
Abstract
We determined the prevalence of mutations in two major functional domains of the leucine-rich repeat kinase 2 gene (LRRK2) in Belgian Parkinson's disease (PD) patients (N=304) of which 18.1% were familial PD patients. Ten patients were heterozygous for five different missense mutations (3.29%) of whom six carried the same mutation p.R1441C (1.97%). All six p.R1441C carriers were familial PD patients explaining 10.7% of familial PD in the Belgian patient group. Moreover, they shared a common disease haplotype of 21 consecutive markers in a region of 438 kb, suggesting that they are distant descendants of a single common ancestor. Clinically, p.R1441C carriers had typical levodopa-responsive parkinsonism with tremor as the most common presenting feature. Their age at onset was highly variable and ranged from 39 to 73 years, suggesting the influence of modifying factors. The remaining four patients were heterozygous each for a novel missense mutation located in the Roc or kinase domain. The pathogenic nature of these mutations remains to be determined, though we have genetic evidence that at least some represent rare but benign variants rather than causal mutations. The latter observation indicates that prudence is needed in diagnostic testing of LRRK2 in PD patients. Functional data should underlie a conclusion on the pathogenic nature of some mutations that have not been conclusively linked to disease.
Similar content being viewed by others

Introduction
Parkinson's disease (PD) is the second most common progressive neurodegenerative brain disorder affecting approximately 2% of the population above the age of 65 years. The pathogenesis of PD is not yet completely understood, however, both genetic and environmental factors contribute to the disease phenotype. To date, six causal genes have been identified1, 2, 3, 4, 5, 6 and more are to be identified in several linked loci. Mutations in the α-synuclein (SNCA), parkin (PARK2), ubiquitin hydrolase L1 (UCH-L1), DJ-1, PTEN-induced kinase 1 (PINK1) and leucine-rich repeat kinase 2 (LRRK2) genes have been implicated in hereditary PD (for recent review see Farrer7). Mutations in LRRK2 were most recently identified and are responsible for PD in at least 5% of familial patients.8, 9 Intriguingly, LRRK2 mutations have also been reported in 1–2% of sporadic PD patients presenting with a clinical phenotype largely indistinguishable from common PD.10
The 51 exons of LRRK2, spanning 144 kb in the chromosome 12q locus, encode an unusually large protein belonging to the ROCO family,11 containing both an Roc-GTPase and a COR domain. In addition, a protein kinase as well as multiple protein interaction domains have been predicted in the C-terminal half of LRRK2.11 Most of these catalytic and protein–protein interaction domains are affected by PD-associated mutations.12 The wide mutational spectrum along LRRK2 is consistent with a gain-of-function disease mechanism, which is supported by three recent functional LRRK2 studies, showing increased levels of kinase activity in vitro for LRRK2 mutations within the kinase domain.13, 14, 15 Further functional analysis of the Roc domain showed that distinct mutations in this domain increase GTP binding.15 Since binding of GTP is essential for kinase activity,16 these data also support a (indirect) gain-of-function mechanism for mutations in the Roc domain.
Thus far, 22 putative pathogenic PD-associated LRRK2 mutations have been identified in genetic studies.12 The most prevalent LRRK2 mutation, p.G2019S, has been observed in more than 250 patients worldwide. In Whites, this mutation explains 0.5–2% of sporadic and 5% of familial PD patients. More recent studies reported that p.G2019S accounted for up to 30% of familial and sporadic PD in Ashkenazi Jews and North African Arabs. Haplotype analyses showed a common Middle Eastern founder haplotype in most p.G2019S carriers, however, at least three European American families carried a distinct disease haplotype suggesting two separate founding events for the p.G2019S mutation in these populations.17, 18, 19, 20 The p.R1441C mutation is the second most frequent LRRK2 mutation and was first identified in a large autosomal dominant kindred from Western Nebraska, family D, presenting with late-onset, levodopa-responsive parkinsonism.6, 21 Since then, p.R1441C mutations were reported in five additional US, three Italian and several single PD families from different nationalities.6, 22, 23, 24, 25, 26, 27, 28, 29 Founder effects have also been reported for the LRRK2 p.R1441 ‘hotspot’ codon. A common disease haplotype containing the p.R1441C mutation was observed in three Italian families,22 while transmission of the p.R1441G mutation from a common ancestor was identified in two neighboring autonomous communities in Northern Spain.30, 31 Although the pathogenicity of many of the reported missense mutations still needs to be proven, it was demonstrated that LRRK2 mutations can also act as potential risk factors. For example, the p.G2385R polymorphism increased the risk for PD in Chinese and Taiwanese populations.32, 33
Materials and methods
Subjects
We studied a large group of 304 Belgian PD patients (Table 1), mainly derived from a retrospective epidemiological study designed to evaluate environmental risk factors in PD (N=181).34 This PD group was supplemented with patients from a prospective Belgian study of neurodegenerative diseases (N=123).35 Patients included in these studies were all residents of Flanders, the Flemish speaking region of Belgium. Overall, the mean onset age in the Belgian PD group was 59.0±11.5 years (range 28–87) and mean age at examination 67.2±10.9 years (range 30–90) with 56.3% male patients. In 18.1% of PD patients, a positive family history of PD with a first-degree relative was reported. The control group consisted of 278 Belgian individuals with a mean age at inclusion of 60.4±12.0 (range 28–89), and included 127 spouses recruited within the epidemiological study34 and 151 community individuals with no clinical evidence or history of PD or another movement disorder.
The local medical ethical committees of the Middelheim General Hospital Antwerp, the University Hospital Antwerp and the University of Antwerp approved the Belgian PD studies and all participating individuals gave written informed consent. Patients were diagnosed using strict diagnostic criteria for PD, requiring three out of four features of bradykinesia, rigidity, tremor and asymmetrical onset with a positive response to levodopa.
Gene sequencing
The coding sequence and exon–intron boundaries of LRRK2 exons 29–31 and 38–44 encoding the Roc and kinase domains, and exons 5 and 34 harboring informative single-nucleotide polymorphisms (SNPs) implemented in the haplotype analysis, were amplified by standard PCR reactions. Primer pairs were designed using Primer3.36 Amplification products were purified and sequenced using the BigDye Terminator Cycle Sequencing v3.1, and analyzed on an ABI3730 DNA analyzer. Novel variants identified in patients were confirmed and tested in a panel of 278 Belgian control individuals by pyrosequencing on a PSQTM pyrosequencer, genescan analysis on an automated ABI3730 DNA analyzer or by SEQUENOM MassARRAY(R) technology (Sequenom Inc.) (primer sequences available upon request). In silico analysis of evolutionary amino-acid conservation between different types of organisms was performed using the UCSC Genome Browser. The effect of mutations at the protein level was predicted using the SIFT program.37
Haplotype analysis
We genotyped 28 LRRK2 intragenic and flanking markers, both microsatellite markers and SNPs that spanned a region of 16 Mb at chromosome 12q in all six Belgian R1441C carriers and in 20 of their relatives for haplotype phase determination. LRRK2 markers were also analyzed in 178 Belgian control individuals to estimate the population frequency of the shared haplotype. Microsatellite markers17 were amplified and fluorescently labeled during PCR amplification. Amplicons were separated on an automated ABI3730 DNA analyzer. Alleles were scored using the ‘Genotyper’ and ‘Pattern automatic allele caller’ programs developed in house. Genotypes of exonic and intronic LRRK2 SNPs were extracted from the sequence trace files.
Results
Mutations in the Roc and kinase domains
Direct sequencing of PCR amplicons of the 10 LRRK2 exons coding for the Roc and kinase domains identified 10 heterozygous LRRK2 mutation carriers in the Belgian PD patients (10/304=3.29%) (Table 1). Six patients carried the same p.R1441C mutation, while each of the other four patients carried a distinct novel missense mutation (Table 2, Figure 1a). We also observed five known SNPs at the same frequencies as previously reported in other European study populations25 (Supplementary Table 1).

(a) Reported and novel coding mutations in the LRRK2 Roc and kinase domains. Symbols: red stars, putative pathogenic missense mutations detected in the Flanders-Belgian PD population; green stars, novel rare variants, putatively not pathogenic; yellow stars, known polymorphisms; cDNA numbering according to AY792511.1 starting at nt 1. Domain boundaries as depicted in Bosgraaf and Van Haastert.11 Protein numbering according to AAV63975.1 starting at nt 1. (b) Data on protein conservation of all coding mutations in the LRRK2 Roc and kinase domains.
p.R1441C founder mutation
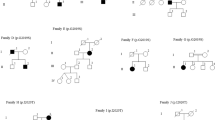
p.R1441C (c.4321C>T in exon 31) was observed in six independently ascertained PD patients (6/304=1.97%; Table 2). Five of the p.R1441C carriers presented with a classical PD phenotype with tremor as the predominant initial symptom, while the sixth patient suffered primarily from rigidity (Table 3). The onset age varied largely among carriers from 39 to 73 years with a mean onset of 56.5 years. A positive family history of PD was reported for all p.R1441C carriers in one or more first-degree (DR75, DR133, DR134, DR135 and DR237) or second-degree (DR99) relatives (Figure 2).

Pedigrees of probands carrying p.R1441C. Symbols: ▪=PD;  =dementia;
=dementia;  =unclear; *=blood drawn; all at-risk individuals are collected in a diamond symbol (carrying wild-type or mutant alleles) at the right of each generation regardless of their age.
=unclear; *=blood drawn; all at-risk individuals are collected in a diamond symbol (carrying wild-type or mutant alleles) at the right of each generation regardless of their age.
On the basis of the segregation data of 28 STR and SNP markers in families DR75, DR99 and DR135, we deduced a disease haplotype that was shared among all three families (Table 4). Further, examination of the individual genotype data from the other three probands DR133.1, DR134.1 and DR237.1 indicated that all three shared alleles for several markers from within the disease haplotype. Together, the allele and haplotype data identified a shared segment of 438 kb between centromeric D12S2194 and telomeric D12S1301 (Table 4). The shared haplotype was absent in 178 Belgian control individuals.
Novel missense mutations
The four novel missense mutations were identified in exons 29, 31 and 44 (Table 2, Figure 1a) and were absent from 278 Belgian control individuals (MAF <0.2%). In silico conservation analyses demonstrated that the mutations in exons 29 and 31 affected evolutionarily conserved amino-acid residues (Figure 1b). For p.Y2189C in exon 44, the in silico conservation analysis showed that the Y residue itself was not conserved through evolution, though the aromatic nature of the respective amino-acid residues was (UCSC Genome Browser Human March 2006 Assembly) (Figure 1b).
Genealogy showed a positive family history of neurodegenerative disease for the respective probands except for DR136.1 (p.K1468E) (Table 3, Figure 3b). A positive family history of PD was reported for the patients carrying p.R1483Q and p.Y2189C, though we have no access to DNA of additional family members of DR241.1 (p.Y2189C; Figure 3d). Two first-degree relatives of DR103.1 (p.R1483Q) were also diagnosed with PD (Figure 3c; I.2 and II.1). The mutation was not detected in an affected sib (II.1) of the proband (II.2), while an older healthy brother (II.3) carried p.R1483Q. The mother (Figure 3a; I.4) of proband DR108.1 (II.1) was not diagnosed with PD though presented with a mild tremor in her late 70s. She turned out to be a homozygous carrier of p.R1325Q. Loss of heterozygosity due to a large genomic deletion of the LRRK2 locus was ruled out by genotyping 18 STR markers. An older unaffected sibling of the proband also carried the mutation but in the heterozygous state.

Pedigrees of probands carrying novel missense mutations. (a) p.R1325Q; (b) p.K1468E; (c) p.R1483Q; (d) p.Y2189C. Symbols: ▪=PD;  =dementia;
=dementia;  =recent tremor; *=blood drawn. All at-risk individuals are collected in a diamond symbol (carrying wild-type or mutant alleles) at the right of each generation regardless of their age.
=recent tremor; *=blood drawn. All at-risk individuals are collected in a diamond symbol (carrying wild-type or mutant alleles) at the right of each generation regardless of their age.
Discussion
LRRK2 is the most frequent mutated PD gene with a prevalence of 5% in familial and 2% in sporadic patients in European populations8, 9, 10, 17 and the majority of the reported pathogenic mutations are located in two major functional LRRK2 domains that is the Roc and kinase domains. In this study, we examined the prevalence of mutations in these domains in a large PD sample of 304 unrelated patients. Although the patient group consisted mainly of sporadic patients (82%), we identified five distinct mutations in 10 patients suggesting an overall LRRK2 frequency of 3.29%. The majority of these mutations were observed in patients with a positive family history of PD (8/56=14.28%). This frequency is higher than what has been reported in other studies,8, 9 however, we should point out that six of eight familial patients carried p.R1441C, indicating that this mutation was the major genetic cause of PD in Belgian patients. All patients were recruited in Flanders, the Flemish speaking region of Belgium. In previous genetic studies of other neurological diseases, we showed that several pathogenic mutations that were identified in Flemish patients had a strong founder effect most likely due to linguistic and geographic isolation of this region.38, 39, 40, 41 Also, in this study, we were able to show that the Flemish-Belgian p.R1441C was present on one single disease haplotype spanning a minimal region of 438 kb, and absent in 178 control individuals. These data strongly support the notion that p.R1441C was transmitted from a single common founder. Comparison of the p.R1441C disease haplotype with that in Italian PD patients22 revealed that in these two European populations, p.R1441C resulted from two independent mutation events. The latter again supports the observation that the p.R1441 codon represents a mutation hotspot since three different missense mutations at this codon have already been reported (p.R1441C, p.R1441G, p.R1441H).6, 26, 30, 31
Interestingly, five out of six R1441C carriers presented with tremor as the dominant symptom, a feature that generally occurs in only one out of three PD patients. Also, most previously reported p.R1441C carriers presented with tremor as the initial symptom,6, 24, 25, 27, 28, 41 though detailed clinical data have not been reported in all studies.22, 23, 26 An extensive clinical comparison of as many carriers as possible will be obligatory to determine the relationship between p.R1441C and this specific phenotype. Previous segregation analyses in families carrying the most prevalent pathogenic LRRK2 mutations, p.G2019S and p.R1441C, highlighted a considerable variation in age at onset of PD within6, 17, 42 and between families17, 43 due to a highly variable disease penetrance. The onset age between the six probands carrying p.R1441C ranged from 39 to 74 years. Within family DR75, the onset age ranged from 39 to 72 years. Moreover, we also observed an older sibling carrying the mutation but not showing any symptoms at the age of 63 years, 24 years later than the onset age of the proband, supporting the idea of reduced disease penetrance for p.R1441C. In contrast, another older sibling of the proband showed disease symptoms, but was not a carrier of p.R1441C. Non-carrier patients have also been reported in other LRRK2 mutation families including p.R1441C (Zimprich et al6; Di Fonzo et al, 200622) and p.G2019S (Hernandez et al43; Khan et al44; Nichols et al9) families. These patients likely represent phenocopies that are not uncommon for frequent diseases as PD.
In addition, we identified four novel missense mutations in the PD group (4/304=1.32%), with each mutation present in a single unrelated patient. Although these rare mutations were not present in 278 Belgian healthy control individuals their pathogenic nature remains obscure. For the three novel mutations identified in the Roc domain, our data do not support a role in PD pathogenesis. For p.K1468E observed in patient DR136.1, no segregation data could be obtained due to lack of other patients or relatives in this small family. In family DR103, p.R1483Q did not segregate with disease since it was absent in another first-degree patient. In family DR108, the patient was diagnosed with early onset PD at age 33 years and was heterozygous for p.R1325Q. His mother, however, had only a mild tremor at age 79 years. She unexpectedly carried two mutant alleles though on a different haplotype. She refused further medical examination preventing a more accurate clinical diagnosis of the parkinsonian nature of her rest tremor. We cannot exclude that the lack of cosegregation in these families was due to reduced age penetrance of PD. However, in silico analyses were supporting a role as benign polymorphism rather than causal mutation for all three novel variants in the Roc domain. The occurrence of benign coding polymorphisms has been reported earlier for LRRK2 (overview in Mata et al25), and supports the idea that some missense mutations do not drastically affect protein function. Indeed, SIFT analyses demonstrated that the three new LRRK2 variants in the Roc domain, p.K1468E, p.R1483Q and p.R1325Q, are most likely tolerable as is true for another previously reported coding polymorphism in the Roc domain, p. R1398H (rs7133914).
In the exons encoding the kinase domain, we identified only one novel missense mutation (p.Y2189C) located 46 amino acids downstream of the kinase domain. Despite the fact that p.Y2189C was allocated a dbSNP ID (rs35658131) based on a screening in carcinoma DNAs, there were no reports on its presence in healthy control individuals. Moreover, p.Y2189C was not found in 556 Belgian control chromosomes and was never reported in relation to neurodegenerative disorders before. In silico analyses implied that this position may be important for the structure of the protein. Indeed, SIFT analyses provided evidence that p.Y2189C is deleterious for LRRK2 function. Together, these data suggest a potential role for p.Y2189C in the etiology of PD. Further functional analyses will be necessary to pinpoint its exact role in the disease mechanism.
More than 20 LRRK2 mutations have been reported to date,12 but pathogenicity of only six has been confirmed through segregation analysis (p.I1122V, p.R1441C, p.R1441G, p.Y1699C, p.G2019S and p.I2020T). This together with our findings indicates that it will be extremely important to obtain functional data establishing the true pathogenic nature of these mutations. This will also contribute to a more accurate estimate of the frequency of pathogenic mutations and benign polymorphisms in LRRK2.
In conclusion, we identified five distinct missense mutations in a large PD group, four of which were novel (p.R1325Q, p.K1468E, p.R1483Q and p.Y2189C). We demonstrated that p.R1441C is a frequent cause of PD in Flanders-Belgium due to a founder effect. Of interest is that we did not observe p.G2019S known as the most common mutation in LRRK2 in PD in most countries. In our opinion, screening of unrelated PD patients for LRRK2 mutations is necessary to increase our understanding of their role in the disease mechanism. Also, a straightforward functional assay will be needed to unambiguously determine the pathogenic character of any novel mutation, particularly when conclusive genetic data are lacking.
References
Polymeropoulos MH, Lavedan C, Leroy E et al: Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997; 276: 2045–2047.
Kitada T, Asakawa S, Hattori N et al: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392: 605–608.
Leroy E, Boyer R, Auburger G et al: The ubiquitin pathway in Parkinson's disease. Nature 1998; 395: 451–452.
Valente EM, Abou-Sleiman PM, Caputo V et al: Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304: 1158–1160.
Bonifati V, Rizzu P, van Baren MJ et al: Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003; 299: 256–259.
Zimprich A, Biskup S, Leitner P et al: Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004; 44: 601–607.
Farrer MJ : Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 2006; 7: 306–318.
Di Fonzo A, Rohe CF, Ferreira J et al: A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet 2005; 365: 412–415.
Nichols WC, Pankratz N, Hernandez D et al: Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease. Lancet 2005; 365: 410–412.
Gilks WP, Abou-Sleiman PM, Gandhi S et al: A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet 2005; 365: 415–416.
Bosgraaf L, Van Haastert PJ : Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta 2003; 1643: 5–10.
Mata IF, Wedemeyer WJ, Farrer MJ et al: LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci 2006; 29: 286–293.
Gloeckner CJ, Kinkl N, Schumacher A et al: The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet 2006; 15: 223–232.
West AB, Moore DJ, Biskup S et al: Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 2005; 102: 16842–16847.
West AB, Moore DJ, Choi C et al: Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 2007; 16: 223–232.
Ito G, Okai T, Fujino G et al: GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry 2007; 46: 1380–1388.
Kachergus J, Mata IF, Hulihan M et al: Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 2005; 76: 672–680.
Lesage S, Leutenegger AL, Ibanez P et al: LRRK2 haplotype analyses in European and North African families with Parkinson disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet 2005; 77: 330–332.
Ozelius LJ, Senthil G, Saunders-Pullman R et al: LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med 2006; 354: 424–425.
Zabetian CP, Hutter CM, Yearout D et al: LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: evidence of two distinct founding events beginning two millennia ago. Am J Hum Genet 2006; 79: 752–758.
Wszolek ZK, Pfeiffer B, Fulgham JR et al: Western Nebraska family (family D) with autosomal dominant parkinsonism. Neurology 1995; 45: 502–505.
Di Fonzo A, Tassorelli C, De Mari M et al: Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson's disease. Eur J Hum Genet 2006; 14: 322–331.
Gaig C, Ezquerra M, Marti MJ et al: LRRK2 mutations in Spanish patients with Parkinson disease: frequency, clinical features, and incomplete penetrance. Arch Neurol 2006; 63: 377–382.
Goldwurm S, Di Fonzo A, Simons EJ et al: The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson's disease and originates from a common ancestor. J Med Genet 2005; 42: e65.
Mata IF, Kachergus JM, Taylor JP et al: Lrrk2 pathogenic substitutions in Parkinson's disease. Neurogenetics 2005; 6: 171–177.
Zabetian CP, Samii A, Mosley AD et al: A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology 2005; 65: 741–744.
Pankratz N, Pauciulo MW, Elsaesser VE et al: Mutations in LRRK2 other than G2019S are rare in a north American-based sample of familial Parkinson's disease. Mov Disord 2006; 21: 2257–2260.
Tan EK, Skipper L, Chua E et al: Analysis of 14 LRRK2 mutations in Parkinson's plus syndromes and late-onset Parkinson's disease. Mov Disord 2006; 21: 997–1001.
Gosal D, Lynch T, Ross OA et al: Global distribution and reduced penetrance: Lrrk2 R1441C in an Irish Parkinson's disease kindred. Mov Disord 2007; 22: 291–292.
Mata IF, Taylor JP, Kachergus J et al: LRRK2 R1441G in Spanish patients with Parkinson's disease. Neurosci Lett 2005; 382: 309–311.
Paisan-Ruiz C, Saenz A, Lopez dM et al: Familial Parkinson's disease: clinical and genetic analysis of four Basque families. Ann Neurol 2005; 57: 365–372.
Di Fonzo A, Wu-Chou YH, Lu CS et al: A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson's disease risk in Taiwan. Neurogenetics 2006; 7: 133–138.
Tan EK, Zhao Y, Skipper L et al: The LRRK2 Gly2385Arg variant is associated with Parkinson's disease: genetic and functional evidence. Hum Genet 2007; 120: 857–863.
Pals P, Van Everbroeck B, Grubben B et al: Case–control study of environmental risk factors for Parkinson's disease in Belgium. Eur J Epidemiol 2003; 18: 1133–1142.
Engelborghs S, Dermaut B, Goeman J et al: Prospective Belgian study of neurodegenerative and vascular dementia: APOE genotype effects. J Neurol Neurosurg Psychiatr 2003; 74: 1148–1151.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Ng PC, Henikoff S : SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814.
Brouwers N, Nuytemans K, van der Zee J et al: Clinical expression of progranulin in an extended founder family includes Alzheimer and Parkinson diagnoses. Arch Neurol 2007; 64: 1436–1446.
Cruts M, Gijselinck I, van der ZJ et al: Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006; 442: 920–924.
van der Zee J, Rademakers R, Engelborghs S et al: A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain 2006; 129: 841–852.
Brouwers N, Sleegers K, Engelborghs S et al: Genetic risk and transcriptional variability of amyloid precursor protein in Alzheimer's disease. Brain 2006; 129: 2984–2991.
Paisan-Ruiz C, Lang AE, Kawarai T et al: LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology 2005; 65: 696–700.
Hernandez DG, Paizan-Ruiz C, McInerney-Leo A et al: clinical and positron emission tomography of Parkinson's disease caused by LRRK2. Ann Neurol 2005; 57: 433–456.
Khan NL, Jain S, Lynch JM et al: Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 2005; 128: 2786–2796.
Acknowledgements
We are grateful to the participants of this study for their kind cooperation and to the personnel of the VIB – Genetic Service Facility (http://www.vibgeneticservicefacility.be) and the Biobank of the Institute Born-Bunge. This research was supported by the Special Research Fund of the University of Antwerp, the Fund for Scientific Research Flanders (FWO-F), the Institute for Science and Technology – Flanders (IWT-F) to CVB, the Medical Research Foundation Antwerp and Neurosearch Antwerp to PPDD and the Interuniversity Attraction Poles program P6/43 of the Belgian Science Policy Office (CVB and PPDD). The IWT-F provided a PhD fellowship to KN; RR, JT and SE are holders of a postdoctoral fellowship of FWO-F, Belgium.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Nuytemans, K., Rademakers, R., Theuns, J. et al. Founder mutation p.R1441C in the leucine-rich repeat kinase 2 gene in Belgian Parkinson's disease patients. Eur J Hum Genet 16, 471–479 (2008). https://doi.org/10.1038/sj.ejhg.5201986
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201986
Keywords
This article is cited by
-
Is inhibition of kinase activity the only therapeutic strategy for LRRK2-associated Parkinson's disease?
BMC Medicine (2012)
-
TMEM106B a Novel Risk Factor for Frontotemporal Lobar Degeneration
Journal of Molecular Neuroscience (2011)
