maitland NJ (2007) Regulation of protein kinase B activity by PTEN and SHIP2 in human prostate-derived lines. Cell Signalling 19: 129–138
Spadaro F, Ramoni C, Mezzanzanica D, Miotti S, Alberti P, Cecchetti S, Iorio E, Dolo V, canevari S, Podo F (2008) Phosphatidylcholine-specific phospholipase C activation in epithelial ovarian cancer cells. Cancer Res 68: 6541–6549
Stewart JR, O’Brian CA (2005) Protein kinase C-α mediates epidermal growth factor transactivation in human prostate cancer cells. Mol Cancer Ther 4: 726–732
Sundaram M, Cook HW, Byers DM (2004) The MARCKS family of phospholipid binding proteins: regulation of phospholipase D and other cellular components. Biochem Cell Biol 82: 191–200
Tomono M, Crilly KS, Kiss Z (1995) Synergistic potentiating effects of choline phosphate and ethanolamine on insulin-induced DNA synthesis in NIH 3T3 fibroblasts. Biochem Biophys Res Commun 213: 980–985
Troyer DA, Gonzalez OF, Padilla RM, Kreisberg JI (1992) Vasopressin and phorbol ester-stimulated phosphatidylcholine metabolism in nesangial cells. Am J Physiol 262: F185–F191
Uberall F, Giselbrecht S, Hellbert K, Fresser F, bauer B, Gschwendt M, Grunicke HH, Baier G (1997) Conventional PKCα, novel PKCɛ and PKCθ, but not atypical PKCλ are MARCKS kinases in intact NIH 3T3 fibroblasts. J Biol Chem 272: 4072–4078
van Blitterswijk WJ, Hilkmann H, de Widt J, van der Bend RL (1991) Phospholipid metabolism in bradykinin-stimulated fibroblasts. J Biol Chem 266: 10344–10350
van Dijk MCM, Muriana FJG, de Widt J, Hilkmann H, van Blitterswijk WJ (1997) Involvement of phosphatidylcholine-specific phospholipase C in platelet-derived growth factor-induced activation of the mitogen-activated protein kinase pathway in Rat-1 fibroblasts. J Biol Chem 272: 11011–11016
Vieira AV, Lamaze C, Schmid SL (1996) Control of EGF receptor signalling by clathrin-mediated endocytosis. Science 274: 2086–2089
Wang T, Li J, Chen F, Zhao Y, He X, Wan D, Gu J (2007) Choline transporters in human lung adenocarcinoma: Expression and functional implications. Acta Biochim Biophys Sinica 39: 668–674
Whiteside TL (2005) Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system? Brit J Cancer 92: 209–211
Yalcin A, Clem B, Makoni S, Clem A, Nelson K, Thornburg J, Siow D, Lane AN, Brock SE, Goswami U, Eaton JW, Telang S, Chesney J (2010) Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene 29: 139–149
Yoshimoto M, Waki A, Obata A, Furukawa T, Yonekura Y, Fujibayashi Y (2004) Radiolabelled choline as a proliferation marker: comparison with radiolabelled acetate. Nucl Med Biol 31: 859–865
Zhong M, Shen Y, Zheng Y, Joseph T, Jackson D, Beychenok S, Foster DA (2003) Phospholipase D prevents apoptosis in v-Src-transformed rat fibroblasts and MDA-MB-231 breast cancer cells. Biochem Biophys Res Comm 302: 615–619
Acknowledgements
This work was funded by grants from the Prostate Cancer Research Foundation, Prostate UK, the Hull-York Medical School and programme support from Yorkshire Cancer Research. We thank Ms Ann Barker and Mrs Jo Farmery for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Rumsby, M., Schmitt, J., Sharrard, M. et al. Human prostate cell lines from normal and tumourigenic epithelia differ in the pattern and control of choline lipid headgroups released into the medium on stimulation of protein kinase C. Br J Cancer 104, 673–684 (2011). https://doi.org/10.1038/sj.bjc.6606077
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6606077
Keywords
This article is cited by
-
Phospholipase D2 in prostate cancer: protein expression changes with Gleason score
British Journal of Cancer (2019)
-
Phospholipase D inhibitors reduce human prostate cancer cell proliferation and colony formation
British Journal of Cancer (2018)
-
Phorbol ester stimulates ethanolamine release from the metastatic basal prostate cancer cell line PC3 but not from prostate epithelial cell lines LNCaP and P4E6
British Journal of Cancer (2014)
-
18F-fluorocholine for prostate cancer imaging: a systematic review of the literature
Prostate Cancer and Prostatic Diseases (2012)
