
Abstract
The aim of this study was to treat carcinoembryonic antigen (CEA)-expressing pancreatic carcinoma cells with tumour necrosis factor alpha (TNFα) and simultaneous radiation therapy (RT), using a bispecific antibody (BAb) anti-TNFα/anti-CEA. TNFα used alone produced a dose-dependent inhibition of the clonogenic capacity of the cultured cells. Flow cytometry analysis of cell cycle progression confirmed the accumulation of cells in G1 phase after exposure to TNFα. When TNFα was added 12 h before RT, the surviving fraction at 2 Gy was 60% lower than that obtained with irradiation alone (0.29 vs 0.73, respectively, P<0.00001). In combination treatment, cell cycle analysis demonstrated that TNFα reduced the number of cells in radiation-induced G2 arrest, blocked irreversibly the cells in G1 phase, and showed an additive decrease of the number of cells in S phase. In mice, RT as a single agent slowed tumour progression as compared with the control group (P<0.00001). BAb+TNFα+RT combination enhanced the delay for the tumour to reach 1500 mm3 as compared with RT alone or with RT+TNFα (P=0.0011). Median delays were 90, 93, and 142 days for RT alone, RT+TNFα, and RT+BAb+TNFα groups, respectively. These results suggest that TNFα in combination with BAb and RT may be beneficial for the treatment of pancreatic cancer in locally advanced or adjuvant settings.
Similar content being viewed by others


Main
Adenocarcinoma of the pancreas remains one of the most difficult malignancies to treat. The incidence has steadily increased over the past four decades (Gudjonsson, 1987), and its prognosis is still dismal, despite tremendous efforts in early diagnosis and therapy. The 5-year survival rate is less than 5% with a complete surgical resection (Gudjonsson, 1987), ranking this cancer fourth among the leading causes of cancer death (Parker et al, 1996). Unfortunately, at the time of diagnosis, the majority of patients (80–90%) have locally or metastatic inoperable tumours. Radiation therapy (RT) alone or in combination with chemotherapy showed modest efficacy in local control and palliation (Andre et al, 2000; Kornek et al, 2000; Azria et al, 2002). Despite these intensive efforts to improve the efficacy of conventional therapy, no satisfactory progress in dealing with this cancer has been made. Accordingly, new treatment modalities are required for this tumour.
Current interest has focused on biological response modifiers as antineoplastic agents. Among them, tumour necrosis factor alpha (TNFα) was originally identified as a tumoricidal protein effecting haemorrhagic necrosis of transplanted solid tumours in mice (Carswell et al, 1975). It is a multipotent cytokine produced mainly by activated macrophages with the ability to mediate cytotoxicity both in vitro (Sugarman et al, 1985) and in vivo (Carswell et al, 1975; Helson et al, 1979). TNFα usually does not kill untransformed cells (Sugarman et al, 1985) but shows an antiproliferative effect on certain tumour cells in vitro by still undefined mechanisms. Recently, Ruegg et al (1998) reported evidence for the involvement of endothelial cell integrin αvβ3 in the disruption of the tumour vasculature induced by the combination of TNFα and IFNγ.
Several in vitro clonogenic assays suggest that an additive or a supra-additive interaction may occur between TNFα and ionising radiation (Hallahan et al, 1990; Gridley et al, 1994a; Azria et al, 2003a) as well as an enhancement of the antitumour effect of radiation in some murine and human tumours in vivo (Sersa et al, 1988; Nishiguchi et al, 1990; Gridley et al, 1997; Azria et al, 2003a). The oxidative damage produced by TNFα (Zimmerman et al, 1989) may enhance cellular damage produced by ionising radiation. In addition, TNFα and radiation can induce apoptosis in target cells (Yamada and Ohyama, 1988; Langley et al, 1993) even if cells are normally highly resistant to the induction of radiation-induced apoptosis (Kimura et al, 1999).
In different clinical trials using systemic injection of TNFα, the results have been disappointing mainly because patients were found to have significantly lower maximum tolerated doses (Abbruzzese et al, 1989; Moritz et al, 1989) as compared with mice (Asher et al, 1987; Havell et al, 1988). These limited results were probably due to the short circulatory half-life of TNFα and its severe systemic side effects. Studies involving regional (Lienard et al, 1992; Mavligit et al, 1992; Lejeune et al, 1998) or intratumoral (van der Schelling et al, 1992) injection of TNFα have demonstrated its potential for cancer therapy, but only when a high enough therapeutic concentration of TNFα was obtained in the tumour with a nontoxic systemic concentration. To overcome this limitation, we used previously a bispecific antibody (BAb) directed against carcinoembryonic antigen (CEA) and TNFα to target this cytokine in human CEA-expressing colorectal carcinoma treated simultaneously with RT (Azria et al, 2003a).
In the present study, we report the results of clonogenic tests of pancreatic (BxPC-3) cell survival, which confirm a superiority of the radiation-TNFα combination as compared with radiation alone. We show here a nonreversible cell cycle arrest of these cells treated by TNFα alone or in combination with ionising radiation. Using nude mice-bearing BxPC-3 xenografts, we showed a significant enhanced tumour growth delay when the BAb+TNFα+RT combination was used as compared with RT alone and with RT+TNFα.
Materials and methods
Cell line and cell culture
The CEA-expressing human pancreatic carcinoma BxPC-3 cell line (Tan et al, 1986) was obtained from the American Type Culture Collection (Rockville, MD, USA). The cells were cultured in RPMI-1640 medium (Gibco Laboratories, France) supplemented with 10% heat-inactivated fetal calf serum (Gibco Laboratories, France), 300 μg ml−1 glutamine, 0.25 μg ml−1 fungizone, 100 μg ml−1 streptomycin, and 100 U ml−1 penicillin G. These cells were adherent and grew as monolayers at 37°C in a humidified 5% CO2 incubator. The BxPC-3 cells were harvested with 0.5 g l−1 trypsin (Gibco Laboratories, France) and 0.2 g l−1 EDTA (Gibco Laboratories, France) for 3 min. Cultures were checked for the absence of mycoplasma every month.
TNFα and BAb
Recombinant human TNFα, kindly provided by Dr GR Adolf (Boehringer Ingelheim, Wien, Austria), was prepared by expression of a synthetic gene in Escherichia coli. The specific activity of TNFα, determined in the presence of actinomycin D, was 5 × 107 U mg−1 protein as determined by cytolysis of murine L929 cells. TNFα (at a concentration of 2.5 mg ml−1) was stored at –80°C until use.
BAb was constructed as previously described (Robert et al, 1996) from the anti-CEA MAb 35A7 (Haskell et al, 1983) and the MAb tnf18 kindly provided by Dr M Brockhaus (Hoffmann-La Roche AG, Basel, Switzerland).
Radiation protocols
Cells were plated in 10 ml RPMI (to ensure homogeneous energy deposition within each dish) using 60-mm Petri dishes and irradiated with a cobalt-60 (60Co) source (γ-irradiation, ELITE 100, Theratronics) in the Radiation Department. The radiation was delivered as a single dose ranging from 2 to 10 Gy in an 11 cm × 11 cm field size at a dose rate of 0.5 Gy min−1. A 3-cm polystyrene block was used under the Petri dishes during each irradiation to allow homogeneous back-scattering γ-rays. Source-half depth distance (SHD) was initially calculated to obtain a constant dose rate of 0.5 Gy min−1 and monthly adapted from the 60Co source radioactivity decrease. Control cells were removed from the incubator and placed for the same period of time under the 60Co source but without radiation treatment. In the combined treatment modality studies, TNFα was added 12 h prior to RT (see Results, ‘TNFα enhances radiosensitivity’).
For in vivo tumour treatment, the radiation was delivered to the flank of five mice simultaneously in a 12.5 cm × 12.5 cm field size at 6 Gy fraction−1 at a dose rate of 0.5 Gy min−1 (SHD of 158 cm), twice a week, for a total dose of 30 Gy. A 6-cm thickness lead block with eight circular apertures, 3 cm in diameter, was used so that only the tumours and the underlying normal tissues were exposed to the radiation. Radiation was measured using dosimetry films (RA711P, Agfa, Belgium).
Immediately prior to irradiation, the mice were anaesthetised by intraperitoneal injection of 233 μg g−1 of tribromoethanol dissolved in an ethanol : saline combination (1 : 10, v v−1). The anaesthetic was given to all mice, regardless of treatment group, to equalise the effects due to stress.
Clonogenic assay
The colony-forming assay and growth curve analyses were used to assess the sensitivity of the BxPC-3 cells to TNFα. Cultures were trypsinised, washed, and cells were plated in quintuplicate at a density of 100 per 60-mm Petri dishes. TNFα was added at concentrations ranging from 0.3 to 5000 U ml−1 12 h after the cells were plated to allow for cell attachment. Cells were incubated at 37°C in a humidified chamber containing 5% CO2 for 12 days. The colonies were then fixed with a 1 : 3 (v v−1) acetic acid : methanol solution and stained with 10% Giemsa (Sigma Chemical Co., St Louis, MO, USA); colonies of more than 50 cells were scored. Plating efficiency was calculated with and without TNFα. The dose–response curves were fitted to a four-parameter logistic model, where the response, R, varies with the dose, D, according to the equation: R=a/(1+(D/b)c)+R, where a is the difference between the maximum and minimum response, b is the concentration of drug needed to obtain 50% of the maximal effect, c is a slope factor, and R is the maximal effect. The cytotoxic effect of irradiation on asynchronous, exponentially growing BxC-3 cells was also determined by the colony-forming assay. Before irradiation, cell density was determined using appropriate dilutions (100, 300, 600, and 1600 cells for 0, 2, 4, and 6 Gy, respectively), and five replicates of each dilution were plated in 60-mm Petri dishes. Cells were irradiated as described above, 24 h after plating to allow for cell attachment prior to the administration of radiation. The TNFα-containing medium was given at a concentration of 625 U ml−1 12 h before irradiation. A dose of 625 U ml−1 of TNFα was chosen because colony-forming assays showed that this dose was sufficient to induce only partial (48% survival) cell growth when the cytokine was used alone. Cultures were irradiated when the drug was in the medium and were immediately returned to the incubator after irradiation. Colonies were counted after 14 days. Experimentally derived data points are the mean of three experiments. The multitarget model survival curves were fit to the data using a least-squares regression to the linear-quadratic model, S=S0 exp (−αD1−βD12), where D1 is the radiation dose, S the surviving fraction, and S0 a normalising parameter.
Flow cytometry
Cells were plated in 60-mm Petri dishes at a density of 5 × 106 cells dish−1. Treatment consisted of TNFα (625 U ml−1) alone at 24 h (H24), RT (4 Gy) at H36, or TNFα (625 U ml−1 at H24)+RT (4 Gy at H36). Cells were collected at 48 and 96 h after cell culture and processed for cell cycle analysis. Cells were harvested by trypsinisation, washed with PBS, and then 1 × 106 cells dish−1 of treatments were fixed in 70% ethanol for 2 min. After removal of ethanol by centrifugation, cells were then stained with a solution containing 40 μg ml−1 propidium iodide (Sigma, St Louis, USA) and 0.1 mg ml−1 RNase A (Roche, Indianapolis, USA). Stained nuclei were analysed for DNA-PI fluorescence using a Becton Dickinson FACScan flow cytometer. Resulting DNA distributions were analysed by the CellQUEST software (Becton Dickinson, Mountain View, CA, USA) for the proportion of cells in sub-G0, G1, S, and G2–M phases of the cell cycle.
In a second series of experiments, cells were treated with TNFα (625 U ml−1) alone at H24 and then cultured for 3 days. Medium was then harvested and replaced by RPMI. Cells were stained at different time points up to 21 days and analysed for DNA content on a FACScan as described above.
In vivo model
All the in vivo experiments were performed in compliance with the French guidelines for experimental animal studies (Agreement No. A34220) and fulfil the UKCCCR Guidelines for the welfare of animals in experimental neoplasia.
Mice
Athymic 7–9-week-old female Swiss nude mice (nu/nu, Iffa Credo, l'Arbresle, France) were housed in self-contained filter-top cages (five mice cage−1) in a facility controlled for temperature, humidity, and a 12 : 12 h light : dark cycle under sterile conditions. The animals were given autoclaved food and water ad libitum.
Experimental protocols
The human pancreatic carcinoma BxPC-3 cells were harvested with 0.25% trypsin solution, washed, and adjusted to 2 × 106 150 μl−1 RPMI-1640 medium without fetal calf serum. Each mouse was injected s.c. in the right flank with 150 μl of the cell suspension. After 35 days, the mice were grouped according to tumour size by measuring tumour diameters with a Vernier caliper to avoid nonhomogeneous groups before beginning treatments. Tumour dimensions were measured twice weekly and volumes (mm3) were estimated by the formula d1 × d2 × d3/2, where d1 is the length, d2 is the width, and d3 is the height of the tumour.
On day 35, the mice were assigned to seven different treatment groups (five mice per group) as follows:
Group 1: 0.9% NaCl i.v. injection alone (200 μl injection−1) for this control group on days 34, 37, 41, 44, and 48.
Group 2: TNFα at 1 μg i.v.−1 injection alone (in 200 μl 0.9% NaCl injection−1) on days 34, 37, 41, 44, and 48.
Group 3: BAb at 25 μg i.v.−1 injection alone (in 200 μl 0.9% NaCl injection−1) on days 33, 36, 40, 43, and 47.
Group 4: BAb+TNFα (ratio 25 μg : 1 μg; molar ratio 12.5 : 1) i.v. injection (in 200 μl 0.9% NaCl injection−1) on days 33, 36, 40, 43, and 47. BAb–TNFα mixture was prepared 24 h before injection.
Group 5: Local radiation as described above delivered on days 34, 37, 41, 44, and 48+0.9% NaCl i.v. injection (200 μl injection−1) 3 h before irradiation.
Group 6: Local radiation as described above delivered on days 34, 37, 41, 44, and 48+TNFα i.v. injection administered using the same time–dose schedules as for group 2 with TNFα injections 3 h before irradiation
Group 7: Local radiation+BAb+TNFα administered using the same time–dose schedules as for group 4 concerning BAb+TNFα and group 5 in regard to radiation.
All i.v. injections were performed in the heat dilated tail vein; the day of tumour implantation was day 0. On the basis of the biodistribution studies of TNFα and BAb–TNFα complexes (Robert et al, 1996), we decided to inject TNFα 3 h prior to RT (group 6) and BAb–TNFα complexes 24 h prior to RT (group 7).
The mice were weighed twice a week and routinely observed for signs of toxicity throughout the study particularly digestive toxicity because of the local flank irradiation.
Statistical analyses
The nonparametric Wilcoxon's signed-rank test was used to compare the surviving fraction between the two groups (RT alone and RT+TNFα). For in vivo experiments, the results were expressed in terms of the time taken for the tumour to reach a volume of 1500 mm3. The Kaplan–Meier method was used to estimate the median time taken to reach a tumour volume of at least 1500 mm3. Differences among treatment groups were tested by the log-rank test. All statistical tests were two-sided with an α level of 0.05. Data were analysed with software STATA 7.0 (Stata Corporation, College Station, TX, USA).
Results
TNFα inhibits BxPC-3 proliferation
The cytotoxic effects of increasing concentrations of TNFα (0.3–5000 U ml−1) on asynchronous, exponentially growing BxPC-3 cells were determined in colony-forming assays. Cell survival followed a dose–response curve fitted to a four-parameter logistic model as described in Materials and Methods.
Cells were killed by concentrations of TNFα as low as 10 U ml−1 (Figure 1A). The LC50, defined as concentration of drug that reduced the cell survival rate to 50% of that of the controls, was 625 U ml−1. Next, BxPC-3 cells were treated with TNFα (625 U ml−1)+BAb (molar ratio of 100 : 1, 1 : 1, or 1 : 100) and plating efficiencies were compared with that obtained with TNFα alone. No difference in the surviving fraction was observed when BAb was added to TNFα at the same or lower molar ratio. In contrast, when BAb was added in a 100-fold excess, the surviving fraction of cells exposed to 2 Gy was 30% greater than that observed with TNFα alone, probably due to competition between the anti-TNFα arm of the BAb in solution and the TNFα receptor on the cell surface.

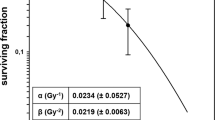
Dose–response curves of the effects of TNFα and irradiation treatment of BxPC-3 cells. (A) Response of BxPC-3 cells to TNFα. Cells were grown in the presence of increasing concentrations of TNFα (0.3–5000 U ml−1). Plating efficiencies were compared with controls grown without TNFα (100% survival). (B) Response of BxPC-3 cells, treated or not with TNFα, to radiation. BxPC-3 cells treated with TNFα (625 U ml−1) added 12 h prior to irradiation showed a surviving fraction at 2 Gy, which was statistically lower (P<0.00001) than when combination treatment was used. When the data were analysed according to the linear quadratic model, the α and β components were, respectively, 0.188±0.08 Gy−1 and 0.017±0.011 Gy−2 without TNFα and 0.39±0.06 Gy−1 and about 0 Gy−2 in combination treatments.
TNFα enhances radiosensitivity
Cell survival following irradiation (Figure 1B) in aerated medium fit a linear quadratic model as described in Materials and Methods. The surviving fraction at 2 Gy (SF2) was 0.73 and a D0 (dose of radiation giving 37% survival rate) of 4 Gy when irradiation was used alone. As shown in Figure 2, TNFα added 12 h before RT (H-12) led to a significant decrease of the surviving fractions as compared with those obtained when TNFα was added at H-1 or H+12 (P=0.02) or when RT was delivered alone. For further experiments using RT with TNFα, we used TNFα at a concentration of 625 U ml−1 added 12 h before RT. In this combination treatment, SF2 and D0 were 0.29 and 1.2 Gy, respectively. SF2 was 60% lower in combination treatment with a significant test result (P<0.00001). When the data were analysed according to the linear quadratic model, the α and β components were 0.188±0.08 and 0.017±0.011 Gy−2, respectively, without TNFα and 0.39±0.06 Gy−1 and nearly 0 Gy−2 in combination treatments. These data indicate that treatment with TNFα results in a steeper decline in cell survival due both to a higher initial slope of the dose–response curve and a major decrease of the quadratic parameter. These results show possible additivity between the two treatments, as confirmed by isobologram analysis (Azria et al, 2003b).

Effect of TNFα on the radiosensitivity of BxPC-3 cells. The influence of TNFα added 12 h before, 1 h before, or 12 h after RT. Results are expressed in terms of the surviving fraction as described in Materials and Methods. Cells were exposed to TNFα (625 U ml−1) and irradiated at the different time points. Control cells were exposed to radiation without TNFα treatment.
TNFα induces G1 cell cycle arrest
The effect of TNFα treatment on cell cycle phase distribution in BxPC-3 cell line was evaluated using flow cytometry (Figure 3). Treatment with 625 U ml−1 TNFα for 24 h induced accumulation of cells in G1 phase (72.8%) with a significant decrease in the percentage of cells in S phase (20.3%) relative to controls (49.6 and 37.8%, respectively) (Figure 3A and B). No cells with subdiploid DNA content was observed, consistent with other results on human colorectal cell line LS174T (Azria et al, 2003b) demonstrating that TNFα does not induce apoptosis in these cell lines. After 3 days of treatment, cells were washed and further cultured for 21 days in the absence of the cytokine. We observed a nonreversible G1 cell cycle arrest (nearly 70% at day 21) without any renewal of activity of the S phase as compared to day 3 after TNFα treatment (Table 1).

Effect of TNFα or/and RT on BxPC-3 cell cycle progression. BxPC-3 were harvested after 36 h exposure to TNFα and compared with control cells ((B) and (A), respectively). In the case of RT treatment, cells were harvested 12 h after RT with or without TNFα (added 12 h before RT) (C and D). Cells were fixed and stained with PI for flow cytometry analysis as described in ‘Materials and Methods.’ Percentages of G0/G1, S, and G2/M were determined by CellQUEST analysis software on the basis of DNA content of the histogram. Data represent mean values of duplicate samples. Similar results were obtained in replicate experiments.
At 1 day after RT alone, we observed a cell cycle arrest in the G2 phase (40%) with a decrease in the percentage of cells in the G0/G1 and S phases as compared with the control (39 vs 50% and 21 vs 38%, respectively). When TNFα was added 12 h before RT, the radiation-induced G2/M arrest decreased as compared with RT alone (31 vs 40%, respectively) with a TNFα-induced G0/G1 blockade and a very low S phase (Figure 3C and D).
BAb+TNFα augments in vivo tumour response to radiation
BxPC-3 tumours growing s.c. in the right flank of nude mice were used to test the antitumour activity of TNFα alone or in combination with RT. TNFα was injected i.v. alone or coinjected with the anti-CEA/anti-TNFα BAb (BAb–TNFα mixture was prepared 24 h before injection at a molar ratio of 12.5 : 1). Median pretreatment tumour volumes (day 35) were 128 (6–135) mm3 with no statistical difference between the groups. Tumour growth was then measured regularly until tumours were larger than 1500 mm3. Radiation alone (group 5), but not TNFα alone (group 2), significantly inhibited tumour progression as compared with the control group (P<0.00001). No difference in growth delay was observed between the control group and groups without RT (groups 2–4). During the same period of observation, treatment with TNFα slowed tumour growth in irradiated groups, particularly when TNFα was coinjected with BAb. At day 93, when mice in all other groups were killed (tumour >1500 mm3), the median value of the tumour volume was 260 mm3 for the RT+BAb+TNFα group.
The results expressed in terms of the time to reach 1500 mm3 are shown in Figure 4. In the control group and the groups treated with TNFα, BAb, or BAb+TNFα, the median delay for the mice to reach a tumour volume greater than 1500 mm3 was 62, 62, 65, and 62 days, respectively, with no statistical difference between the groups. In the RT-treated groups, the median delays were 90, 93, and 142 days for the RT alone, the RT+TNFα, and the RT+BAb+TNFα groups, respectively. No statistical difference was observed between the RT and RT+TNFα groups. However, in the presence of the BAb, the curve for group 7 was shown to be statistically different from the growth curves for tumours treated with RT alone or RT+TNFα (P=0.0011).

Kaplan–Meier survival curves obtained as a function of time for all groups: group 1: dotted line (○) no treatment (62 days); group 2: dotted line (⋄) TNFα (62 days); group 3: dotted line (X) BAb (65 days); group 4: dotted line (△) BAb+TNFα (62 days); group 5: solid line (X) RT (90 days); group 6: solid line (⋄) RT+TNFα (93 days); group 7: solid line (□) RT+TNFα+BAb (142 days). The number in parentheses corresponds to the median delay (time taken for the tumour to reach a volume of 1500 mm3 in 50% of the mice).
At the end of all treatments, no significant differences were found in mouse body weight between the seven groups. The mean±s.e.m. were 23.1±0.47, 22.4±0.87, 23.6±0.61, 24±0.65, 24±0.37, 24.4±0.41, 23.7±0.54 for groups 1, 2, 3, 4, 5, 6, 7, respectively. No diarrhoea was observed in any group, suggesting the absence of digestive toxicity. No significant fluid retention, respiratory distress, or other signs of toxicity were observed in any of the animals during the course of the study.
Discussion
Pancreatic carcinoma is the fourth leading cause of cancer deaths. Patient survival of this devastating disease is bleak with less than 5% of patients surviving 5 years after the time of diagnosis (Greenlee et al, 2000). The current treatment includes a combination of surgery, chemotherapy, and radiation without any major improvement in survival (Azria et al, 2002). Over 10 years ago, it was hypothesised that TNFα could increase tumour response to radiation through stimulation of the host antitumour immune responses, direct tumour-cell kill, or through the increase in tumour-cell sensitivity to radiation (Sersa et al, 1988; Hallahan et al, 1990; Gridley et al, 1994a, 1994b; Kimura et al, 1999; Azria et al, 2003a). However, early clinical trials were generally disappointing, with hypotension and vascular leakage frequently being the dose-limiting side effects (Chapman et al, 1987; Sherman et al, 1988). To overcome these limitations, we used a BAb directed against CEA and human TNFα to target this cytokine to the human pancreatic carcinoma cells BxPC-3 treated simultaneously with RT.
In the first part of our study, we demonstrated direct cytotoxicity of TNFα on BxPC-3 cells in culture using a clonogenic assay: TNFα-treated BxPC-3 cells showed reduced plating efficiency (Figure 1), confirming that TNFα can be tumoristatic or tumoricidal as described for a variety of neoplastic cell types (Hallahan et al, 1990; Manetta et al, 1990; Gridley et al, 1994a; Kimura et al, 1999; Azria et al, 2003a). In a time-course experiment (Figure 2), we demonstrated that maximal cell killing increase was obtained when TNFα was added to the cells 12 h before RT as compared with 1 h before and 12 h after RT. These data confirmed those published by Hallahan et al (1990), who demonstrated that addition of TNFα 4 to 12 h prior to irradiation maximally increases cell killing.
We also observed that TNFα induced a G1 cell cycle arrest and that cell exposure for 24 h to TNFα was sufficient to obtain this effect, which could be considered as irreversible since the G1 arrest was maintained up to 21 days after elimination of TNFα from the culture medium (Table 1). This effect can probably be explained by modifications of the expression of cell-cycle-related proteins (ongoing research), as described for other cytokine such as interferon γ (Matsuoka et al, 1999; Gooch et al, 2000), and by the fact that TNFα induces BxPC-3 cycle distribution modification which may render the cells more radiosensitive. In the RT–TNFα combination treatment, we observed a 25% decrease of BxPC-3 cells arrested in the G2 phase as compared with RT alone, a proportional redistribution in the G1 phase, and an interrupted synthesis phase. We did not observe any induction of apoptosis in BxPC-3 cells, as previously suggested in another model (Gridley et al, 1994a) and recently described in a human prostate carcinoma cell line (Kimura et al, 1999). This cell cycle redistribution phenomenon may also explain the decrease in the surviving fraction in the combination treatment presented in the present study (Figure 1B). To our knowledge, these results are the first to confirm that TNFα is a biological cell cycle modifier, which is responsible for a cell cycle redistribution in the more radiosensitive (G1) phase rather than in the S phase. Recently, Dormond et al (2002) described that TNFα alone or in combination with IFNγ induced a G1 arrest in endothelial cells (HUVEC), which was associated with reduced levels of cyclin D1 and cdk2, and with increased expression of the cdk inhibitors p16INK4a, p21WAF, and p27Kip1.
In the present study, the in vitro growth-inhibitory effect of TNFα was accompanied by a marked enhancement of the radioresponse of the tumour in vivo, particularly, when TNFα was concentrated in the tumour xenografts thanks to our BAb. In addition to the in vitro cytotoxic effect of TNFα, indirect in vivo mechanisms could be responsible for this synergistic rather than additive effect of the combination (Ruegg et al, 1998). Several studies have demonstrated the antitumour activity of RT+TNFα, but this treatment was given before the tumours reached a palpable volume, making a comparison with our results difficult (Gridley et al, 1994b, 1997). In mammary carcinoma and sarcoma models, TNFα was shown to significantly increase tumour radiocurability even when TNFα was injected 3 h after RT (Sersa et al, 1988; Nishiguchi et al, 1990). Our data demonstrate the interest of targeting TNFα to tumours to improve RT and finally to keep a large differential effect between tumour and normal tissues. Various methods have recently tried to concentrate TNFα into tumour such as Cu2+-dextran (Tabata et al, 1999), TNFα-biotin conjugates (Moro et al, 1997; Gasparri et al, 1999), or liposomal encapsulated-TNFα (Kim et al, 2001) which are less specific targeting than our BAb and were not tested with concomitant radiotherapy.
Another approach currently in clinical evaluation uses an adenoviral vector that contains radio-inducible DNA sequences from the early growth response gene (EGR1) promoter and cDNA for the gene encoding human TNFα. While avoiding the systemic side effects of TNFα, this method involves injections in or near the tumour, which might be difficult to perform in the case of pelvic or retroperitoneal tumours (Weichselbaum et al, 2002).
Concerning the immunotargeting strategy, two attractive methods have been recently described. Cooke et al (2002) tested a genetic fusion of human recombinant TNFα with MFE-23, a single-chain Fv antibody fragment directed against CEA. Radiolabelled fusion protein binds both human and mouse TNF receptor 1 in vitro and in vivo and is able to localise effectively in nude mice-bearing human LS174T xenografts with a tumour/tissue ratios of 21 : 1 and 60 : 1 achieved 24 and 48 h after i.v. injection, respectively. The maximum % injected dose (ID) g−1 LS174T tumour (4.33) was obtained 6 h postinjection. At that time, in T380 human colon carcinoma nude mice, our BAb was able to concentrate up to 7.15% ID g−1 of tumour as compared to 2.2% when BAb was injected alone (Robert et al, 1996). Wüest et al (2002) described a TNFα fusion protein designated TNF-Selectokine, which is a homotrimeric molecule comprised of a single-chain antibody (scFv) targeting molecule, a trimerisation domain and TNFα. Membrane targeting dependent immobilisation of this TNF-Selectokine induced cell death in TNFR1 and TNFR2 dependent manner. The authors constructed, also, a TNF-Selectokine prodrug by insertion of a TNFR1 fragment separated from TNF by a protease-sensitive linker in order to restrict TNF activity to the tumour. Both studies suggest interests but are in the early phase of development without any indications of their capacity of radiation enhancement.
The results of our study should be of potential clinical interest. They provide a rational for the combination of TNFα, BAb, and RT in the treatment of adenocarcinoma of the pancreas. One of the advantages of our BAb strategy, namely, the potential decrease of TNFα systemic toxicity, cannot be addressed in our nude mice model, which lacks T cells. The difference between the TNFα +RT and the BAb+TNFα+RT combination treatments will probably be even more evident in an immunocompetent model or in a clinical setting. Such an immunocompetent situation is also needed for the entire expression of TNFα antitumour action, including immunological (production of IL-1 and IFNγ, activation of macrophages, and NK cells; Dinarello et al, 1986; Ostensen et al, 1987; Talmadge et al, 1987) and nonimmunological mechanisms such as damage to the tumour vasculature (Gamble et al, 1985; Sato et al, 1986; Cavender et al, 1987; Ruegg et al, 1998).
In conclusion, we demonstrated that an anti-CEA/anti-TNFα BAb can markedly enhance the radioresponse of pancreatic tumour xenografts in nude mice. Presently, we are testing the antitumour effect of BAb, TNFα, and RT combination in an immunocompetent CEA-transgenic mice transplanted with a syngenic CEA-expressing tumour in which all the effects of the targeted cytokine can be analysed. The next step will be the opening of a phase I clinical study in locally advanced pancreatic cancer.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abbruzzese JL, Levin B, Ajani JA, Faintuch JS, Saks S, Patt YZ, Edwards C, Ende K, Gutterman JU (1989) Phase I trial of recombinant human gamma-interferon and recombinant human tumor necrosis factor in patients with advanced gastrointestinal cancer. Cancer Res 49: 4057–4061
Andre T, Balosso J, Louvet C, Hannoun L, Houry S, Huguier M, Colonna M, Lotz JP, De Gramont A, Bellaiche A, Parc R, Touboul E, Izrael V (2000) Combined radiotherapy and chemotherapy (cisplatin and 5-fluorouracil) as palliative treatment for localized unresectable or adjuvant treatment for resected pancreatic adenocarcinoma: results of a feasibility study. Int J Radiat Oncol Biol Phys 46: 903–911
Asher A, Mule JJ, Reichert CM, Shiloni E, Rosenberg SA (1987) Studies on the anti-tumor efficacy of systemically administered recombinant tumor necrosis factor against several murine tumors in vivo. J Immunol 138: 963–974
Azria D, Dorvillius M, Gourgou S, Martineau P, Robert B, Pugnière M, Delard R, Ychou M, Dubois JB, Pèlegrin A (2003a) Enhancement of radiation therapy by tumor necrosis factor alpha in human colon cancer using a bispecific antibody. Int J Radiat Oncol Biol Phys 55: 1363–1373
Azria D, Larbouret C, Martineau P, Robert B, Aillères N, Ychou M, Dubois JB, Pèlegrin A (2003b) A bispecific antibody against tumor necrosis factor alpha and carcinoembryonic antigen (CEA) to enhance radiation therapy in CEA-expressing digestive tumors. Int J Radiat Oncol Biol Phys, in press
Azria D, Ychou M, Jacot W, Thezenas S, Lemanski C, Senesse P, Prost P, Delard R, Masson B, Dubois JB (2002) Treatment of unresectable, locally advanced pancreatic adenocarcinoma with combined radiochemotherapy with 5-fluorouracil and cisplatin. Pancreas 25: 360–365
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA 72: 3666–3670
Cavender D, Saegusa Y, Ziff M (1987) Stimulation of endothelial cell binding of lymphocytes by tumor necrosis factor. J Immunol 139: 1855–1860
Chapman PB, Lester TJ, Casper ES, Gabrilove JL, Wong GY, Kempin SJ, Gold PJ, Welt S, Warren RS, Starnes HF, Sherwin SA, Old LJ, Oettgen HF (1987) Clinical pharmacology of recombinant human tumor necrosis factor in patients with advanced cancer. J Clin Oncol 5: 1942–1951
Cooke SP, Pedley RB, Boden R, Begent RH, Chester KA (2002) In vivo tumor delivery of a recombinant single chain Fv::tumor necrosis factor-alpha fusion [correction of factor: a fusion] protein. Bioconjug Chem 13: 7–15
Dinarello CA, Cannon JG, Wolff SM, Bernheim HA, Beutler B, Cerami A, Figari IS, Palladino Jr MA, O'Connor JV (1986) Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J Exp Med 163: 1433–1450
Dormond O, Lejeune FJ, Rüegg C (2002) Modulation of cdk2, cyclin D1, p16INK4a, p21 WAF and p27 Kip1 expression in endothelial cells by TNF/IFNγ. Anticancer Res 22: 3159–3163
Gamble JR, Harlan JM, Klebanoff SJ, Vadas MA (1985) Stimulation of the adherence of neutrophils to umbilical vein endothelium by human recombinant tumor necrosis factor. Proc Natl Acad Sci USA 82: 8667–8671
Gasparri A, Moro M, Curnis F, Sacchi A, Pagano S, Veglia F, Casorati G, Siccardi AG, Dellabona P, Corti A (1999) Tumor pretargeting with avidin improves the therapeutic index of biotinylated tumor necrosis factor alpha in mouse models. Cancer Res 59: 2917–2923
Gooch JL, Herrera RE, Yee D (2000) The role of p21 in interferon gamma-mediated growth inhibition of human breast cancer cells. Cell Growth Differ 11: 335–342
Greenlee RT, Murray T, Bolden S, Wingo PA (2000) Cancer statistics, 2000. CA Cancer J Clin 50: 7–33
Gridley DS, Archambeau JO, Andres MA, Mao XW, Wright K, Slater JM (1997) Tumor necrosis factor-alpha enhances antitumor effects of radiation against glioma xenografts. Oncol Res 9: 217–227
Gridley DS, Glisson WC, Uhm JR (1994a) Interaction of tumour necrosis factor-alpha and radiation against human colon tumour cells. Ther Immunol 1: 25–31
Gridley DS, Hammond SN, Liwnicz BH (1994b) Tumor necrosis factor-alpha augments radiation effects against human colon tumor xenografts. Anticancer Res 14: 1107–1112
Gudjonsson B (1987) Cancer of the pancreas. 50 years of surgery. Cancer 60: 2284–2303
Hallahan DE, Beckett MA, Kufe D, Weichselbaum RR (1990) The interaction between recombinant human tumor necrosis factor and radiation in 13 human tumor cell lines. Int J Radiat Oncol Biol Phys 19: 69–74
Haskell CM, Buchegger F, Schreyer M, Carrel S, Mach JP (1983) Monoclonal antibodies to carcinoembryonic antigen: ionic strength as a factor in the selection of antibodies for immunoscintigraphy. Cancer Res 43: 3857–3864
Havell EA, Fiers W, North RJ (1988) The antitumor function of tumor necrosis factor (TNF). I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J Exp Med 167: 1067–1085
Helson L, Helson C, Green S (1979) Effects of murine tumor necrosis factor on heterotransplanted human tumors. Exp Cell Biol 47: 53–60
Kim DW, Andres ML, Li J, Kajioka EH, Miller GM, Seynhaeve AL, Ten Hagen TL, Gridley DS (2001) Liposome-encapsulated tumor necrosis factor-alpha enhances the effects of radiation against human colon tumor xenografts. J Interferon Cytokine Res 21: 885–897
Kimura K, Bowen C, Spiegel S, Gelmann EP (1999) Tumor necrosis factor-alpha sensitizes prostate cancer cells to gamma-irradiation-induced apoptosis. Cancer Res 59: 1606–1614
Kornek GV, Schratter-Sehn A, Marczell A, Depisch D, Karner J, Krauss G, Haider K, Kwasny W, Locker G, Scheithauer W (2000) Treatment of unresectable, locally advanced pancreatic adenocarcinoma with combined radiochemotherapy with 5-fluorouracil, leucovorin and cisplatin. Br J Cancer 82: 98–103
Langley RE, Palayoor ST, Coleman CN, Bump EA (1993) Modifiers of radiation-induced apoptosis. Radiat Res 136: 320–326
Lejeune FJ, Ruegg C, Lienard D (1998) Clinical applications of TNF-alpha in cancer. Curr Opin Immunol 10: 573–580
Lienard D, Ewalenko P, Delmotte JJ, Renard N, Lejeune FJ (1992) High-dose recombinant tumor necrosis factor alpha in combination with interferon gamma and melphalan in isolation perfusion of the limbs for melanoma and sarcoma. J Clin Oncol 10: 52–60
Manetta A, Lucci J, Soopikian J, Granger G, Berman ML, DiSaia PJ (1990) In vitro cytotoxicity of human recombinant tumor necrosis factor alpha in association with radiotherapy in a human ovarian carcinoma cell line. Gynecol Oncol 38: 200–202
Matsuoka M, Nishimoto I, Asano S (1999) Interferon-gamma impairs physiologic downregulation of cyclin-dependent kinase inhibitor, p27Kip1, during G1 phase progression in macrophages. Exp Hematol 27: 203–209
Mavligit GM, Zukiwski AA, Charnsangavej C, Carrasco CH, Wallace S, Gutterman JU (1992) Regional biologic therapy. Hepatic arterial infusion of recombinant human tumor necrosis factor in patients with liver metastases. Cancer 69: 557–561
Moritz T, Niederle N, Baumann J, May D, Kurschel E, Osieka R, Kempeni J, Schlick E, Schmidt CG (1989) Phase I study of recombinant human tumor necrosis factor alpha in advanced malignant disease. Cancer Immunol Immunother 29: 144–150
Moro M, Pelagi M, Fulci G, Paganelli G, Dellabona P, Casorati G, Siccardi AG, Corti A (1997) Tumor cell targeting with antibody–avidin complexes and biotinylated tumor necrosis factor alpha. Cancer Res 57: 1922–1928
Nishiguchi I, Willingham V, Milas L (1990) Tumor necrosis factor as an adjunct to fractionated radiotherapy in the treatment of murine tumors. Int J Radiat Oncol Biol Phys 18: 555–558
Ostensen ME, Thiele DL, Lipsky PE (1987) Tumor necrosis factor-alpha enhances cytolytic activity of human natural killer cells. J Immunol 138: 4185–4191
Parker SL, Tong T, Bolden S, Wingo PA (1996) Cancer statistics, 1996. CA Cancer J Clin 46: 5–27
Robert B, Mach JP, Mani JC, Ychou M, Folli S, Artus JC, Pelegrin A (1996) Cytokine targeting in tumors using a bispecific antibody directed against carcinoembryonic antigen and tumor necrosis factor alpha. Cancer Res 56: 4758–4765
Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Lejeune FJ (1998) Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat Med 4: 408–414
Sato N, Goto T, Haranaka K, Satomi N, Nariuchi H, Mano-Hirano Y, Sawasaki Y (1986) Actions of tumor necrosis factor on cultured vascular endothelial cells: morphologic modulation, growth inhibition, and cytotoxicity. J Natl Cancer Inst 76: 1113–1121
Sersa G, Willingham V, Milas L (1988) Anti-tumor effects of tumor necrosis factor alone or combined with radiotherapy. Int J Cancer 42: 129–134
Sherman ML, Spriggs DR, Arthur KA, Imamura K, Frei III E, Kufe DW (1988) Recombinant human tumor necrosis factor administered as a five-day continuous infusion in cancer patients: phase I toxicity and effects on lipid metabolism. J Clin Oncol 6: 344–350
Sugarman BJ, Aggarwal BB, Hass PE, Figari IS, Palladino Jr MA, Shepard HM (1985) Recombinant human tumor necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science 230: 943–945
Tabata Y, Noda Y, Matsui Y, Ikada Y (1999) Targeting of tumor necrosis factor to tumor by use of dextran and metal coordination. J Control Release 59: 187–196
Talmadge JE, Tribble HR, Pennington RW, Phillips H, Wiltrout RH (1987) Immunomodulatory and immunotherapeutic properties of recombinant gamma-interferon and recombinant tumor necrosis factor in mice. Cancer Res 47: 2563–2570
Tan MH, Nowak NJ, Loor R, Ochi H, Sandberg AA, Lopez C, Pickren JW, Berjian R, Douglass Jr HO, Chu TM (1986) Characterization of a new primary human pancreatic tumor line. Cancer Invest 4: 15–23
van der Schelling GP, Ijzermans JNM, Kok TC, Scheringa M, Marquet RL, Splinter TA, Jeekel J (1992) A phase I study of local treatment of liver metastases with recombinant tumour necrosis factor. Eur J Cancer 28: 1073–1078
Weichselbaum RR, Kufe DW, Hellman S, Rasmussen HS, King CR, Fischer PH, Mauceri HJ (2002) Radiation-induced tumour necrosis factor-alpha expression: clinical application of transcriptional and physical targeting of gene therapy. Lancet Oncol 3: 665–671
Wüest T, Gerlach E, Banerjee D, Gerspach J, Moosmayer D, Pfizenmaier K (2002) TNF-Selectokine: a novel prodrug generated for tumor targeting and site-specific activation of tumor necrosis factor. Oncogene 21: 4257–4265
Yamada T, Ohyama H (1988) Radiation-induced interphase death of rat thymocytes is internally programmed (apoptosis). Int J Radiat Biol Relat Stud Phys Chem Med 53: 65–75
Zimmerman RJ, Chan A, Leadon SA (1989) Oxidative damage in murine tumor cells treated in vitro by recombinant human tumor necrosis factor. Cancer Res 49: 1644–1648
Acknowledgements
This study was supported by the comité de l'Hérault de la Ligue Nationale Contre le Cancer. David Azria was supported by the Association de Recherche sur le Cancer.
We thank Genevieve Heinz, Sabine Bousquié, Celine Passet, and Philippe Gauthier for excellent technical assistance; Michel Brissac for help in performing animal experiments; Dr Jacques Dornand for the TNFα cytotoxicity assays; and Dr SL Salhi for critical comments and excellent editorial assistance. This work was presented in part at the Second International Conference of Translational Research and Preclinical Strategies in Radio-oncology, 16–19 March 2003 in Lugano, Switzerland.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Azria, D., Larbouret, C., Garambois, V. et al. Potentiation of ionising radiation by targeting tumour necrosis factor alpha using a bispecific antibody in human pancreatic cancer. Br J Cancer 89, 1987–1994 (2003). https://doi.org/10.1038/sj.bjc.6601362
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601362
Keywords
This article is cited by
-
Novel agents for pancreatic ductal adenocarcinoma: emerging therapeutics and future directions
Journal of Hematology & Oncology (2018)
-
Recent advances of bispecific antibodies in solid tumors
Journal of Hematology & Oncology (2017)
