
Abstract
PUMA, a key mediator of p53-induced apoptosis, is a BH3-only domain proapoptotic protein that localizes to mitochondria and interacts with antiapoptotic Bcl-2 and Bcl-XL. Recent evidence implicates Bax to be an important mediator of PUMA-activated apoptotic signals. We have previously demonstrated that Bax deficiency significantly affects thapsigargin (TG)-mediated endoplasmic reticulum calcium pool depletion-induced apoptosis. We now present evidence that TG upregulates PUMA expression and that although Bax-deficient cells exhibit resistance to TG, Bax deficiency does not attenuate TG upregulation of PUMA expression. Furthermore, TG transcriptionally upregulates PUMA expression in a p53-independent manner and that PUMA-deficient cells are more resistant to undergo TG-induced apoptosis than the PUMA-proficient counterparts. Thus, our results demonstrate that TG engages PUMA and Bax for full transduction of apoptotic signals and both PUMA and Bax appear to exist in the same TG-activated apoptotic pathway in which PUMA may reside upstream of Bax.
Similar content being viewed by others


Introduction
PUMA (p53 upregulated modulator of apoptosis), also known as bbc3 (Bcl-2 binding component 3), is a recently identified p53 downstream target gene.1, 2, 3 It is a direct transcriptional target of p53 that is also regulated by DNA damaging agents in a p53-dependent manner and has also been found to be regulated in response to serum withdrawal as well as treatments with dexamethasone, tunicamycin and thapsigargin (TG).3, 4, 5 PUMA encodes a BH3 only domain proapoptotic protein that localizes to mitochondria and interacts with antiapoptotic Bcl-2 and Bcl-XL (1-3). It is now well established that the tumor suppressor p53 predominantly functions as a transcription factor and mediates its effects by inducing growth arrest and/or apoptosis (reviewed in Sheikh and Fornace Jr,6 Vousden and Lu,7 and Hofseth et al.8). p53 mediates its apoptotic effects by transcriptionally activating the expression of proapoptotic genes, some of which include Bax, NOXA, PUMA and DR5, whose products modulate the intrinsic and extrinsic pathways of apoptosis (reviewed in Hofseth et al.8). Although each p53-regulated proapoptotic protein is believed to play some role in mediating p53-induced apoptosis, PUMA has garnered particular attention in this regard. For example, gene knockout studies have revealed that DNA damage-induced p53-dependent apoptosis is severely diminished in PUMA-deficient cells.9, 10, 11 Based on these findings, it has been proposed that PUMA is a key mediator of p53-dependent apoptosis.
Several lines of recent evidence suggests that Bcl-2 family members such as Bcl-2, Bak and Bax appear to modulate apoptosis that is induced in response to alterations in intracellular Ca2+ homeostasis (Kim et al.,12 Nutt et al.13 and reviewed in Sheikh and Huang14). According to some studies, Bcl-2 by decreasing the ER Ca2+ load and mitochondrial Ca2+ uptake appears to protect cells from apoptosis.15, 16, 17 By contrast, Bax and Bak overexpression-coupled increases in the ER Ca2+ load promote Ca2+ transfer from the ER to mitochondria, and thus favor apoptosis.13, 18 Consistent with these data, Bax and/or Bak-deficient murine cells are found to exhibit lower ER Ca2+ load and relative resistance to apoptosis induced by certain agents.18 Bax and Bak double knockout (DKO) cells albeit proficient in transferring Ca2+ to mitochondria appear to exhibit low ER Ca2+ contents.19 It is noteworthy that although Bcl-2 is believed to protect from apoptosis by decreasing the ER Ca2+ load and Bax/Bak to promote apoptosis by increasing the ER Ca2+ load, excessive ER Ca2+ pool depletion also proves detrimental. For example, mutual interactions between cytochrome c and inositol1, 4, 5 triphosphate receptor (InsP3R) have been reported that are believed to amplify the apoptotic signals.20 InsP3R is an ER membrane protein responsible for promoting ER Ca2+ release into cytosol. It has been proposed that a feed-forward mechanism exists such that early in apoptosis cytochrome c and InsP3R interactions promote further releases of Ca2+ from the ER into cytosol and subsequent excessive release of cytochrome c from mitochondria, thereby further amplifying the apoptotic effects.20
PUMA is a newer member of the Bcl-2 family and, thus, very little is known about its role in apoptosis that is induced in response to alterations in intracellular Ca2+ homeostasis. TG is a sesqueterpene lactone that perturbs calcium (Ca2+) homeostasis by inhibiting the ER Ca2+ ATPases.21 Inhibition of the ER Ca2+ ATPases following TG treatment interrupts the re-uptake of cytosolic Ca2+ into the ER, and thereby promotes increased levels of Ca2+ in cytoplasm and depletion of Ca2+ within the ER.21 It is now well documented that perturbations in intracellular Ca2+ homeostasis due to alterations in the ER Ca2+ contents and Ca2+ movement between the ER and mitochondria profoundly affect cellular sensitivity to apoptotic stimuli.15, 20, 22, 23 In fact, excessive ER Ca2+ pool depletion mediated by TG is believed to induce ‘ER stress’ that triggers apoptosis.
We have recently reported that TG activates the death receptor 5 (DR5) pathway by increasing DR5 expression, and that DR5 and mitochondrial pathways crosstalk via TG-induced Bid cleavage.24 We also used Bax-proficient and -deficient HCT116 human colon cancer cells, and found that Bax deficiency confers resistance against TG-induced apoptosis.25 We further noted that although Bax was not absolutely required for DR5-dependent signals, it was nevertheless a key molecule in TG-regulated mitochondrial events.25 Recent evidence implicates Bax to be a critical mediator of PUMA-dependent apoptosis.11, 26 For example, PUMA has been reported to promote Bax translocation from cytosol towards mitochondria, where Bax affects mitochondrial permeability transition and consequent cytochrome c release.26 Furthermore, overexpression of exogenous PUMA does not induce apoptosis in Bax-deficient cells.11 Together these findings implicate PUMA to reside upstream of Bax and that Bax could be a key downstream effector of PUMA-mediated apoptotic signals. In view of our recent findings that Bax plays an important role during TG-induced apoptosis25 and the finding that PUMA and Bax appear to be intimately linked,11, 26 we undertook this study to explore the molecular basis of mutual interactions between Bax and PUMA during TG-mediated apoptosis.
Results
We utilized Bax/PUMA-proficient, Bax-deficient and PUMA-deficient HCT116 human colon cancer cells11, 27 to investigate TG-mediated apoptosis and as shown in Figure 1, PUMA-deficient or Bax-deficient cells are more resistant to undergo TG-induced apoptosis than the Bax/PUMA-proficient counterparts. The extent of resistance to TG-induced apoptosis displayed by PUMA-deficient cells is somewhat similar to that noted for Bax-deficient cells (Figure 1; He et al.25). Next, we investigated TG regulation of PUMA and Bax, and as shown in Figure 2, TG upregulates PUMA protein levels in both Bax-proficient and -deficient cells, but the effect is more pronounced in Bax-deficient cells. TG on the other hand does not affect Bax levels in either Bax and PUMA-proficient or PUMA-deficient cells. As expected, PUMA-deficient and Bax-deficient cells do not exhibit PUMA and Bax expression, respectively (Figure 2) (Bax/PUMA-proficient refers to cells with Bax and PUMA intact; Bax-deficient cells have PUMA intact whereas PUMA-deficient cells have Bax intact). Together, these results suggest that (i) Bax or PUMA deficiency confers resistance to TG; (ii) TG upregulates PUMA expression but does not affect Bax levels and (iii) although Bax-deficient cells exhibit resistance to TG, Bax deficiency does not prevent TG upregulation of PUMA expression.

Thapsigargin (TG)-induced apoptotic effects in Bax/PUMA-proficient and Bax-deficient and PUMA-deficient cells. Bax/PUMA-proficient or -deficient HCT116 cells were treated with 100 nM TG for approximately 24 h and processed for apoptosis detection as we have reported previously.23, 24 The values represent mean±S.E.M. of three independent experiments

Western blot analyses to show the effects of thapsigargin (TG) on PUMA and Bax protein levels. Bax and PUMA-proficient, Bax-deficient and PUMA-deficient cells were either left untreated or treated with 100 nM TG for approximately 24 h and then harvested for Western blot analysis. Western blotting was performed as described in Materials and Methods and same blot was sequentially probed with anti-PUMA and anti-Bax antibodies. The same blot was also probed with anti-β-actin antibody to ensure comparable loading
As mentioned above, we have previously shown that Bax deficiency conferring resistance to TG also affects TG regulation of intracellular apoptotic molecules.25 Since PUMA-deficient cells exhibit resistance to TG-induced apoptosis, we therefore investigated the effect of PUMA deficiency on TG regulation of extrinsic and intrinsic pathways of apoptosis. To this end, we used PUMA-deficient and proficient cells and the results shown in Figure 3 indicate that TG clearly induces caspases 8, 3 and 9 activation as well as Bid cleavage in PUMA-proficient cells (Figure 3a, lane 2), but its effect on these molecules is less pronounced in PUMA-deficient cells (Figure 3a, lane 4). The release of cytochrome c from mitochondria into cytosol is an important step in the activation of intrinsic pathway of apoptosis (reviewed in Wang28). Next, we investigated the effect of TG on the release of cytochrome c from mitochondria into cytosol in PUMA-proficient and -deficient cells. The cytosolic fractions from untreated and TG-treated PUMA-proficient and -deficient cells were prepared and analyzed and as shown in Figure 3b, TG clearly induces the release of cytochrome c into cytosol in PUMA-proficient cells, but its effect is significantly blunted in PUMA-deficient cells. Overall, the TG effects on PUMA-deficient cells are similar to those obtained for Bax-deficient cells except that Bax-deficient cells exhibit complete abrogation of cytochrome c release into cytosol.25 Together these results coupled with our findings that TG upregulates PUMA expression even in Bax-deficient cells (Figure 2) would support the notion that TG modulates mitochondrial apoptotic events via PUMA and Bax, and that PUMA may reside upstream of Bax in this pathway.

(a) Western blot analyses to show the effects of thapsigargin (TG) on intracellular apoptotic molecules in PUMA-proficient and PUMA-deficient cells. Cells were either left untreated or treated with 100 nM TG for approximately 24 h and then harvested for Western blot analyses as described in Materials and Methods. Same blot was sequentially probed with anticaspases-8, -9, -3, anti-Bid and anti-β-actin antibodies. (b) To analyze the effect of TG on cytochrome c release from mitochondria into cytosol, PUMA-proficient and -deficient cells were either left untreated or treated with 100 nM TG for approximately 24 h and then harvested to prepare cytosolic fractions as described in Materials and Methods. Western blot analyses were performed using anticytochrome c antibodies. The same blot was sequentially probed with anticytochrome c and anti-β-actin antibodies
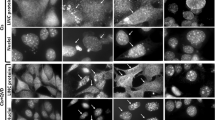
PUMA has been reported to promote Bax translocation from cytosol towards mitochondria where Bax affects mitochondrial permeability transition and consequent cytochrome c release.26 Although our results implicate PUMA to reside upstream of Bax during TG-induced apoptosis, the effect of TG on intracellular Bax translocation is not known. Next, we sought to investigate how TG would affect intracellular Bax distribution. To this end, we transiently transfected MCF-7 cells with pEGFP-Bax expression vector that expresses GFP-tagged Bax. Following overnight expression of GFP-tagged Bax, a majority of the cells exhibited a diffuse-type distribution pattern for Bax (Figure 4a, panel 3, arrowheads) while some cells also displayed a punctate distribution pattern (Figure 4a, panel 3, arrow). These GFP-Bax distribution patterns were consistent with those previously reported for Bax using same vector.29 Cells transfected with GFP-only vector displayed only diffuse staining pattern different from that for GFP-Bax, which further highlights the staining specificity for Bax (Figure 4a, panel 1). Coexpression of GFP-Bax with RFP-mito showed that the punctate pattern of Bax distribution colocalized with the mitochondrial staining (data not shown). To test the effect of TG, cells transfected with pEGFP-Bax or pEGFP constructs were treated with TG and GFP-Bax distribution was evaluated. TG treatment significantly decreased the number of cells exhibiting diffuse GFP-Bax staining pattern as a function of time (Figure 4b) while the number of cells displaying punctate GFP-Bax pattern was increased (Figure 4a, panel 4, arrows), indicating that TG significantly enhanced Bax distribution towards mitochondria. TG did not affect the staining pattern of GFP-only transfected cells, which further highlights the specificity of TG effect on Bax distribution (Figure 4a, panel 2).

Thapsigargin (TG) effect on intracellular Bax distribution. (a) MCF-7 cells were transiently transfected with pEGFP alone or pEGFP-Bax construct using Mirus TransIT-LT1 transfection reagent, and then were left untreated or treated with 100 nM TG. After 24 h incubation, cells were washed three times with PBS, fixed with 4% paraformaldehyde, and stained with DAPI. Cells were analyzed using Olympus AX 70 fluorescent microscope and MagnaFire SP model S99810 digital camera was used to take photomicrographs. Arrowheads show diffuse distribution of GFP-Bax while arrows point to punctate distribution corresponding to mitochondrial localization. (b) MCF-7 cells were transiently transfected with pEGFP-Bax construct or pEGFP vector alone and then were either left untreated or treated with 100 nM TG for 6, 12 or 24 h. Green fluorescent cells were readily visible and counted using a fluorescent microscope. For each time point, approximately 500–1600 cells were randomly counted to determine the percentage of cells exhibiting diffused distribution of Bax. The values represent mean±S.E.M. of three independent experiments
TG enhances the accumulation of PUMA protein levels (Figure 2) but the mechanism is not known. TG could enhance PUMA protein levels via upregulation of PUMA gene expression and/or via alterations in translational/post-translational controls. To investigate whether TG upregulates PUMA gene expression, we performed Northern blot analyses. Results shown in Figure 5 clearly indicate that TG upregulates PUMA mRNA levels in both Bax-proficient and -deficient cells and consistent with results noted at the protein levels, the effect is more pronounced in Bax-deficient cells (Figure 2). We also investigated TG regulation of PUMA in other cell lines including MCF-7, and H1299 human breast and lung cancer cells respectively and as shown in Figure 6a and b, TG also upregulates PUMA mRNA as well as protein levels in these cell lines. MCF-7 cells harbor wild-type p53 whereas H1299 are p53-null cells. The finding that TG upregulates PUMA levels in these cell lines would suggest that TG regulates PUMA expression in a p53-independent manner. To further confirm that TG upregulates PUMA in a p53-independent manner, we utilized p53 wild type and p53-null HCT116 isogenic cells that differ only with respect to p53. As shown in Figure 6c and d, TG also upregulates PUMA mRNA and protein levels in p53 wild type and null HCT116 isogenic cells which further confirm that TG upregulates PUMA expression in a p53-indepenent manner. Etoposide is known to upregulate PUMA expression in a p53-dependent manner and the fact that it upregulated PUMA expression only in p53 wild-type cells but not in p53 null cells (Figure 6c) further highlights the specificity of TG effect on PUMA regulation.

A Northern blot showing TG regulation of PUMA mRNA in Bax-proficient and -deficient HCT116 cells. Cells were either left untreated or treated with the indicated concentrations of TG for approximately 24 h. RNA extraction and Northern analysis were performed by standard procedures as described in Materials and Methods using a PCR amplified human PUMA cDNA fragment as a probe; ethidium bromide staining of the gel (prior to transfer) shows RNA integrity and comparable loading

Thapsigargin (TG) upregulates PUMA expression in p53 wild type and null cells. (a) A representative Northern blot showing TG regulation of PUMA mRNA in p53 wild-type MCF-7 and p53 null H1299 cells. Cells were either not treated or treated with 100 nM TG for 24 h and Northern blotting was performed as described in Materials and Methods; ethidium bromide staining of the gel (prior to transfer) shows RNA integrity and comparable loading. (b) A representative Western blot showing TG regulation of PUMA protein expression in MCF-7 and H1299 cells. Cells were treated with 100 nM TG for 24 h and Western blotting was performed with anti-PUMA antibody. Same blot was also probed with anti-β actin antibody to show comparable loading in each lane. (c) Thapsigargin (TG) upregulates PUMA expression in p53 wild type and null HCT116 isogenic cells. Isogenic p53+/+ and p53−/− HCT116 cells were either not treated or treated with TG at 50, 75, or 100 nM, for approximately 24 h. Cells were also treated with 30 μM etoposide (ETOP) for 24 h. Northern blot analysis was performed using a human PUMA cDNA probe. Ethidium bromide staining of the gel shows RNA integrity and comparable loading. (d) A representative Western blot showing TG regulation of PUMA protein levels in isogenic p53+/+ and p53−/− HCT116 cells. Cells were either not treated or treated with TG 100 nM for 24 h and Western blotting was performed using an anti-PUMA antibody as described in Materials and Methods. The same blot was also probed with anti-β actin antibody to show comparable loading in each lane
p73 is a p53 homolog that has been reported to regulate a number of p53 target genes. p73 has also been reported to upregulate PUMA expression,26 and thus it was possible that in p53-null cells TG could promote PUMA upregulation via p73. To rule in or rule out this possibility, we utilized p53-null Saos-2 cells, which are also believed to lack p73.26 Saos-2 cells were treated with TG and Northern blot analyses were performed to evaluate the effect on PUMA expression. As shown in Figure 7a, TG also upregulates PUMA expression in Saos-2 cells that lack both p53 and p73, a finding that confirms that TG-mediated PUMA upregulation occurs in a p53 and p73-independent manner.

(a) TG upregulates PUMA expression in p53 and p73 negative Saos-2 cells. Cells were either not treated or treated with TG at 1 or 2 μM for 24 h. Northern blot analysis was performed using human PUMA cDNA probe. Ethidium bromide staining of the gel shows RNA integrity and comparable loading in each lane. (b) Ionomycin upregulates PUMA expression. MCF-7 cells were either left untreated or treated with ionomycin at 1, 5, or 10 μM for 24 h. Northern blot analysis was performed using human PUMA cDNA probe; ethidium bromide staining of the gel shows RNA integrity and comparable loading in each lane
TG is known to increase intracellular Ca2+ levels by depleting the ER Ca2+ pools. Ionomycin on the other hand is believed to promote increases in intracellular Ca2+ levels via a different mechanism. We therefore sought to investigate whether ionomycin an agent different from TG affecting intracellular Ca2+ homeostasis would also regulate PUMA expression. Results shown in Figure 7b indicate that indeed ionomycin also upregulates PUMA expression. Taken together our results indicate that PUMA upregulation is not restricted only to TG but is a general cellular response to alterations in intracellular Ca2+ homeostasis.
Given that TG upregulates PUMA mRNA levels, it is possible that TG may increase PUMA gene transcription or modulate PUMA mRNA stability or both. To gain further insight into the mechanism of TG regulation of PUMA mRNA levels, we sought to investigate TG regulation of PUMA promoter expression. To this end, we sequenced the 2104 bp 5′-flanking region of PUMA gene promoter and as illustrated in Figure 8a, the 2104 bp 5′-flanking region corresponding to PUMA promoter harbors five NFAT and two CREB (cAMP response element-binding protein) binding sites. The transcription factors NFAT and CREB are known to be activated via calmodulin (CaM)-dependent signaling pathways in response to increases in the intracellular Ca2+ levels.30, 31 We therefore reasoned that TG could transcriptionally upregulate PUMA expression and that TG-mediated increases in the intracellular Ca2+ levels might regulate PUMA gene expression via activation of these transcription factors. To further explore this issue, we investigated the TG effect on PUMA gene promoter activity. We utilized a reporter construct carrying the 2104 bp PUMA promoter sequence placed upstream of a promoter-less luciferase gene hereafter denoted as pGL-Full. We also generated various deletions of the PUMA promoter region as schematically illustrated in Figure 8b. Next, we used p53-negative DU145 prostate cancer cells to transiently transfect with PUMA promoter luciferase reporter constructs and evaluated TG effect on PUMA promoter-luciferase activity. Results shown in Figure 8c indicate that TG clearly increases PUMA promoter-luciferase activity from pGL-Full construct. Deletion of the distal 1094 nucleotides removing four NFAT and one CREB binding sites (pGL-1010) does not affect TG regulation of PUMA promoter activity (Figure 8c). Removal of the remaining CREB and NFAT sites (pGL-697) only partly reduces TG responsiveness but does not completely abolish TG enhancement of PUMA promoter regulation (Figure 8c). These results thus indicate that TG transcriptionally upregulates PUMA expression and may involve NFAT and/or CREB-dependent and -independent mechanisms.


(a) Nucleotide sequence of the 5′-flanking region corresponding to PUMA promoter. Boxed nucleotides are potential transcription factor binding sites predicted by blast search of TRANSFAC 4.0 database. p53 binding site has been reported by Yu et al.2 and Han et al.3 Underlined nucleotides point to exon 1b as reported by Nakano et al.1 (b) Schematic illustration of PUMA promoter and its deletions used in this study. (c) TG increases PUMA promoter activity. p53-negative DU145 human prostate cancer cells were transiently transfected with indicated PUMA promoter luciferase constructs and were either not treated or treated with 50 nM TG. Cells were harvested approximately 24 h after TG treatment and luciferase assays were performed by standard procedures as described in Materials and Methods. The values are expressed as means±S.E.M of three independent experiments
Discussion
Recently, PUMA has garnered significant interests as a key mediator of DNA damage and p53-induced apoptosis.9, 10, 11 Overexpression of exogenous PUMA in Bax-deficient cells does not induce apoptosis, a finding that has led to the proposal that engagement of Bax is an obligatory step in PUMA-mediated apoptosis and that PUMA appears to reside upstream of Bax.11 PUMA has been reported to promote Bax translocation from cytosol towards mitochondria where Bax affects mitochondrial permeability transition and consequent cytochrome c release.26 These findings also implicate PUMA to reside upstream of Bax. Our current results indicate that PUMA appears to also play an important role in TG-mediated Ca2+ pool depletion-induced apoptosis. For instance, PUMA-deficient cells are less sensitive to TG-induced apoptosis than the PUMA-proficient counterparts, and that the extent of TG-resistance in PUMA-deficient cells is similar to that noted for Bax-deficient cells.25 Our results also indicate that TG upregulates PUMA expression in both Bax-proficient and -deficient cells and although Bax-deficient cells exhibit resistance to TG, Bax deficiency does not attenuate TG upregulation of PUMA expression. In fact, Bax-deficient cells exhibit a stronger TG-induced upregulation of PUMA than the Bax-proficient counterparts. It is possible that cells by mounting a stronger upregulation of PUMA may attempt to compensate for Bax deficiency in response to TG-induced apoptosis. Thus, mutual interactions between Bax and PUMA seem to also exist during TG-induced apoptosis. Taken together, our results indicate that Bax and PUMA appear to exist in the same apoptotic pathway activated by TG and support the notion that PUMA seems to reside upstream of Bax in this pathway.
Our present results demonstrate that TG upregulates PUMA expression in a p53 and p73-independent manner. Our results further indicate that TG upregulates PUMA expression, at least in part, at the transcriptional level. PUMA promoter harbors several NFAT and CREB binding sites. The transcription factors NFAT and CREB are known to be activated in response to increases in the intracellular Ca2+ levels.30, 31 However, the finding that the removal of all NFAT and CREB sites from the PUMA promoter (pGL-697) only partly reduces TG responsiveness but does not completely abolish TG enhancement of PUMA promoter activity (Figure 8b and c) indicates that TG transcriptional upregulation of PUMA expression may involve NFAT and/or CREB-dependent and -independent mechanisms.
Transcriptional upregulation of the mammalian glucose-regulated protein (grp) genes including grp78, grp94 and ERp72 has also been reported to occur following TG-induced depletion of intracellular ER Ca2+ stores.32 The promoters of these genes have been found to harbor unique cis-acting elements named ERSEs (ER stress response elements) that confer TG regulation.32 ERSE is a tripartite conserved sequence CCAAT(N9)CCACG (where N is GC rich 9 bp region).32 A second cis-acting element carrying the nucleotide sequence ATTGG-N-CCACG has also been identified.33 Named ERSE-II, this element is present in the promoter of human Herp gene and is also responsive to TG.33 Sequence analysis of the TG-regulated minimal PUMA promoter does not identify any ERSE-like elements, indicating that TG may also regulate PUMA promoter activity via a novel mechanism.
A wealth of information now suggests that defects in apoptosis regulation confer upon cancer cells a growth advantage that appears to underlie malignant progression (reviewed in Johnstone et al.,34 Nicholson,35 and Reed36). A better understanding of the molecular mechanisms via which tumor cells acquire roadblocks to apoptosis would likely facilitate the development of better anticancer strategies. In this context, our current results show that PUMA, an important apoptotic molecule, is activated in response to TG, an agent that perturbs intracellular Ca2+ homeostasis. TG, transcriptionally upregulates PUMA expression and engages PUMA and Bax for full transduction of apoptotic signals. Furthermore, both Bax and PUMA appear to exist in the same TG-activated apoptotic pathway in which PUMA seems to resides upstream of Bax.
Materials and Methods
Cell lines, culture conditions and apoptosis analysis
MCF-7, the human breast cancer cells, H1299, the human lung cancer cells, and Saos-2, the human osteosarcoma cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Cellgro, Meidatech, Herndon, VA, USA) supplemented with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA, USA). Human colon cancer cells, HCT116(Bax+/−), HCT116(Bax−/−), and HCT116(PUMA−/−) (kindly provided by Dr. Bert Vogelstein, Johns Hopkins University, Baltimore, MD, USA) were maintained in MoCoy's 5A medium (Cellgro, Meidatech, Herndon, VA, USA) supplemented with 10% fetal bovine serum. For apoptosis analyses, logarithmically growing cells were treated with TG for approximately 24 h, then floating and adherent cells that exhibited morphologic features of apoptosis were detected and quantitated as previously reported.24, 25
Northern and Western blot analyses
RNA extraction and Northern blot analyses were performed as we have previously described.24, 37, 38 Briefly, logarithmically growing cells were either not treated or treated with TG or ionomycin for approximately 24 h and then harvested. Total RNA was extracted with Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA), according to the manufacturer's instructions. For each sample, 20 μg of total RNA were fractionated on 1.2% agrose gel, and transferred onto Nytran SuperCharge membranes (Schleicher & Schuell, Keene, NH, USA). RNA was crosslinked with membranes via UV irradiation at 1200 J/m2. Prehybridizations and hybridizations were performed in QuikHyb Solution (Stratagene, La Jolla, CA, USA) at 65°C. PUMA mRNA was detected using a cDNA probe specific for exon 4 of human PUMA gene, which was generated by a pair of primers: sense primer 5′ ATCAATCCCATTGCATAGGTTTAG 3′ and antisense primer 5′ ACTAAGGCTGGGGCGGCTTC 3′. The probe was labeled with 32P by a random primer method using Prime-It RmT Random Primer Labeling Kit (Stratagene, La Jolla, CA, USA). The 32P-labeled probe was further purified by using Centricon filter cartridges (Millipore, Bedford, MA, USA). After hybridization, membranes were washed with appropriate buffer and the signals were visualized using autoradiographic films (Marsh Bio Products, Rochester, NY, USA). Western blot analyses were performed according to the standard procedures as we previously described.24, 37 Primary antibodies, anti-PUMA raised against a synthetic peptide PLPRGHRAPEMEPN corresponding to the C-terminal end of PUMA (AXXORA LLC, San Diego, CA, USA), Bax (Santa Cruz, CA, USA), Bid (R&D Systems, Minneapolis, MN, USA), procaspase 9 (Stressgen Biotechnologies, Victoria, BC, Canada), procaspase 8 (Stressgen Biotechnologies), procaspase 3 (Transduction Laboratory, Lexington, KY, USA), cytochrome c (PharMingen), and β-actin (Sigma Chemical, St. Louis, MO, USA) were used in detection of these proteins.
Preparation of cytosolic fractions
Cytosolic fractions were prepared as we have previously reported.39 Briefly, PUMA proficient and deficient cells were either not treated or treated with TG for 24 h. After the treatment, cells were harvested by centrifugation at 1000 r.p.m. (GLC-2B centrifuge, SORVALL) for 5 min and washed with 1 × cold phospate-buffered saline (PBS) once. Then cells were resuspended in 400 μl buffer A (20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 250 mM sucrose, 1 mM sodium EGTA, 1 mM dithiothreitol, 0.4 mM phenylmethylsulfonylfluoride, 10 μg/ml aprotinin) and homogenized with 40 strokes of a Dounce Homogenizer using a B-type pestle. Cell homogenates were first centrifuged at 750 × g for approximately 15 min at 4°C, and then the recovered supernatants were centrifuged at 10 000 × g for approximately 15 min at 4°C. The resulting supernatants were centrifuged one more time at 100 000 × g for 1 h at 4°C. After this step of centrifugation, the supernatants were recovered and analyzed for cytochrome c by Western blotting.
Detection of TG induced Bax translocation
MCF-7 human breast cancer cells were transiently transfected with pEGFP-Bax29 or pEGFP contructs using transfection reagent TransIT-LT1 (Mirus, Madison, WI, USA) as per the manufacturer's protocol. After overnight transfection, cells were washed with PBS once, and cultured with or without 100 nM TG. Cells were then washed three times with PBS and fixed with 4% paraformaldehyde at various time points. Fixed cells were further incubated with DAPI to stain the nuclei. Cells were analyzed using Olympus AX 70 fluorescent microscope and MagnaFire SP model S99810 digital camera was used to capture photomicrographs. To determine the percentage of diffuse cells, 500–1600 GFP-Bax expressing cells were randomly counted under microscope. The values represent mean±s.e.m. of three independent experiments.
Generation of PUMA promoter deletion constructs
PUMA promoter luciferase construct, pGL-Full was kindly provide by Dr. Thomas Chittenden (ImmunoGen Inc., Cambridge, MA, USA) and contains a 2 kb 5′-flanking region of human PUMA gene placed upstream of the promoterless luciferase gene in pGL3Luc-Basic luciferase reporter vector (Promega, Madison, WI, USA). To generate pGL-Δ1132, we digested the pGL-Full plasmid with SmaI restriction enzyme, which is specific to the 2 kb PUMA promoter region, and obtained a PUMA promoter deletion construct lacking 1132 nucleotides corresponding to 3′-end of the 2 kb full-length PUMA promote region. The pGL-1010 PUMA promoter deletion construct was also generated by digestion of the pGL-Full plasmid with SpeI and EcoRI restriction enzymes, which are specific to the multi-cloning site of pGL3Luc-Basic luciferase reporter vector and the 2 kb PUMA promoter region, respectively. The pGL-1010 deletion construct lacks 1094 nucleotides corresponding to the 5′-end of 2 kb full-length PUMA promoter and contains one NFAT and one CREB sites. The pGL-697 PUMA reporter construct was generated by site-directed mutagenesis approach using QuikChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). Briefly, 20 ng of pGL-Full and 125 ng of each primer were mixed with reaction buffer, which contains dNTPs and Pfu DNA polymerase. Reaction was performed on a thermal cycler (Mastercycler personal, Eppendorf, Hamburg, Germany) using the following cycling parameters: 95°C 1 min, 1 cycle; 95°C 50 s, 60°C 50 s, 68°C 4 min and 40 s, 18 cycles; 68°C 7 min 1 cycle. After amplification, 1 μl of the DpnI restriction enzyme (10 U/μl) was added to digest the template pGL-Full plamsids at 37°C for 1 h and 2 μl of digested reaction solution was used to transform into bacteria. Positive clones were selected in the presence of antibiotic ampicillin and confirmed by sequencing. The pGL-697 construct lacks the 1407 nucleotides that correspond to the 5′-end of the 2 kb full-length promoter. Following primers pair was used for site-directed mutagenesis: sense primer 5′-TTCACAAACAACCCTACCGCGGCTGCAGTTCTAGAGCGGCCGCT-3′, and antisense primer 5′-AGCGGCCGCTCTAGAACTGCAGCCGCGGTAGGGTTGTTTGTGAA-3′.
Transfections and Luciferase assays
DU145 human prostate cancer cells were transiently transfected with each of PUMA promoter luciferase reporter construct using transfection reagent TransIT-LT1 (Mirus, Madison, WI, USA) as per the manufacturer's protocol. Cells were either not treated or treated with 50 nM TG and approximately 24 h post-treatment harvested for luciferase assays. Luciferase assays were performed by standard procedures as we have previously described.24, 38 Briefly, harvested cells were washed with 1 × cold PBS once, then re-suspended in 50–70 μl of K3PO4 solution (100 mM, pH 7.8) and lysed by three freeze and thaw cycles. Supernatants were separated from pellets by centrifugation at 16 000 × g for 30 min and protein concentrations were measured by Bradford method. Approximately 20 μg of total protein per sample were added into 100 μl Luciferase Assay Reagent (Promega, Madison, WI, USA) and luciferase activity was measured using a luminometer (LUMAT LB 9507, Berthold Technologies, Germany).
Abbreviations
- Ca2+:
-
calcium
- CaM:
-
calmodulin
- CREB:
-
cAMP response element-binding protein
- bbc3:
-
Bcl-2 binding component 3
- DMEM:
-
Dulbecco's modified Eagle's medium
- DKO:
-
double knockout
- DR5:
-
death receptor 5
- ER:
-
endoplasmic reticulum
- ERSEs:
-
ER stress response elements
- ETOP:
-
etoposide
- InsP3R:
-
inositol (1,4,5) triphosphate receptor
- PBS:
-
phospate-buffered saline
- grp:
-
glucose-regulated protein
- PUMA:
-
p53 upregulated modulator of apoptosis
- TG:
-
thapsigargin
References
Nakano K and Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7: 683–694
Yu J, Zhang L, Hwang PM, Kinzler KW and Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 7: 673–682
Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L and Chittenden T (2001) Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 98: 11318–11323
Reimertz C, Kogel D, Rami A, Chittenden T and Prehn JH (2003) Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 162: 587–597
Futami T, Miyagishi M and Taira K (2005) Identification of a network involved in thapsigargin-induced apoptosis using a library of small interfering RNA expression vectors. J. Biol. Chem. 280: 826–831
Sheikh MS and Fornace Jr AJ (2000) Role of p53 family members in apoptosis. J. Cell Physiol. 182: 171–181
Vousden KH and Lu X (2002) Live or let die: the cell's response to p53. Nat. Rev. Cancer 2: 594–604
Hofseth LJ, Hussain SP and Harris CC (2004) p53: 25 years after its discovery. Trends Pharm. Sci. 25: 177–181
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland Jl and Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM and Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038
Yu J, Wang Z, Kinzler KW, Vogelstein B and Zhang L (2003) PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 100: 1931–1936
Kim BC, Kim HT, Mamura M, Ambudkar IS, Choi KS and Kim SJ (2002) Tumor necrosis factor induces apoptosis in hepatoma cells by increasing Ca2+ release from the endoplasmic reticulum and suppressing Bcl-2 expression. J. Biol. Chem. 277: 31381–31389
Nutt LK, Chandra J, Pataer A, Fang B, Roth JA, Swisher SG, O'Neil RG and McConkey DJ (2002) Bax-mediated Ca2+ mobilization promotes cytochrome c release during apoptosis. J. Biol. Chem. 277: 20301–20308
Sheikh MS and Huang Y (2004) TRAIL death receptors, Bcl-2 protein family, and endoplasmic reticulum calcium pool. Vitam. Horm. 67: 169–188
Foyouzi-Youssefi R, Arnaudeau S, Broner C, Kelley WL, Tschopp J, Lew DP, Demaurex N and Krause KH (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 97: 5723–5728
Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, DiVirgilio F, Pozzan T and Rizzuto R (2000) Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol. 148: 857–862
Demaurex N and Distelhorst C (2003) Cell biology. Apoptosis – the calcium connection. Science 300: 65–67
Nutt LK, Pataer A, Pahler J, Fang B, Roth JA, McConkey DJ and Swisher SG (2002) Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277: 9219–9225
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T and Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139
Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T and Snyder SH (2003) Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 5: 1051–1061
Sagara Y and Inesi G (1991) Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J. Biol. Chem. 266: 13503–13506
Pinton P, Ferrari D, Rapizzi E, DiVirgilio FD, Pozzan T and Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20: 2690–2701
Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE and Rizzuto R (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159: 613–624
He Q, Lee DI, Rong R, Yu M, Luo X, Klein M, El-Deiry WS, Huang Y, Hussain A and Sheikh MS (2002) Endoplasmic reticulum calcium pool depletion-induced apoptosis is coupled with activation of the death receptor 5 pathway. Oncogene 21: 2623–2633
He Q, Montalbano J, Corcoran C, Jin W, Huang Y and Sheikh MS (2003) Effect of Bax deficiency on death receptor 5 and mitochondrial pathways during endoplasmic reticulum calcium pool depletion-induced apoptosis. Oncogene 22: 2674–2679
Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C and Vousden KH (2004) p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J. Biol. Chem. 279: 8076–8083
Zhang L, Yu J, Park BH, Kinzler KW and Vogelstein B (2000) Role of BAX in the apoptotic response to anticancer agents. Science 290: 989–992
Wang X (2001) The expanding role of mitochondria in apoptosis. Gene Dev. 15: 2922–2933
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG and Youle RJ (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139: 1281–1292
Hogan PG, Chen L, Nardone J and Rao A (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17: 2205–2232
Hook SS and Means AR (2001) Ca2+/CaM-dependent kinases: from activation to function. Annu. Rev. Pharmocol. Toxicol. 41: 471–505
Roy B and Lee AS (1999) The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 27: 1437–1443
Kokame K, Kato H and Miyata T (2001) Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 276: 9199–9205
Johnstone RW, Ruefli AA and Lowe SW (2002) Apoptosis: a link between cancer genetics and chemotherapy. Cell 108: 153–164
Nicholson DW (2000) From bench to clinic with apoptosis-based therapeutic agents. Nature 407: 810–816
Reed JC (2002) Apoptosis-based therapies. Nat. Rev. Drug Dev. 1: 111–121
Huang Y, He Q, Rong R, Hillman MJ and Sheikh MS (2001) Sulindac sulfide-induced apoptosis involves death receptor 5 and the caspase 8-dependent pathway in human colon and prostate cancer cells. Cancer Res. 61: 6918–6924
Luo X, Huang Y and Sheikh MS (2003) Cloning and characterization of a novel gene PDRG that is differentially regulated by p53 and ultraviolet radiation. Oncogene 22: 7247–7257
He Q, Luo X, Huang Y and Sheikh MS (2004) Apo2L/TRAIL differentially modulates the apoptotic effects of sulindac and a COX-2 selective non-steroidal anti-inflammatory agent in Bax-deficient cells. Oncogene 21: 6032–6040
Acknowledgements
We thank Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD, USA) for kindly providing the Bax and PUMA-proficient and -deficient HCT116 cells, and Dr. Allen (SUNY Upstate Medical University, Syracuse, NY, USA) and Dr. Oscar Colamonici (The University of Illinios at Chicago, Chicago, IL, USA) for Saos-2 cells. We are also thankful to Dr. Chittenden (ImmunoGen, Cambridge, MA, USA) for providing the human PUMA/bbc promoter-luciferase construct, and Dr. Richard J Youle (National Institutes of Health, Bethesda, MD, USA) for providing the pEGFP-Bax construct. This work was partly supported by the NIH Grants CA86945, DK062136, DK067271 and funds from the Sinsheimer Scholar Award by the Alxandrine and Alexander Sinsheimer Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by V De Laurenzi
Rights and permissions
About this article
Cite this article
Luo, X., He, Q., Huang, Y. et al. Transcriptional upregulation of PUMA modulates endoplasmic reticulum calcium pool depletion-induced apoptosis via Bax activation. Cell Death Differ 12, 1310–1318 (2005). https://doi.org/10.1038/sj.cdd.4401659
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401659
Keywords
This article is cited by
-
A metabolic associated fatty liver disease risk variant in MBOAT7 regulates toll like receptor induced outcomes
Nature Communications (2022)
-
Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis
Cell Death & Disease (2021)
-
Non-apoptotic functions of BCL-2 family proteins
Cell Death & Differentiation (2017)
-
Overexpression of a dominant-negative mutant of SIRT1 in mouse heart causes cardiomyocyte apoptosis and early-onset heart failure
Science China Life Sciences (2014)
-
Bcl-2 family in inter-organelle modulation of calcium signaling; roles in bioenergetics and cell survival
Journal of Bioenergetics and Biomembranes (2014)
