
Abstract
With uniparental disomy (UPD), the presence in a diploid genome of a chromosome pair derived from one genitor carries two main types of developmental risk: the inheritance of a recessive trait or the occurrence of an imprinting disorder. When the uniparentally derived pair carries two homozygous sequences (isodisomy) with a duplicated mutant, this ‘reduction to homozygosity’ determines a recessive phenotype solely inherited from one heterozygote. Thus far, some 40 examples of such recessive trait transmission have been reported in the medical literature and, among the current 32 known types of UPDs, UPD of chromosomes 1, 2, and 7 have contributed to the larger contingent of these conditions. Being at variance with the traditional mode of transmission, they constitute a group of ‘Mendelian outlaws’. Several imprinted chromosome domains and loci have been, for a large part, identified through different UPDs. Thus, disomies for paternal 6, maternal 7, paternal 11, paternal and maternal 14 and 15, maternal 20 (and paternal 20q) and possibly maternal 16 cause as many syndromes, as at the biological level the loss or duplication of monoparentally expressed allele sequences constitutes ‘imprinting rights infringements’. The above pitfalls represent the price to pay when, instead of a Mendelian even segregation and independent assortment of the chromosomes, the fertilized product with a nondisjunctional meiotic error undergoes correction (for unknown or fortuitous reasons) through a mitotic adjustment as a means to restore euploidy, thereby resulting in UPD. Happily enough, UPDs leading to the healthy rescue from some chromosomal mishaps also exist.
Similar content being viewed by others


Part 1
Preamble
Mendelian recessive outlaws are traits inherited outside the undisputed Mendel laws, which are the even segregation and independent assortment of alleles in germ cells. In uniparental disomy (UPD), by contrast, the meiotic mis-segregation of alleles on a chromosome pair is followed, in general, by a revised early mitotic balancing reassortment. Such a reassortment, to be conducive to UPD, resorts to the loss of the normally inherited member of a trisomy or, more rarely, to the duplication of the lone member of a monosomy. If all ends well, the euploid status is restored but one of the 23 pairs lacks the other parent's partner. In rarer situations, instead of a dual, that is meiotic and mitotic compensating mistake, two meiotic errors, one in the fertilizing germ cell of each sex, complement each other. Although hypothetical, the latter probability served as the basis of the UPD concept, by taking into account the high rate of gametal aneuploidy in humans.
Introduction
In situations of grief, people who consult genetic clinics deserve as much truth and solace as relevant scientific information permits. When in consultation with couples suffering from repeated pregnancy loss, I found that the would-be parents were relieved to learn that fully half of first trimester miscarriages are the result of a chromosomal aberration incompatible with life or with normal development. Sitting around the table with a karyotype at hand, I would then unwearyingly, time and again, spell out the nature of such random mishaps, one-fifth being found to be 45,X; one-half with a trisomy, chiefly for chromosomes 16, 15, 21 and 22; some others with polyploidy, all nearly lethal except for a contingent of trisomies 21 and a few 45,X.
So often had I heard myself telling the habitual-aborters consulting me about this high incidence of certain anomalies in their type of problem that, in the end, a question dawned on me: what if, at fertilization, on occasion, a gamete with a disomy would meet one with a nullisomy for a same chromosome? Meiotic nondisjunction, whether it occurs in the ovary or in the testis, should produce complementary germ cells, one nullisomic and the other disomic for the chromosome in question. It follows that two abnormal germ cells, aneuploid for the same chromosome, might occasionally complement each other at fertilization. If viable, defective but complementary gametes could then, by chance, fertilize one another and proceed towards development with one of the 23 chromosome pairs derived from one parent. With this in mind, I called UPD this presumed and occult presence of a pair inherited from only the mother or father, in a diploid conceptus.1
For a long time, the above thoughts fascinated me and haunted my sleep. So much so that in the end, one evening in June 1979, a Saturday night, I sat home at my dining room table to tell that story. However, no need then for any handy reprint, since abortion studies alone, among them the work of Hassold et al,2 were to serve as the groundwork of this early summer dream. It so happened that, at the end of that night, on Sunday, I was still sitting at that table, putting the final dot on an initial draft. Months later, after the due process of peer review, the final draft would be downsized to the point that the thrust of the remaining idea was that complementary errors could result in a seemingly euploid, normal zygote, with one whole pair derived from one parent (UPD). Such a pair could potentially carry lots of homozygous sequences (a case of so-called isodisomy), depending on the rate and level of crossing over and the meiotic stage at which mis-segregation had occurred. If so, by inheriting a (paternal) XY pair, male-to-male transmission of an X-linked mutant could happen; duplication of an autosomal recessive allele could cause a trait to be inherited from a single carrier parent and so on (see below). I dismissed the thought that, besides complementation, the secondary loss, at mitosis, of the normally inherited member of a trisomy would also result in a pattern of diploidy including a uniparental pair because, in most instances, this should have led, in my view, to a detectable mosaic pattern. Such a mechanism, however, later proved to be true, with the aneuploid cell line apparently often confined to the placenta and somatically undetected or inexistent (confined placental mosaicism, CPM).3
Another thought that was initially put down in the draft was obscurely present in my mind. In hindsight, I realize that it was implicitly relying on the notion of genomic imprinting, at that time totally alien to my mind. As is now well known, the process of genomic imprinting ends up in a selectively biased expression of maternal or paternal alleles at some loci or domains of the offspring genome. This evidence stemmed from the work of Lyon,4 Searle and Beechey5 and was indeed known from publications in ‘mouse language’. Thus, in 1978, one could read the following apodictical statement:5 ‘The possibility that haploid expression of particular maternal or paternal genes is important for normal mouse development is discussed’. However, I was not then personally conversant with that murine language and I remained basically ignorant of the above notion. Yet, I had my own questions regarding the somatic effects of a pair of parental chromosomes not properly channelled to a zygote. Sex has always fascinated me and I was then wondering what could developmentally occur were a boy to get his X, as well as the Y chromosome from his father. Or a girl, her X pair from only her father. It had also occurred to me, and I had referred to it in my initial drafts that, maybe, an ultimately undue process of uniparental autosomal transmission might account as well for some not yet explained idiopathic syndromes, for instance the Cornelia-de-Lange syndrome, whose mode of transmission was then so poorly understood. However,, at the time, I did remove this statement from the article, as a Reviewer objected that the condition did not result from a pair of recessive alleles and also because I found myself unable to process the proposed thought into a scientifically referenced statement. Yes, indeed, Professor ten Kate, there also is fascination in being a reviewer of a scientific article for a noted journal!
Publication of the UPD article
Once masterly edited by John Opitz, and accepted after a long wait for publication in the American Journal of Medical Genetics in 1980, the paper1 slept on a shelf for several years, for want of the molecular developments which would make it possible to tell the parental origin of the chromosomes from their DNA polymorphisms. Some of the main techniques now in use have been reviewed in our book.6
The broad features of a genotype with UPD are as follows:
-
1)
It has 46 chromosomes which can be sorted out in 23 normal looking pairs, baring a translocation;
-
2)
22 of the 23 pairs have been inherited normally;
-
3)
One pair has been derived from one parent only;
-
4)
This exceptional pair may result from three main mechanisms, namely:
-
a)
trisomy rescue, through early mitotic loss of one of the three chromosomes;
-
b)
monosomy duplication, through doubling of the lone chromosome of a monosomy;
-
c)
gamete complementation, when a germ cell, by chance, has the surplus of what lacks in the other;
-
a)
-
5)
Alleles at the uniparental loci may all be heterozygous (heterodisomy), homozygous (isodisomy) or a mix of both, depending on the nondisjunctional mechanisms and the mode of chromosome reassortment restoring the 2n state;
-
6)
Segmental UPD involving only some part of a chromosome pair can occur as a result of equal somatic crossing over.
UPD types currently documented
The first clinically recognized case of UPD was presented at the 1987 ASHG annual meeting, published in 1988 by Arthur Beaudet's group7 and commented upon in an editorial.8 This was the story of a mentally normal 16-year-old girl, menarch at 14, who measured only 130 cm, with some physical asymmetry suggestive of the Silver–Russell syndrome. She suffered from cystic fibrosis, although the father was homozygous for alleles distinct from hers on chromosome 7. The use of a number of RFLPs and centromeric alphoid probes for number 7 confirmed an absence of paternal alleles and showed full homozygosity of the patient's DNA. In short, ‘…the uniparental origin of the centromere, the lack of heterozygosity and failure to demonstrate mosaicism led to the conclusion that there had been duplication of a maternal chromosome 7 in a monosomic conception’ of biologically unquestionable parentage. This first report of a case considered as most exceptional8 was to be followed by a number of others dealing with this and a number of other chromosome pairs as well.
In theory, uniparental derivation of an entire chromosome pair offers a chance of 47 possibilities made of the 22 autosomes and X in each sex plus the XY paternal duplet. From the 1987 to 1988 period that started with the Spence et al,7 initial publication and a remarkable case of Créau-Goldberg et al,9 some three to six new types of UPDs were biannually described until 1997–1998, comprising from two to four paternal and/or one to five maternal ones (Figure 1 and Table 1). In all, they represent the 32 (out of 47) known examples of their type for a whole pair (holo-uniparental disomy) as opposed to the untold number of biparental pairs having acquired a monoparental segment (see below).

Chronology of the first identification of each of the 32 known maternal and paternal UPD types.
Table 2 displays the chromosome pairs with UPD, identified or not yet identified in either sex, and gives a rough estimate of the relative clinical frequencies for the various pairs involved.
Reduction to homozygosity: the ‘Mendelian outlaws’!
These outlaws are a by-product of isodisomy and will only cause trouble when one parental duplicated allele is a mutant. In heterodisomy, the whole length or a portion of a uniparentally inherited pair is heterozygous. In isodisomy, the loci of both pair members are homozygous on the entire length or on a part of the pair. At the extremes, complete heterodisomy will occur when one parental pair, not reshuffled by crossing over (nulli-chiasmatic) and nondisjoined at the first meiotic division, is inherited as such, after a normal meiosis 2. In wholesale isodisomy, meiosis 2 nondisjunction of a meiosis 1 (me1) nulli-chiasmatic pair, as well as the duplication of a monosomic chromosome at early mitosis, may cause both members to be carbon copies of each other. If so, all loci of such pairs are homoallelic, homozygous and thus isodisomic. Ruling out the above mechanisms, a number of uniparental pairs are a mix of hetero- and iso-disomy segments. In any event, as crossing over does not take place in juxta-centromeric areas, the latter remain heterodisomic in UPD pairs nondisjoined at me1 and isodisomic in pairs not disjoined at meiosis 2 (me2), which allows them to be distinguished from each other. It is thus obvious that the genotypic risk of isodisomy is the transmission of a clinically significant mutant allele on a duplicated chromosome or chromosome segment. As a result, some 40 examples of homozygosity for a recessive mutant present in only one parent have been published in medical literature and are presented in chromosome numerical order in Table 3.
It can be seen that isodisomy as a cause for homozygosity of a recessive mutant is less rare for numbers 1, 7 and 2. With respect to chromosome 1, five maternal and eight paternal such instances are on record. Chromosome 7 provides six examples of clinically significant reduction to homozygosity – four maternal, two paternal. As to chromosome 2, it is seen five times, caused by two maternal and three paternal UPDs. Most other examples of such intimidating recessive outlaws are singular! Their high recurrence on chromosome 1 is fascinating because one hardly knows of a single case of trisomy 1 among first trimester abortuses so that all such derived number 1 disomies must result from a very early rescue of almost unviable conceptuses, as is notoriously the case for nearly all monosomic ones as well, hardly seen in abortion products, with an exception for some X monosomies.
In general, one might presume that, with a higher number of loci, the larger chromosomes such as 1 and 2, in isodisomy, contribute to a commensurate display of their recessive mutations. Yet, one remains puzzled by the very fact that the (nonsense!) mutations of the LAMB3 locus on chromosome 1q32.2, encoding the subunit polypeptide gamma 2 of lamin 5, account for three of the 13 recorded instances of reduction to homozygosity of that member!31, 45, 48 The relatively high incidence of cystic fibrosis, for which four of six UPDs seven are responsible7, 55, 56, 57 may be biased as a result of the many segregation studies carried out for counselling in CF families, increasing the chances of detecting such events. All these disparate remarks point to the fact that the nondisjunctional meiotic mechanisms, central to the occurrence of the isodisomy process, vary strongly among the different chromosome members as shown for some on Table 4, inspired from Jacobs and Hassold.64
Suffice it to say that key differences exist in the ways our chromosomes may go astray. Thus, so common among early abortuses, trisomies 16 feature an exclusive, almost totalitarian display of ovarian me1 errors. In contrast, trisomies 18 are not always of maternal origin and are caused, in the majority, by an me2 nondisjunction (reviewed by Jacobs and Hassold64). The implications for UPD16 may be straightforward: the noted frequency of trisomy 16, its unique and constant maternal origin, and the reduced recombination going along with me1-generated disomies64 should explain the relatively high frequency of UPD16 mat void of isodisomy (reviewed by Engel and Antonarakis6). And, UPD16pat20, 65 should mainly and perhaps constantly be the rare by-product of maternal 16 meiotic nullisomy. If so, holo-isodisomy would prevail by early mitotic duplication of the lone paternal 16 and could carry the risk of homozygosity for recessive mutants.20
A somewhat similar commentary could hold true for the UPD's 21 derived from a trisomy 21 where the trend seems as follows: the risk of isodisomy is lowered and the chance of heterodisomy maintained if a lack or a dearth of crossing over recombination is a cause for me1 nondisjunction, as is indeed the case66; such a risk is lessened as well if a (proximal) increase in recombination at me1 favours me2 nondisjunction, which has also been well documented.67 We see therefore that the mood of chromosome 21, in pathology, is against isodisomic deviancy and does not at all favour the works of ‘recessive outlaws’! In addition, UPD21 is clinically innocuous at the difference of UPD16 mat (reviewed by Engel and Antonarakis6) and the pair presents a relatively small number of genes up for recessive mutations. Detecting UPD21 with present means will thus remain quite hard, except in cases where an isochromosome 21, de novo or transmitted, calls attention to it.9 A different pattern of conversion to UPD is seen for other chromosome members of somewhat different pathologic behaviour. Our review of the situation in UPD1, maternal or paternal, emphasizes again the major role of maternal meiosis errors. This is no surprise since most known cases have been uncovered on the basis of a reduction to homozygosity leading mainly to recessive traits (Table 3).
From this clinically biased sample, it would appear that the chromosome1 Mendelian outlaws stem not only from the rescue of trisomies caused by maternal meiotic nondisjunction31, 40, 42 but also from maternal nullisomies complemented by paternal monosomy duplications,38, 44, 45, 46, 47 a mechanism much rarer for chromosomes 18 and 21, as already discussed. Besides, a paternal meiotic involvement at the origin of these cases is also rather surprising, with 2n genotypes presumably arising from the correction of paternally induced trisomies35, 49 or from mitotic complementations of the monosomies resulting from a meiotic loss of the paternal member.41, 43
A survey of and a personal contribution to published maternal UPD7 cases68 produced as many cases of heterodisomy after presumed maternal trisomy rescue (12) as cases of holo-isodisomy (11). In my view, although other mechanisms may be considered, the isodisomy group is best explained by maternal complementation of paternal gamete nullisomy. If so, one should also assume that paternal germ cells with chromosome 7 nondisjunction and disomy are as numerous as their nullisomic counterparts and will contribute to trisomy 7 conceptuses, which they may well do, although survival to birth is nil.64 Cases of paternal UPD7 appear rarely in medical literature and may show isodisomy, to make up for maternal nullisomy. The redundance of isodisomy among UPD7 pairs, the size of the chromosome, the presence of a CF mutation on one for every 40–50 such members and the contribution of maternal UPD7 to the Silver–Russell phenotype all guarantee that this UPD will be relatively frequently met in the clinical field when looked for in the laboratory.
As a final example, we shall also trace some of the pathologic features of chromosome 15 aneuploidy to examine their bearing on the allelic assortment of the uniparental derivatives. Trisomies 15 are not so uncommon in spontaneous abortuses, being next in frequency to trisomies 16, 21 and 22.64 They are sublethal. Three-quarters of them result from maternal me1 nonsegregation events and, as for chromosome 21, are aided by a lack or a dearth of recombination. Close to 10% are of mitotic origin,69 so that roughly one-third, or 3% of all 15 trisomies may contribute to maternal or paternal isodisomy with the implied risk of a recessive trait from a mutant. Given the high contribution of maternal meiotic nondisjunction to trisomy 15, most detected rescues will have to represent cases of maternal UPD. It is not therefore surprising that quite a few cases of Prader–Willi syndrome (PWS), caused by the deficit of a paternal 15 imprinted domain, come from this relatively common disomy. Cases of paternal UPD15 are much rarer and result in the Angelman syndrome (AS), owing to the deficit of a maternally active domain on that number. As seen for other members, mitotic duplication of a paternal chromosome is a palliative for a maternal nullisomy owing to a meiotic loss; it is conducive to isodisomy and can cause, as such, a reduction to homozygosity of any present recessive allele. Some cases of UPD15 also occur from a rescued paternal 15 trisomy caused by an me1 error but, all in all, 75–80% UPD15 pat stem from a mitotic segregation error or from a centromeric misdivision with isochromosome formation. Thus, quite evidently, most holo-isodisomies for chromosome 15 are paternal in origin, leading potentially to a recessive trait, aside from causing AS.
The point of this discussion of the ways and means by which chromosomes 1, 21, 7 and 15, taken as examples, behave in pathology is to show that, in the process, their ‘skin changes’ from hetero- to iso-disomy – should UPD occur – depend highly on the chromosome at stake and reflect both the mode of formation involved and, oddly enough, something of the very essence of the particular chromosome involved. This implies that, under the circumstances, some chromosome individuals are much more prone than others to breed Mendelian outlaws. It is hard therefore to state the level of contribution of the various uniparental pairs to the overall occurrence of recessive disorders. As an indication, among some series of recessive conditions studied, such an aetiology was found in one of 61 cases of junctional epidermolysis bullosa of Herlitz, nearly 2%,31 in one of 55 cases of cystic fibrosis, again 2%55 and in two of 54 cases of cartilage–hair hypoplasia, about 4%.33
Segmental UPD
Cases with this type of defect are difficult to expose, as a segment only of an otherwise biparental pair is affected. This segment may be terminal or interstitial and results from a balanced somatic crossing over. A terminal exchange occurs with one single symmetrical break, whereas, when interstitial, the balanced homologous exchange requires two such breaks. The reasons for these types of somatic reshuffling are unknown and hot spots to this effect must exist. Once such a remodelling has happened, a mosaic chromosomal state is achieved with the simultaneous presence of the initial genotype and the genotypes of the two new reciprocal clones issued from the balanced interchange. The archetype of a relatively frequent somatic, mitotic interchange is to be seen in the terminal uniparental paternal segment at or close to 11p15.5, a hallmark in a proportion of patients with the Wiedemann–Beckwith syndrome (WBS).70 Oddly enough, in this case, the maternal 11p15.5 homologous segment, as the counterpart of this exchange, has not been identified and may well be selected against from the tissues and organs in development. So intriguing is the underlying cause of the exchange that, as a rule, tissues of only one of identical twins will undergo this change in early gestation and suffer from the WBS.
Table 5 lists clinical instances of reported segmental UPDs in congenital disorders. Just like complete uniparental pairs, partial ones may be responsible for recessive traits or for various imprinting syndromes. Segmental UPD research probably has a great future as it occasionally happens in association with another holo-disomic UPD pair within a same genome83, 84 or with some other rearrangements,85 and as a part of the clonal evolution of malignancies.86, 87, 88 A full review of these instances would be beyond the scope of this article.
Part 2
Genomic imprinting amounts to a silencing of the expression of certain genes or domains through a reversible methylation process and other secondary biochemical changes of the DNA blueprint, as a mark of the parental sex. The end result is a functional hemizygosity (maternal or paternal) for the loci of some domains. ‘Imprinting copyrights infringements’ are lawless acts of nature whereby allelic expression contravenes the expected course of the conventional biological printing press medium! The complex normal printing machine is under the control of an imprinting centre and a relaxation of the imprint normally occurs in early gametogenesis, to be selectively reinstated afterwards as a function of the parental sex, according to some prescribed developmental timing for different tissues and organs. The chromosomal by-pass of one of the two parental channels of transmission in UPD perturbs the above functional adjustments of allelic expression where relevant. In such cases, the presence of a second homologue from the same genitor is no substitute for that missing from the other genitor, as far as the specifically imprinted domains are concerned.
Besides reduction to homozygosity, the genotypic risk of carrying UPD pairs with imprinted domains results from the eventual loss or duplication of allelic expression by the offspring. Just as had happened with the first clinical report of UPD7, the interest in the subject matter took another turn with the report of the first case of UPD as an imprinting disorder.10 This initial observation was that connecting UPD15 mat to the PWS.
The fascination for the genetics of UPD grew further with this and other reports confirming an intimate link between certain pairs and disturbances of the ordinances of genomic imprinting. It had been known for some years that a tiny deletion in 15q11–q13 was a frequent cause of PWS.89 Sometime later, it appeared that such a deletion would cause two distinct syndromes, AS versus PWS, when affecting, respectively, the maternal or paternal chromosome 15.90 It was now being seen that the paternal counterpart for UPD15 mat also confirmed this dichotomy, by causing AS, not PWS. In short, a second maternal or paternal 15 in a pair could not substitute for the missing partner! Undoubtedly, even intact, chromosomes 15 mat and 15 pat did not express the same genetic message: they thus were imprinted and the hemizygously expressed domain must also lie in the area where the deletion could occur and cause a similar damage. We now know that chromosome 15 has two adjacent imprinted domains, one of several complex expressed alleles on paternal 15, the other more distal with active alleles on the maternal contiguous sequence. With time it would be learned that the lack of expression of paternal alleles in UPD15 mat accounted for some 25% of PWS cases, whereas UPD, as a rule with isodisomy, for the paternal 15 segment at q11–q13, accounts for about 2% of AS only.6 After these essential findings, other UPD pairs or segments causing various imprinted syndromes were documented, and the main ones shown on Table 6.
These various pairs appear to delineate two types of situations, one where other genomic alterations may also cause the condition, and another type in which the newly identified UPDs point to syndromes until then not recognized, as is seen for the maternal and paternal variants of UPD14 and maternal UPD16.
As shown on Table 6, today nine full-blown UPDs cause ‘imprinting copyrights infringements’. Wholesale UPD20 mat and (segmental) 20q pat specifically alter the activities of the Neuronatin96 and GNAS97 alleles, respectively, paternally and maternally expressed. Curiously, at the difference of other chromosomes such as maternal and paternal 15 or paternal 11, etc., the imprinted genes of chromosome 20 are not part of domains and represent isolated exceptions amidst biparentally active loci, the so-called microimprinting.96
On Table 7 are shown the frequencies of UPD contributing to the aetiologies of these syndromes.
The approximate population frequencies of UPD pairs in the aetiology of major imprinted syndromes are detailed in Table 8.
Making some assumptions, an approximate population frequency of UPD as an aetiology of major imprinted syndromes is presented. As an example, let us refer to the PWS phenotype, which is in general viable and may know a clinical frequency of one in 20 000 live births. Considering that maternal UPD15 serves as an aetiology for some 25% of the cases, one may infer that this maternal UPD occurs around once every 80 000 live births. The same type of calculation can also help to approach an estimate for several other UPDs, at the origin of a proportion of syndromic conditions of rather well-documented frequencies.
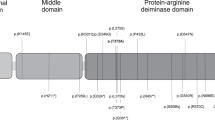
It would be beyond the scope of this clinical overview to inventory the specific loci subject to the imprinting of paternal or maternal chromosomes of major clinical interest, but Figure 2 is an attempt at showing some of the main ones involved. The interested reader may refer to some attuned reviews available on the Internet.

Major imprinted areas involved in clinical disorders (NB: GRB10 and MEG1 are localized on 7p).
I have not so far written about some cytogenetic suggestive oddities met in the laboratory search to unmask UPD, namely translocations, isochromosomes or markers, such as, to quote but a few isolated examples:
-
a)
one paternal 13/14 balanced translocation which as a back-up brings about a 14/14 maternal isochromosome in the offspring, as a substitute for the paternal nullisomy segregating from this rearrangement98;
-
b)
a monosomy rescue which resorts to a paternal isochromosome of one arm – i(7p) pat – and a maternal one for the other – i(7q) mat – to restore euploidy, thereby setting up a mixed (paternal and maternal) segmental UPD in a same genome99;
-
c)
a tiny centromeric marker which represents either a minimal leftover of the rescued nearly nullisomic culprit or a remnant of the not quite vanished member of a trisomy!6 The result is a flag for either trisomy or monosomy rescue!
Rescue
The word rescue comes back quite often in any discussion of the mitotic twists involved in the return to a cytogenetically balanced development imposing a uniparental pair! Poor salvage, indeed, when it relies on biological outlaws or imposters that too often disseminate recessive traits or ontogenic derailments. Indeed, on the path to euploid rescue, four things may happen:
-
a)
rescue at the cost of a recessive trait,
-
b)
rescue at the cost of an imprinting disorder,
-
c)
rescue at the cost of both (rarely!),
-
d)
true, healthy rescue.
Leo, with my wholehearted best wishes to a distinguished retiree, the following quotes hopefully constitute two fascinating offerings for your appreciation. In one, a pattern of homologous centric fusion for chromosome 22 is found in a woman who aborts 10 times in a row before producing a normal female offspring who will in turn miscarriage several times,100 Figure 3.

Habitual abortion and translocation (22q;22q): unexpected transmission from a mother to her phenotypically normal daughter (published with permission of the Authors).
In another example, a homologous 13/13 centric fusion (or an isochromosome 13q) is found in a balanced woman without an inherited maternal 13,24 Figure 4.

Trisomy 13 rescue or disomic gamete 13 complementation over two generations.
In this figure, the habitual aborter exemplifies a case of paternal UPD13. In the end, she produces a balanced male offspring, born after five spontaneous abortions. Her balanced offspring, with the inherited 13/13 fusion of the mother and no paternal 13 is an example of maternal UPD13,29 the second case over two generations of this family! A true miracle.
I shall now close with some nagging (and fascinating) questions to be resolved in the areas of UPD and genomic imprinting6: What is the spectrum of all possible UPD types? What is the frequency of UPD? What is the extent and role of segmental UPD? How can the cytologic mechanisms involved in UPD be elucidated? What is the level of implication of UPD in recessive disorders? How can we improve the diagnostic tests for fast detection of UPD? What is the evolutionary significance of genomic imprinting?
References
Engel E : A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet 1980; 6: 137–143.
Hassold TJ, Matsuyama A, Newlands IM et al: A cytogenetic study of spontaneous abortions in Hawaii. Ann Hum Genet (London) 1978; 41: 443–454.
Kalousek DK, Barrett IJ, Gartner AB : Spontaneous abortion and confined chromosomal mosaicism. Hum Genet 1992; 88: 642–646.
Lyon MF, Glenister PH : Factors affecting the observed number of youngs resulting from adjacent-2 disjunction in mice carrying a translocation. Genet Res 1977; 29: 83–92.
Searle AG, Beechey CV : Complementation studies with mouse translocations. Cytogenet Cell Genet 1978; 20: 282–303.
Engel E, Antonarakis SE : Genomic Imprinting and Uniparental Disomy in Medicine (Clinical and Molecular Aspects). Wiley-Liss Inc.: New York, 2002, pp 1–285.
Spence JE, Perciaccante RG, Greig GM et al: Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet 1988; 42: 217–225.
Warburton D : Uniparental disomy: a rare consequence of the high rate of aneuploidy in human gametes. Am J Hum Genet 1988; 42: 215–216.
Créau-Goldberg N, Gegonne A, Delabar J et al: Maternal origin of a de novo balanced t(21q21q) identified by ets-2 polymorphism. Hum Genet 1987; 76: 396–398.
Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M : Genetic imprinting suggested by maternal heterodisomy in non deletion Prader–Willi syndrome. Nature 1989; 342: 281–285.
Vidaud M, Kitzis A, Ferec C et al: Father to son transmission of hemophilia A due to uniparental disomy. Am J Hum Genet 1989; 59: A 226 (abstract).
Welch TR, Beischel LS, Choi E, Balakrishnan K, Bischof NA : Uniparental isodisomy 6 associated with deficiency of the fourth component of complement. J Clin Invest 1990; 86: 675–678.
Grundy P, Telzerow P, Paterson MC et al: Chromosome 11 uniparental isodisomy predisposing to embryonal neoplasms. Lancet 1991; 338: 1079–1080 (letter).
Lindenbaum RH, Woods CG, Norbury CG, Povey S, Rysiecki G : An individual with maternal disomy of chromosome 4 and iso4p, iso4q. Am J Hum Genet 1991; 49: 285 (abstract).
Temple IK, Cockwell A, Hassold T, Pettey D, Jacobs P : Maternal uniparental disomy for chromosome 14. J Med Genet 1991; 28: 511–514.
Wang JC, Passage MB, Yen PH, Shapiro LJ, Mohandas TK : Uniparental heterodisomy for chromosome 14 in a phenotypically abnormal familial balanced 13/14 Robertsonian translocation carrier. Am J Hum Genet 1991; 48: 1069–1074.
Malcolm S, Clayton-Smith J, Nichols M et al: Uniparental paternal disomy in Angelman's syndrome. Lancet 1991; 337: 694–697.
Bennett P, Vaughan J, Henderson D, Longhna S, Moore G : Association between confined placental trisomy, fetal uniparental disomy and early intra-uterine growth retardation. Lancet 1992; 340: 1284–1285 (letter).
Blouin JL, Avramopoulos D, Pangalos C, Antonarakis SE : Normal phenotype with paternal uniparental isodisomy for chromosome 21. Am J Hum Genet 1993; 53: 1074–1078.
N'go KY, Lee J, Dixon B, Liu D, Jones OW : Paternal uniparental iso-disomy in a hydrops fetalis, alpha-thalassemia fetus. Am J Hum Genet 1993; 53: A1207 (abstract).
Schinzel AA, Basaran S, Bernasconi F, Karaman B, Yuksel-Apak M, Robinson WP : Maternal uniparental disomy 22 has no impact on the phenotype. Am J Hum Genet 1994; 54: 21–24.
Brzustowicz LM, Allitto BA, Matseoane D et al: Paternal isodisomy for chromosome 5 in a child with spinal muscular atrophy. Am J Hum Genet 1994; 54: 482–488.
Höglund P, Holmberg C, de la Chapelle A, Kere J : Paternal isodisomy for chromosome 7 is compatible with normal growth and development in a patient with congenital chloride diarrhea. Am J Hum Genet 1994; 55: 747–752.
Slater H, Shaw JH, Dawson G, Bankier A, Forrest SM : Maternal uniparental disomy of chromosome 13 in a phenotypically normal child. J Med Genet 1994; 31: 644–646.
Slater H, Shaw JH, Bankier A, Forrest M, Dawson G : UPD 13: no indication of maternal or paternal imprinting of genes of chromosome 13. J Med Genet 1995; 32: 493.
Harrison K, Eisenger K, Anyane-Yelboa K, Brown S : Maternal uniparental disomy of chromosome 2 in a baby with trisomy 2 mosaicism in amniotic fluid culture. Am J Med Genet 1995; 58: 147–151.
Jones C, Booth C, Rita D et al: Identification of a case of maternal uniparental disomy of chromosome 10 associated with confined placental mosaicism. Prenat Diagn 1995; 15: 843–848.
Miny P, Koppers B, Bogadanova N, Schulte-Vallentin M, Horst J, Dworniczak B : Paternal uniparental disomy 22. Am J Med Genet 1995; 7: 216 (abstract).
Benlian P, Foubert L, Gagné BL et al: Complete paternal isodisomy for chromosome 8 unmasked by lipoprotein lipase deficiency. Am J Hum Genet 1996; 59: 431–436.
Van Den Berg-Loonen EM, Savelkoul P, Van Hooff H, van Eede P, Riesewijk W, Geraedts J : Uniparental disomy 6 in a renal transplant patient. Hum Immunol 1996; 45: 46–51.
Pulkkinen L, Bullrich F, Czarnecki P, Weiss L, Uitto J : Maternal uniparental disomy of chromosome 1 with reduction to homozygosity of the LAMB 3 locus in a patient with Herlitz junctional epidermolysis bullosa. Am J Hum Genet 1997; 61: 611–619.
Piantanida M, Dellavecchia C, Floridia G : Ataxic gait and mental retardation with absence of the paternal chromosome 8 and an idic(8p23.3): imprinting effect or nullisomy for distal 8p genes? Hum Genet 1997; 99: 766–771.
Sulisalo T, Makitie O, Sistonen P et al: Uniparental disomy in cartilage–hair hypoplasia. Eur J Hum Genet 1997; 5: 35–42.
Quan F, Janas J, Toth-Fejel S, Johnson DB, Wolford SK, Pojowich PW : Uniparental disomy of the entire X chromosome in a female with Duchenne muscular dystrophy. Am J Hum Genet 1997; 60: 160–165.
Gelb BD, Willner JP, Dunn TM et al: Paternal uniparental disomy for chromosome 1 revealed by molecular analysis of a patient with pycnodysostosis. Am J Hum Genet 1998; 62: 448–454.
Chudoba I, Franke Y, Senger G et al: Maternal UPD 20 in a hyperactive child with severe growth retardation. Eur J Hum Genet 1999; 7: 533–540.
Genuardi M, Tozzi C, Pomponi MG et al: Mosaic trisomy 17 in amniocytes: phenotypic outcome, tissue distribution, and uniparental disomy studies. Eur J Hum Genet 1999; 7: 421–426.
Thomson DA, McHenry CL, Li Y et al: Retinal dystrophy due to paternal isodisomy for chromosome 1 or chromosome 2, with homoallelism for mutations in RPE65 or MERTK respectively. Am J Hum Genet 2002; 70: 224–229.
Von Eggling F, Hoppe C, Bartz U et al: Maternal uniparental disomy 12 in a healthy girl with a 47,XX, +der(12) (:p11–q11:)/46,XX karyotype. J Med Genet 2002; 39: 519–521.
Field LL, Tobias R, Robinson WP, Paisey R, Bain S : Maternal uniparental disomy of chromosome 1 with no apparent phenotypic effects. Am J Hum Genet 1998; 63: 1216–1220.
Dufourcq-Lagelouse R, Lambert N, Duval M et al: Chediak–Higashi syndrome associated with maternal uniparental disomy of chromosome 1. Eur J Hum Genet 1999; 7: 633–637.
Lebo RV, Shapiro LR, Yosumkaya Tenerci E et al: Rare etiology of autosomal recessive disease in a child with noncarrier parents. Am J Hum Genet 2000; 67: 750–754.
Rothlisberger B, Zerova TE, Kotzot D, Buzhievskaia TI, Balmer D, Schinzel A : Supernumerary marker chromosome (1) of paternal origin and maternal uniparental disomy 1 in a developmentally delayed child. J Med Genet 2001; 38: 885–888.
Chen H, Young R, Mu X et al: Uniparental isodisomy resulting from 46,XX,i(1p) i(1q) in a woman with short stature, ptosis, micro/retrognathia, myopathy, deafness and sterility. Am J Med Genet 1999; 82: 215–218.
Takizawa Y, Pulkkinen L, Chao SE, Nakajima H, Nakano Y, Shimizu Y : Mutation report: complete paternal uniparental isodisomy of chromosome 1: a novel mechanism for Herlitz junctional epidermolysis bullosa. J Invest Dermatol 2000; 115: 307–311.
Miura Y, Hiura M, Torigoe K et al: Complete paternal uniparental isodisomy for chromosome 1 revealed by mutation analyses of the TRKA (NTRK1) gene encoding a receptor tyrosine kinase for nerve growth factor in a patient with congenital insensitivity to pain with anhydrosis. Hum Genet 2000; 107: 205–209.
Indo Y, Mardy S, Miura Y et al: Congenital insensitivity to pain with anhydrosis (CIPA): novel mutations of the TRKA (NTRK1) gene, a putative uniparental disomy and a linkage of the mutant TRKA and PKLR genes in a family with CIPA and pyruvate kinase deficiency. Hum Mutat 2001; 4: 308–318.
Fassihi H, Wessagowit W, Ashton GH et al: Complete paternal uniparental isodisomy of chromosome 1 resulting in Herlitz junctional epidermolysis. Clin Exp Dermatol 2005; 30: 71–74.
Rivolta C, Berson EL, Dryja TR : Paternal uniparental heterodisomy with partial isodisomy of chromosome 1 in a patient with retinitis pigmentosa without hearing loss and a misense mutation in the Usher syndrome type II gene USH2A. Arch Ophtalmol 2002; 120: 1566–1571.
Spiekerkoetter U, Eedes A, Yue Z, Haines J, Strauss AV, Summar M : Uniparental disomy of chromosome 2 resulting in lethal trifunctional protein deficiency due to homozygous alpha-subunit mutations. Hum Mutat 2002; 20: 447–451.
Chavez B, Valdez E, Vilchis F : Uniparental disomy in steroid 5-alpha-reductase2 deficiency. J Clin Endocrinol Metab 2000; 85: 3147–3150.
Petit FM, Gajdos V, Parisot F et al: Paternal isodisomy for chromosome 2 as the cause of Crigler–Najjar type I syndrome. Eur J Hum Genet 2004; 13: 278–282.
Spena S, Duga S, Asselta R et al: Congenital afibrinogenaemia caused by uniparental isodisomy of chromosome 4 containing a novel 15-kb deletion involving fibrinogen alpha-chain gene. Eur J Hum Genet 2004; 12: 891–898.
Spiro RP, Christian SL, Ledbetter DH et al: Intra-uterine growth retardation associated with maternal uniparental disomy for chromosome 6 unmasked by congenital adrenal hyperplasia. Pediatr Res 1999; 46: 510–513.
Voss R, Ben-Simon E, Avital A et al: Isodisomy of chromosome 7 in a patient with cystic fibrosis: could uniparental disomy be common in human? Am J Hum Genet 1989; 45: 373–380.
Spotila ID, Sereda L, Prockop DJ et al: Partial disomy for maternal chromosome 7 and short stature in an individual with a mutation at the Col1A2 locus. Am J Hum Genet 1992; 51: 1396–1405.
Hehr U, Dorr S, Hagemann M, Hansmann I, Preiss U, Bromme S : Silver–Russell syndrome and cystic fibrosis associated with maternal uniparental disomy 7. Am J Med Genet 2000; 91: 237–239.
Pan Y, Christopher D, McCaskill K et al: Paternal isodisomy of chromosome 7 associated with complete situs inversus and immotile cilia. Am J Hum Genet 1998; 62: 1551–1555 (letter).
Tiranti V, Lamantea E, Uziel G et al: Leigh syndrome transmitted by uniparental disomy of chromosome 9. J Med Genet 1999; 36: 927–928 (letter).
Beldjord C, Henry I, Bennani C et al: Uniparental disomy: a novel mechanism for thalasemia major. Blood 1992; 80: 287–289.
Alvarez A, del Castillo E, Pera A et al: Uniparental disomy of chromosome 13q causing homozygosity for the 35delG mutation in the gene encoding connexin26 (GJB2) results in prelingual hearing impairment in two unrelated Spanish patients. J Med Genet 2003; 40: 636–639.
Pentao L, Lewis RA, Ledbetter DH, Patel PI, Lupski JR : Maternal uniparental isodisomy of chromosome 14: association with autosomal recessive red monochromacy. Am J Hum Genet 1992; 50: 690–699.
Woodage T, Prasad M, Dixon JW : Bloom syndrome and maternal uniparental disomy for chromosome 15. Am J Hum Genet 1994; 55: 74–80.
Jacobs PA, Hassold TJ : The origin of numerical chromosome abnormalities. Adv Hum Genet 1995; 33: 101–133.
Kohlhase J, Janssen B, Weidenauer K, Harms K, Bartels I : First confirmed case with paternal uniparental disomy of chromosome 16. Am J Med Genet 2000; 91: 190–191.
Sherman SL, Petersen MB, Freeman SB et al: Non-disjunction of chromosome 21 in maternal meiosis I: evidence for a maternal age-dependent mechanism involving reduced recombination. Hum Mol Genet 1994; 3: 1529–1535.
Lamb NE, Freeman SB, Savage-Austin A et al: Susceptible chiasmate configurations of chromosome 21 predispose to non-disjunction in both maternal meiosis I and meiosis II. Nat Genet 1996; 14: 400–405.
Mergenthaler S, Burger B, Eggermann K et al: Formation of uniparental disomy 7 delineated from new cases and a UPD7 case after trisomy 7 rescue. Presentation of own results and review of the literature. Ann Genet (Paris) 2000; 43: 15–21.
Robinson WP, Kuchinka BD, Bernasconi F et al: Maternal meiosis 1 non disjunction of chromosome 15: dependence of the maternal age effect on level of recombination. Hum Mol Genet 1998; 7: 1011–1019.
Henry I, Bonaiti-Pellie C, Chehensse V et al: Uniparental paternal disomy in a genetic cancer predisposing syndrome. Nature 1991; 351: 665–667.
Erikson M, Brown WT, Gordon LB et al: Recurrent de novo point mutations in Lamin A cause Hutchinson–Gilford progeria syndrome. Nature 2003; 423: 293–297 (letter).
Chevalier-Porst F, Rolland MO, Cochat P, Bozon D : Maternal isodisomy of the telomeric end of chromosome 2 is responsible for a case of primary hyperoxaluria type 1. Am J Med Genet 2005; 132: 80–84.
Yang XP, Inazu A, Yagi K, Kajimani K, Koizumi J, Malbuchi H : Abetalipoproternemia caused by maternal isodisomy of chromosome 4q containing an intron 9 splice acceptor mutation in the microsomial triglyceride transfer protein gene. Artérioscler Thromb Vasc Biol 1999; 19: 1950–1955.
Tomson SWJ, Ruiz-Perez VI, Wright MJ et al: Ellis-van Creveld syndrome resulting from segmental uniparental disomy of chromosome 4. J Med Genet 2001; 38: E18 (letter).
Lopez-Gutierrez AU, Riba L, Ordonez-Sanchez ML, Ramorez-Jimenez S, Cerrillo-Hinojasa M, Tusie-Luna MT : Uniparental disomy for chromosome 6 results in steroid 21-hydroxylase deficiency: evidence of different genetic mechanisms involved in the production of the disease. J Med Genet 1998; 35: 1014–1019.
Das S, Lese M, Song M et al: Partial paternal uniparental disomy of chromosome 6 in an infant with neonatal diabetes, macroglossia, and craniofacial abnormalities. Am J Hum Genet 2000; 67: 1586–1591.
Hannula K, Lipsanen-Nyman M, Kontiokari T, Kere J : A narrow segment of maternal uniparental disomy of chromosome 7q31-qter in Silver–Russell syndrome delimits a candidate gene region. Am J Hum Genet 2001; 68: 247–253.
Eggermann T, Mergenthaler S, Eggermann K, Ranke MB, Wollmann RA : Segmental uniparental disomy of 7q31-qter is rare in Silver–Russell syndrome. Clin Genet 2001; 60: 395–396.
Kotzot D, Rothlisberger B, Riegel M, Schinzel A : Maternal uniparental isodisomy 11q13-qter in a dysmorphic and mentally retarded female with partial trisomy mosaicism 11q13-qter. J Med Genet 2001; 38: 876–881.
Towner D, Yang SP, Shaffer LG : Prenatal ultrasound findings in a fetus with paternal uniparental disomy 14q12-qter. Ultrasound Obstet Gynecol 2001; 18: 268–271.
Nazarenko S, Sazhenova E, Baumer A, Schinzel A : Segmental maternal heterodisomy of the proximal part of chromosome 15 in an infant with Prader–Willi syndrome. Eur J Hum Genet 2004; 12: 411–414.
Bastepe M, Lane AH, Jüppner H : Paternal uniparental isodisomy of chromosome 20q – and the resulting changes in GNAS1 methylation – as a plausible cause of pseudohypoparathyroidism. Am J Hum Genet 2001; 68: 1283–1289.
Eggermann T, Mergenthaler S, Eggermann K et al: Identification of interstitial maternal uniparental disomy (UPD)(14) and complete maternal UPD(20) in a cohort of growth retarded patients. J Med Genet 2001; 38: 86–89.
Tsai AC, Gibby T, Beischel L, McGavran L, Johnson JP : A child with Angelman syndrome and trisomy 13 findings due to associated paternal UPD 15 and segmental UPD 13. Am J Med 2004; 126: 208–212.
de Ravel TJ, Matthijs G, Fryns J : An unbalanced translocation 46, XX,+der(18)t(18;21)(q12.2;q11.2)mat,−21 associated with maternal isodisomy 18pter → 18q12.2. Ann Genet (Paris) 2001; 44: 63–66.
Rousseau-Merck M-F, Versteege I, Legrand I et al: HSNF5/IN11 inactivation is mainly associated with homozygous deletions and mitotic recombinations in rhabdoid tumors. Cancer Res 1999; 59: 3152–3156.
Kralovics R, Guan Y, Prchal JT : Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycytemia vera. Exp Hematol 2002; 30: 229–236.
Raghavan M, Lillington DM, Skoulakis S et al: Genome-wide single nucleotide polymorphism analysis reveals frequent partial uniparental disomy due to somatic recombination in acute myeloid leukemias. Cancer Res 2005; 65: 375–378.
Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD : Deletions of chromosome 15 as a cause of the Prader–Willi syndrome. N Engl J Med 1981; 304: 325–329.
Magenis RE, Toth-Fejel S, Allen LJ et al: Comparison of the 15q deletions in Prader–Willi and Angelman syndrome: specific regions, extent of deletions, parental origin and clinical consequences. Am J Med Genet 1990; 35: 333–349.
Temple IK, Gardue RJ, Robinson DO et al: Further evidence for an imprinted gene for neonatal diabetes localized to chromosome 6q22–q23. Hum Mol Genet 1996; 5: 1117–1121.
Kotzot D, Schmitt S, Bernasconi F : Uniparental disomy 7 in Silver–Russell syndrome and primordial growth retardation. Hum Mol Genet 1995; 4: 583–587.
Antonarakis SE, Blouin JL, Maher J, Avramopoulos D, Thomas G, Talbot CCG : Maternal uniparental disomy for human chromosome 14, due to loss of a chromosome 14 from somatic cells with t(13;14) trisomy 14. Am J Hum Genet 1993; 52: 1145–1152.
Abu-Amero SN, Ali Z, Abu-Amero KK, Stanier P, Moore GE : An analysis of common isodisomic regions in five mUPD 16 probands. J Med Genet 1999; 36: 204–207.
Yong PJ, Marion SA, Barrett IJ, Kalousek DK, Robinson WP : Evidence for imprinting on chromosome 16: the effect of uniparental disomy on the outcome of mosaic trisomy 16 pregnancies. Am J Med Genet 2002; 12: 123–132.
Evans HK, Wylie AA, Murphy SK, Jirtle RL : The neuronatin gene resides in a ‘micro-imprinted’ domain on human chromosome 20q11.2. Genomics 2001; 77: 99–104.
Liu J, Litman D, Rosenberg MG, Yu S, Biesecker LG, Weinstein S : A GNAS imprinting defect in pseudo hypoparathyroidism type 1B. J Clin Invest 2001; 106: 1167–1174.
Tomkins DJ, Roux AF, Waye JL, Freeman VC, Cox DW, Whelan DT : Maternal uniparental isodomy of human chromosome 14 associated with a paternal t(13q14q). Eur J Hum Genet 1996; 4: 153–159.
Eggerding FA, Schonberg SA, Chehab FF, Norton ME, Cox VA, Epstein CJ : Uniparental isodisomy for paternal 7p and maternal 7q in a child with growth retardation. Am J Hum Genet 1994; 55: 253–265.
Kirkels VJ, Hustinx TW, Scheres JM : Habitual abortion and translocation (22q;22q): unexpected transmission from a mother to her phenotypically normal daughter. Clin Genet 1980; 18: 456–461.
Acknowledgements
The author is most indebted to Ms Fabienne Stassen, Ms Dominique Sutter and Mr J-C Malgouyres for technical assistance and gratefully acknowledges his successor, Professor SE Antonarakis for his valued friendship and constant encouragements.
Author information
Authors and Affiliations
Corresponding author
Additional information
This text is the elaboration of a lecture delivered on March 17, 2005, in Amsterdam, in the honour of Professor Leo ten Kate on the occasion of his retirement.
Rights and permissions
About this article
Cite this article
Engel, E. A fascination with chromosome rescue in uniparental disomy: Mendelian recessive outlaws and imprinting copyrights infringements. Eur J Hum Genet 14, 1158–1169 (2006). https://doi.org/10.1038/sj.ejhg.5201619
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201619
Keywords
This article is cited by
-
A cohort study of neurodevelopmental disorders and/or congenital anomalies using high resolution chromosomal microarrays in southern Brazil highlighting the significance of ASD
Scientific Reports (2024)
-
Prenatal diagnosis of mosaic chromosomal aneuploidy and uniparental disomy and clinical outcomes evaluation of four fetuses
Molecular Cytogenetics (2023)
-
Uniparental disomy is a chromosomic disorder in the first place
Molecular Cytogenetics (2022)
-
Prenatal diagnosis of 7 cases with uniparental disomy by utilization of single nucleotide polymorphism array
Molecular Cytogenetics (2021)
-
Long contiguous stretches of homozygosity detected by chromosomal microarrays (CMA) in patients with neurodevelopmental disorders in the South of Brazil
BMC Medical Genomics (2019)
